

Background: Inorganic phosphate (Pi) buffers matrix Ca2+, but its impact on mitochondrial Ca2+ handling is often overlooked.
Results: Mitochondrial Ca2+ uptake and buffering strictly depend on anion transport rates; ATP accelerates Pi-independent Ca2+ uptake.
Conclusion: Maximal Ca2+ uniporter rate and Ca2+ buffering are anion transport limited; ATP alters influx without being hydrolyzed.
Significance: Pi transport is fundamentally important in controlling mitochondrial Ca2+ signals.
Keywords: ATP, bioenergetics, calcium, metabolism, mitochondrial transport, calcium buffer, calcium uniporter, phosphate carrier
Abstract
The large inner membrane electrochemical driving force and restricted volume of the matrix confer unique constraints on mitochondrial ion transport. Cation uptake along with anion and water movement induces swelling if not compensated by other processes. For mitochondrial Ca2+ uptake, these include activation of countertransporters (Na+/Ca2+ exchanger and Na+/H+ exchanger) coupled to the proton gradient, ultimately maintained by the proton pumps of the respiratory chain, and Ca2+ binding to matrix buffers. Inorganic phosphate (Pi) is known to affect both the Ca2+ uptake rate and the buffering reaction, but the role of anion transport in determining mitochondrial Ca2+ dynamics is poorly understood. Here we simultaneously monitor extra- and intra-mitochondrial Ca2+ and mitochondrial membrane potential (ΔΨm) to examine the effects of anion transport on mitochondrial Ca2+ flux and buffering in Pi-depleted guinea pig cardiac mitochondria. Mitochondrial Ca2+ uptake proceeded slowly in the absence of Pi but matrix free Ca2+ ([Ca2+]mito) still rose to ∼50 μm. Pi (0.001–1 mm) accelerated Ca2+ uptake but decreased [Ca2+]mito by almost 50% while restoring ΔΨm. Pi-dependent effects on Ca2+ were blocked by inhibiting the phosphate carrier. Mitochondrial Ca2+ uptake rate was also increased by vanadate (Vi), acetate, ATP, or a non-hydrolyzable ATP analog (AMP-PNP), with differential effects on matrix Ca2+ buffering and ΔΨm recovery. Interestingly, ATP or AMP-PNP prevented the effects of Pi on Ca2+ uptake. The results show that anion transport imposes an upper limit on mitochondrial Ca2+ uptake and modifies the [Ca2+]mito response in a complex manner.
Introduction
Mitochondrial Ca2+ plays a central role in regulating cellular energy supply, redox balance and cell death (1, 2) yet many questions remain about the determinants of mitochondrial Ca2+ flux, including the full complement of molecular components (2), signaling pathways affecting influx or efflux (2), and mitochondrial free Ca2+ concentrations during low and high work conditions. Accurate determination of matrix Ca2+ is particularly important in the heart, where workload continuously varies, accompanied by dynamic changes in ATP hydrolysis rate, Ca2+, and phosphate (Pi).2
Pi transport through the phosphate carrier (PiC) is fundamental for oxidative phosphorylation and affects the pH gradient across the inner membrane with the effective co-transport of protons. A less appreciated function of Pi is its obligatory role in modulating mitochondrial Ca2+ fluxes. The earliest descriptions of mitochondrial Ca2+ uptake/binding (3–5) noted that ATP, Pi, and Mg2+ could potentiate Ca2+ accumulation by mitochondria, but the mechanism by which these modulators acted was unclear. Pi has been reported to affect mitochondrial Ca2+ in several ways, including modulation of Ca2+ influx, efflux, and buffering. The conventional view is that Pi transport facilitates Ca2+ influx by dissipating the pH gradient that builds up during Ca2+ entry as the protonmotive force redistributes from ΔΨm to ΔpH upon stimulation of proton pumping (6). After entry, Pi then participates in dynamically buffering matrix Ca2+ by the formation of Ca-Pi complexes (7), to clamp mitochondrial Ca2+ ([Ca2+]mito) at a fixed level (8). This not only permits further Ca2+ uptake but also renders efflux through the mitochondrial Na+/Ca2+ exchanger (mNCE) independent of total matrix Ca2+ load (7). The complex nature and impact of Pi, nucleotides, or other anions on mitochondrial Ca2+ dynamics is incompletely understood. Because of the restricted volume of the mitochondrial matrix, compensation for the electrophoretic entry of cations is a thermodynamic requirement. The PiC is regarded as an antiporter of H2PO4−/OH−, equivalent to a symport of H2PO4−/H+ (9, 10) and co-transport of protons has been noted to be an important feature of anions that are most effective at facilitating mitochondrial Ca2+ uptake (11, 12).
In a recent study in which PiC (SLC25A3) was knocked out in a cardiac-specific manner (13), mitochondrial permeability transition pore (mPTP) activation by Ca2+ was decreased and Ca2+-induced cell death was blunted. However, in that study, some residual Pi uptake was present, and it was not determined if the maximal mitochondrial Ca2+ uniporter (MCU) rate in the absence of mPTP opening was altered. In contrast, another recent study showed no effect of knockdown of SLC25A3 on mitochondrial Ca2+ loading capacity or mPTP sensitivity but an increased cardiac hypertrophic response to pressure overload (14). Other Pi transporters also exist on the mitochondrial inner membrane. The Ca2+-activated ATP-Mg/Pi carrier, SLC25A23, exchanges MgATP for Pi (15), and, in a recent study, it was proposed that SLC25A23 interacts with MCU and another accessory protein, MICU1, to regulate mitochondrial Ca2+ uptake, as evidenced by reduced mitochondrial Ca2+ uptake after knockdown of SLC25A23 (16).
Several modes of mitochondrial Ca2+ uptake have been reported in previous work. In addition to the main MCU-mediated uptake, a rapid uptake mode (RaM), activating and inactivating during rapid increases in Ca2+ near the mitochondria has been reported (17, 18). In addition, we have noted two slow MCU modes of uptake in cardiac mitochondria that displayed different Ca2+ affinities and Ru360 sensitivities (8), which were also modulated differentially by Pi. Mode 1 had a high Ca2+ affinity and low Ru360 sensitivity, while mode 2, comprising the bulk of mitochondrial Ca2+ uptake, was inhibited by low Ru360 concentrations (8). Extramitochondrial Pi enhanced mitochondrial Ca2+ accumulation through mode 1 but resulted in almost complete buffering of Ca2+ entry through mode 2. Interestingly, we found that mPTP opening was independent of [Ca2+]mito but was dependent on total mitochondrial Ca2+ load, leading us to postulate that Ca-Pi complex accumulation was the trigger for activation of the pore.
To better understand the role of Pi in modulating mitochondrial Ca2+ dynamics, here we utilize Pi-depleted, isolated cardiac mitochondria to directly study the role of anions in mitochondrial Ca2+ uptake and buffering while monitoring changes in ΔΨm. We show that mitochondrial Ca2+ uptake occurs through both Pi-independent and Pi-dependent pathways, the latter of which could be blocked by inhibition of the PiC. Substitution of other anions led to differential effects on Ca2+ uptake and buffering, and revealed a novel action of adenine nucleoside trisphosphates in a non-hydrolytic role.
Experimental Procedures
Guinea pig heart mitochondria were isolated using a homogenization protocol on ice, as previously described (19). The homogenized heart suspension was centrifuged in the isolation solution (75 mm sucrose, 225 mm mannitol, 1 mm HEPES, and 1 mm EGTA, pH 7.4) at 480 g for 10 min at 4 °C, and the resulting supernatant was centrifuged at 7,700 × g for 10 min. The mitochondrial pellet was washed at 7,700 × g for 6 min and re-suspended in isolation solution at a concentration of ∼20 mg/ml. The protein concentration of isolated mitochondria was determined using the BCA (bicinchoninic acid assay) method.
Endogenous phosphate in the isolated cardiac mitochondria was depleted by utilizing the hexokinase reaction with 0.75 units/ml hexokinase, 1 mm glucose, 0.5 mm ADP, 1 mm MgCl2, and 5 mm glutamate/malate in 37 °C for 5 min, as previously described (20).
Mitochondria (0.5 mg) were suspended in 2 ml of a potassium-based buffer solution without phosphate (137 mm KCl, 20 μm EGTA, 20 mm HEPES, 5 mm NaCl, 1 μm CsA, and 5 mm glutamate/malate at pH 7.2). Multiple mitochondrial parameters were simultaneously monitored using a wavelength-scanning fluorometer (QuantaMaster; Photon Technology International) at room temperature. The extra-mitochondrial calcium (Ca2+out) was measured with 50 μm CaGreen-5N, hexapotasssium salt (505 nm ex.: 535 nm em.). Fura-FF-AM (20 μm fura-FF was incubated for 30 min at room temperature beforehand) was used to monitor intra-mitochondrial calcium (Ca2+mito) (340 nm/380 nm ex.: 510 nm em.). Mitochondrial membrane potential (ΔΨm) was monitored by the ratiometric method with 300 nm tetramethylrhodamine methyl ester (TMRM) (546 nm/573 nm ex.: 590 nm em.), and mitochondria swelling was monitored by 90 degree light scattering (540 nm ex.), as previously described (8, 21).
To measure mitochondrial pH, the succinimidyl ester form of carboxy-SNARF-1 (Life Technologies, Inc.) was loaded into Pi-depleted isolated mitochondria (incubating 30 μm for 30 min at room temperature), then washed twice and resuspended in sucrose-based isolation solution. During the measurement (520 nm ex.: 580/680 nm em.), 0.5 mg of mitochondria were suspended in 2 ml of KCl-based buffer solution without phosphate. The SNARF-1 signal was calibrated in buffer solutions with different pH values (pH 6–8) in the presence of 1 μm FCCP, 20 μg oligomycin, and 10 μm nigericin.
Results
Effects of Acute Pi Addition on Mitochondrial Ca2+ Dynamics
In zero Pi buffer solution, mitochondrial Ca2+ uptake from the cuvette upon addition of 35 μm Ca2+ (which increases free Ca2+ in the cuvette to 7 μm after binding to Ca2+ buffers present in the solution; Fig. 1A; 120–240 s) was slow and incomplete; nevertheless, [Ca2+]mito increased monotonically to 50 μm. Acute addition of 1 mm phosphate to the buffer (at 240 s; black trace) resulted in an immediate acceleration of Ca2+ uptake from the cuvette, but, paradoxically, a decrease in [Ca2+]mito to ∼27 μm (Fig. 1B; black trace). The Pi effect on Ca2+ uptake was concentration-dependent and approached a maximum rate at 0.1 mm phosphate. The EC50 for the Pi effect on the initial linear Ca2+ uptake rate was 0.087 mm (Fig. 1E, blue trace). The Pi effect on buffering, reflected in an increase in the ratio of bound/free matrix Ca2+ (Fig. 1D), had a similar Pi dependence, as did the rate of formation of the Ca2+-Pi complex (EC50 = 0.096 mm; Fig. 1E, green trace). Mitochondrial membrane potential (ΔΨm) was depolarized from ∼215 mV to 170 mV in the presence of Ca2+ without Pi (Fig. 1C), but when phosphate was added, the decline in [Ca2+]out was paralleled by restoration of ΔΨm to 210mV. The rate of recovery of ΔΨm was linearly related to mitochondrial Ca2+ uptake rate (Fig. 1F). Mitochondrial light scattering decreased slightly upon Pi addition, indicating low amplitude mitochondrial swelling but no activation of the mitochondrial permeability transition pore (mPTP) during the experiments (data not shown).
FIGURE 1.

Concentration-dependent effects of Pi on mitochondrial Ca2+ uptake and buffering in cardiac mitochondria. A single Ca2+ addition (35 μm total CaCl2 or ∼7 μm free calcium) to a suspension of Pi-depleted guinea pig cardiac mitochondria results in a slow and limited uptake from the cuvette (A:[Ca2+]out), while matrix Ca2+ (B:[Ca2+]mito) rises with fast and slow components. Addition of various Pi concentrations (0.001–1 mm NaH2PO4) accelerated Ca2+ uptake (A) but decreased [Ca2+]mito (B), reflecting an increase in matrix bound:free Ca2+ (D). Mitochondrial inner membrane potential (ΔΨm) depolarizes and recovers in parallel with the Ca2+ and Pi additions, respectively (C). E, concentration dependence of the Pi effect on Ca2+ uptake (blue) and buffering (green) rates, with saturation at ∼0.1 mm Pi. F, linear relationship between the rate of change of [Ca2+]out and ΔΨm measured as the first derivative of the signals for first 15 s after the Pi addition. (Data in panels A-E represent mean ± S.E. for n = 3 experiments; panel F, n = 5).
Next, we examined the Pi effect on Ca2+ uptake for different Ca2+ additions (20–40 μm) and after adding a fixed concentration of Pi (0.1 mm; Fig. 2). Recordings of [Ca2+]out (Fig. 2A), [Ca2+]mito (Fig. 2B), and ΔΨm (Fig. 2C) reveal that the Pi-dependent buffering effect takes effect above a certain threshold of matrix Ca2+ is reached. In the absence of Pi, [Ca2+]mito rises to a range of 20–80 μm for total Ca2+ additions between 20 and 40 μm (extramitochondrial Ca2+ range of 1–7 μm). When phosphate is added, [Ca2+]mito is clamped to the same steady state level (∼30 μm) for any [Ca2+]out larger than 2 μm (Fig. 2B). The degree of membrane potential depolarization was proportional to the amount of Ca2+ added to the cuvette, in accord with the size of the electrochemical gradient for Ca2+ across the inner membrane, the major determinant of Ca2+ current (Fig. 2C). Phase plane plots of [Ca2+]mito versus[Ca2+]out illustrates the effect of Pi to lower matrix Ca2+ to a similar level, despite markedly different loading histories (Fig. 2D, upper panel). In parallel, Pi transport permitted full ΔΨm repolarization after various Ca2+ loads were accommodated (Fig. 2D, lower panel).
FIGURE 2.

Pi uptake is required to clamp [Ca2+]mito to a setpoint. A, Ca2+ additions to Pi-depleted mitochondria in the range of 20–40 μm (at 150 s) were followed by 0.1 mm Pi (at 300 s) to initiate Ca2+ uptake and buffering. B, increases in [Ca2+]mito exceeding 30 μm in the absence of Pi were quickly clamped back to 30 μm upon Pi addition, regardless of total mitochondrial Ca2+ load. C, ΔΨm changes during the protocol. D, phase-plane analysis of [Ca2+]out versus [Ca2+]mito (upper) and [Ca2+]out versus ΔΨm (lower) before and after Pi addition.
Mersalyl, a thiol reagent that inhibits the mitochondrial phosphate carrier (22, 23), was used to test whether phosphate regulation of mitochondrial Ca2+ uptake required transport through PiC (Fig. 3). Mersalyl (10 μm; blue line) partially inhibited Ca2+ uptake in the absence of Pi, possibly due to a partial depolarization of ΔΨm, yet [Ca2+]mito still increased to the same plateau, but at a slightly slower rate. With PiC blocked, when 1 mm phosphate was added, depletion of cuvette [Ca2+] was not accelerated, and [Ca2+]mito buffering did not occur, confirming that PiC-mediated Pi entry into the matrix was required for both processes.
FIGURE 3.
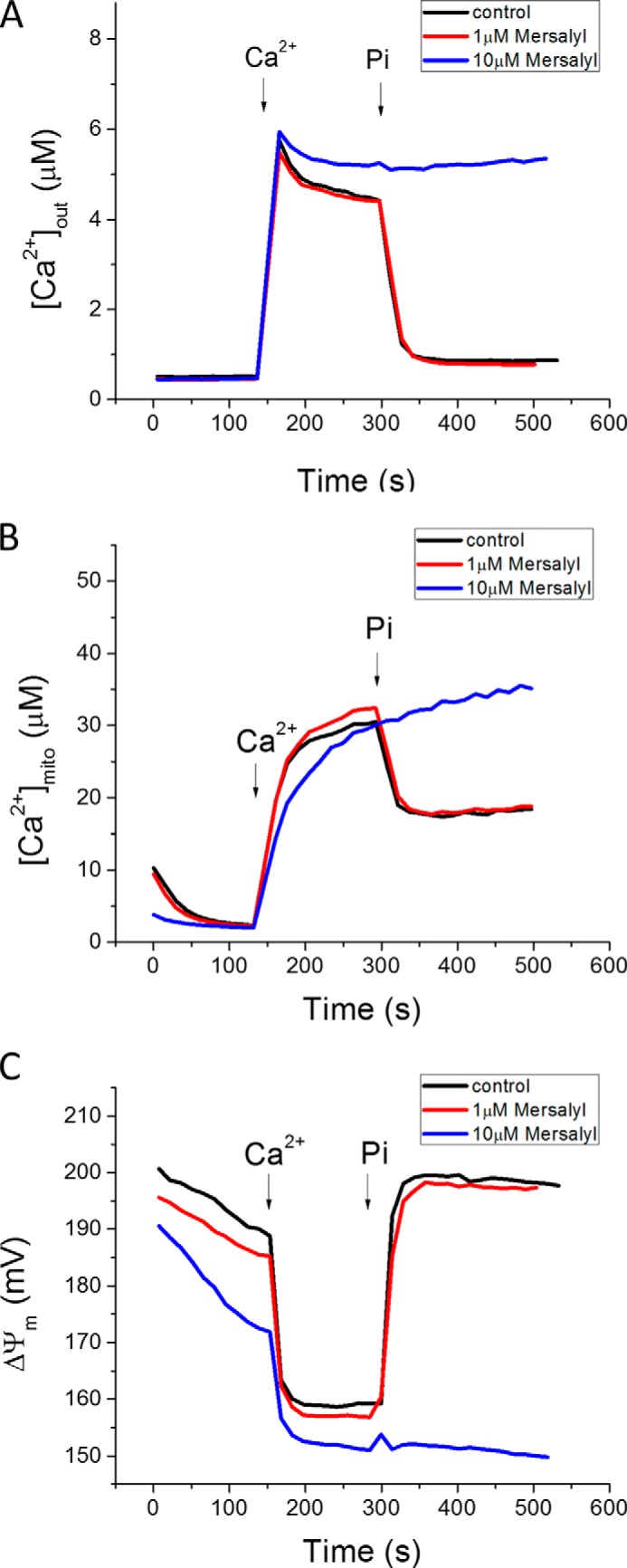
PiC inhibition blocks acceleration of Ca2+ uptake and matrix Ca2+ buffering. 10 μm (blue), but not 1 μm (red), mersalyl inhibited the Pi effect on mitochondrial Ca2+ uptake (A) and buffering (B). C, ΔΨm was partially depolarized in the presence of 10 μm mersalyl, which may account for the somewhat slower initial Pi-independent Ca2+ uptake kinetics.
As we previously reported (8), high capacity (mode 2) Ca2+ uptake is blocked by a low concentration (50 nm) of Ru360 (Fig. 4A), leaving only mode 1 uptake, detected by the rise in matrix Ca2+ (Fig. 4B). Inhibiting PiC with mersalyl (10 μm) had little effect on the rate of mode 1 Ca2+ uptake, suggesting that mode 1 uptake is Pi-independent; however, mersalyl still inhibited the effect of Pi on matrix Ca2+ buffering and ΔΨm recovery (Fig. 4C).
FIGURE 4.
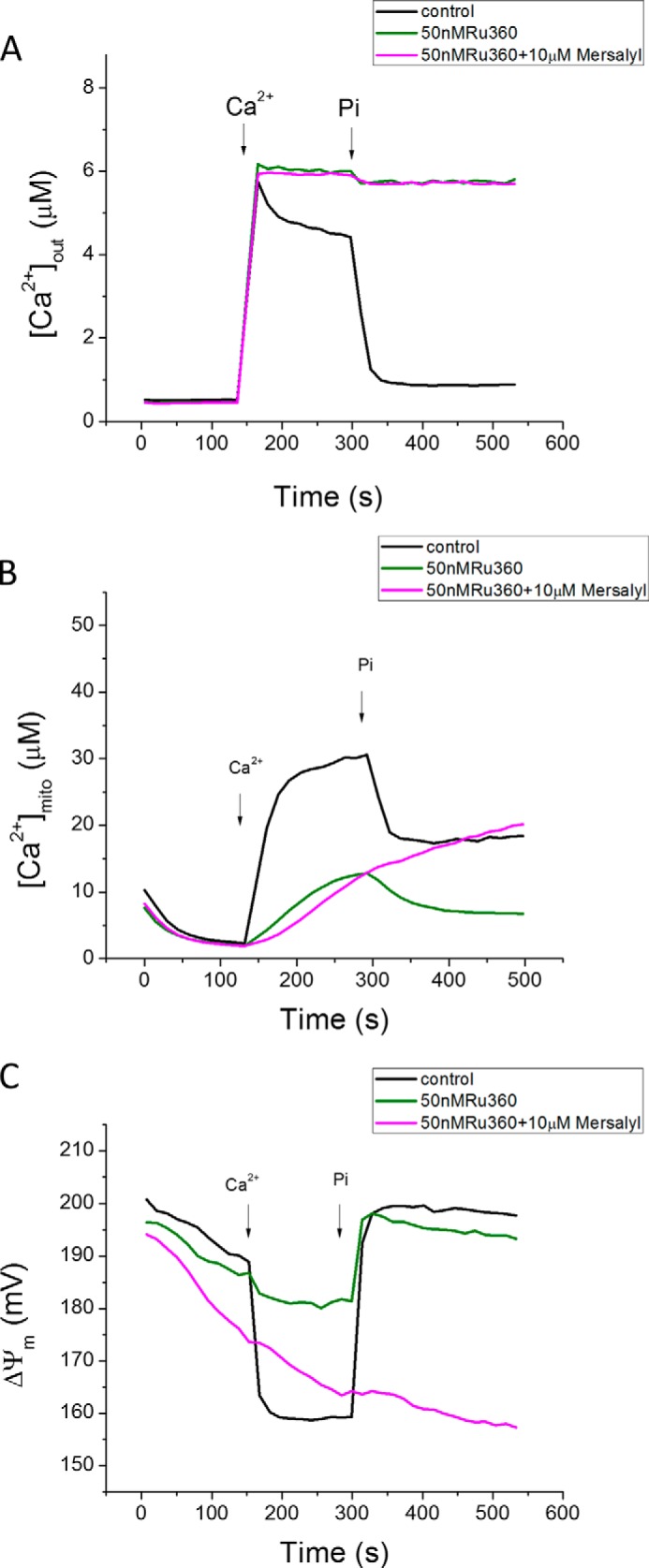
Effects of Pi and mersalyl on mitochondrial Ca2+ transport in the presence of Ru360. A, with bulk Ca2+ uptake (mode 2) inhibited with Ru360 (50 nm), remaining [Ca2+]mito increase (mode 1) was not inhibited by mersalyl, although the Pi effect on buffering was still blocked (B). C, ΔΨm responses.
Lehninger (11) previously reported that respiration-dependent Ca2+ uptake by mitochondria depends on anion transport across the inner mitochondrial membrane, supported only by permeant anions that were also capable of transporting a proton. Acetate anion is transported as the weak acid by passive diffusion with the concomitant release of a proton in the matrix. Acetate (1 mm) was capable of increasing the Ca2+ uptake rate in Pi-depleted mitochondria (Fig. 5, blue traces), albeit at a slower rate than a similar concentration of Pi, most likely due to the slow rate of acetate diffusion across the inner membrane. Increasing the acetate concentration to 30 mm accelerated the initial Ca2+ uptake rate to a level close to that of Pi (Fig. 5, red traces), with much smaller effects on matrix Ca2+ buffering. The acetate effect on Ca2+ uptake was not sustained as well as that of Pi, and was associated with slow depolarization following the initial repolarization phase. The acetate effect was not inhibited by mersalyl treatment (data not shown). Vanadate, a Pi analog, accelerated Ca2+ uptake in a manner similar to phosphate, and also strongly supported matrix Ca2+ buffering to decrease [Ca2+]mito (Fig. 5, green traces). Notably, mitochondrial Ca2+ uptake in the presence of vanadate was not accompanied by restoration of ΔΨm, probably due to inhibition of OxPhos by vanadate (24). This finding argues against the idea that the anion-dependent acceleration of Ca2+ uptake is secondary to an increase in electrochemical driving as ΔpH is converted to ΔΨm (20).
FIGURE 5.

Anions acetate, vanadate, and phosphate show different effects on mitochondrial Ca2+ dynamics. Phosphate (0.5 mm Pi, black), acetate (1, blue or 30 mm Acet., red), or vanadate (1 mm Vi, green) were added at 240 s to phosphate-depleted mitochondria after a 35 μm Ca2+ addition. [Ca2+]out (A), [Ca2+]mito (B), ΔΨm (C), Ca2+ bound-to-free ratio (D).
To explore this in more detail, we measured mitochondrial matrix pH ratiometrically (SNARF-1) using the same protocol of Ca2+ addition followed by anion addition in Pi-depleted mitochondria (Fig. 6). Prior to Ca2+ addition, baseline matrix pH was ∼7.75 with extramitochondrial pH buffered at 7.2. Ca2+ addition in the absence of Pi resulted in matrix acidification to ∼7.6. Therefore, ΔpH decreased, rather than increased, during Pi-independent Ca2+ uptake, which could have been due to displacement of protons from matrix Ca2+ binding sites or the coupling of Ca2+ entry to Na+/Ca2+ and Na+/H+ exchange activity. The addition of Pi, Vi, or acetate led to a further decrease in pH to 7.35 during the phase of ΔΨm recovery (Fig. 6). The lack of increase in ΔpH in Pi-depleted mitochondria during Ca2+ uptake, and the similar effects of the 3 anions on matrix pH despite differences in the extent of ΔΨm recovery (Fig. 5C), provides further evidence against interconversion of ΔpH to ΔΨm as the primary explanation for the acceleration of MCU rate. It should also be noted that there was still ample membrane potential to drive MCU current both in the absence and presence of Pi (−170 mV versus −190 mV). Hence, anion entry, not matrix pH change, was the primary facilitator of Ca2+ uptake.
FIGURE 6.
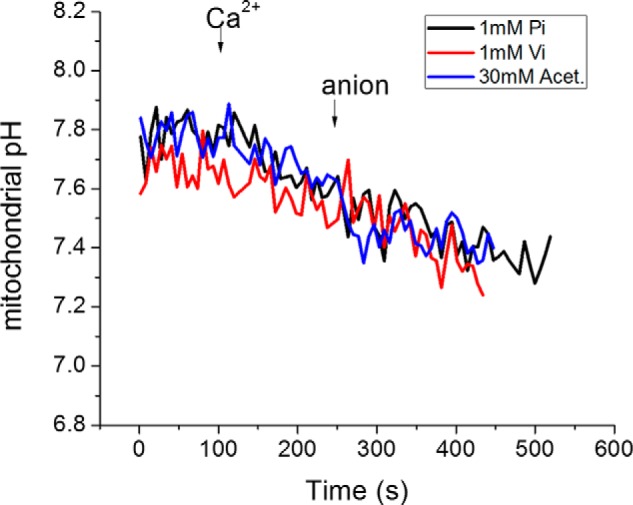
Mitochondrial pH measurement in Pi-depleted mitochondria. Mitochondrial pH was measured using the ratiometric pH indicator carboxy-SNARF-1. Mitochondrial pH decreased to a similar extent after the Ca2+ addition and after anion addition regardless which anion was used. 40 μm Ca2+ was first added, followed by 1 mm Pi (or 1 mm Vi or 30 mm acetate), as indicated.
Nucleotides represent another major physiological anion, hence we investigated their possible effects on mitochondrial Ca2+ uptake. MgATP (1 mm) accelerated mitochondrial Ca2+ uptake in the absence of Pi when added after (Fig. 7A) or prior to (Fig. 7B) the Ca2+ addition. [Ca2+]mito level was also decreased, indicating that MgATP could also buffer matrix Ca2+. Additional experiments were carried out to further investigate the mechanism of the MgATP effect on Pi-independent mitochondrial Ca2+ uptake. The addition of MgCl2 (1 mm) alone showed only a partial inhibitory effect on mitochondrial Ca2+ uptake, either in the absence or presence of Pi, and no effect on matrix buffering (Fig. 7B). Treating the mitochondria with oligomycin (Fig. 8A) did not prevent the MgATP effect, ruling out the possibility that ATP hydrolysis by the FOF1 ATPase could be providing matrix Pi. The ATP effect was also not impacted by blocking the adenine nucleotide translocase with bongkrekic acid (Fig. 8B) or carboxyatractyloside (data not shown), and ADP was incapable of accelerating the Ca2+ uptake rate (Fig. 8B). PiC inhibition also had no effect on the MgATP facilitation of Ca2+ uptake (Fig. 8C).
FIGURE 7.

MgATP accelerates mitochondrial Ca2+ uptake in Pi-depleted mitochondria. A, acute addition of either 0.5 mm Pi (black), 1 mm MgATP(red), or 1 mm Li+-AMP-PNP/MgCl2 (blue), added after Ca2+, accelerates mitochondrial Ca2+ uptake. Ca2+ (35 μm) was added at 100 s to initiate Pi-independent Ca2+ uptake (i); 1 mm Pi was added at 270 s to initiate the Pi-dependent phase (ii). B, Ca2+ uptake rate after pre-incubation with MgATP, AMP-PNP or MgCl2, added 120 s before Ca2+. Notably, Pi (1 μm) did not accelerate Ca2+ uptake when mitochondria were preincubated with MgATP or MgAMP-PNP. C, summary of the effects of MgATP, AMP-PNP or MgCl2 on Ca2+ uptake. Data in C are presented as mean ± S.E. n = 10, 8, 4, and 4 for the 4 groups shown. *: p < 0.05. The kinetics of extra-mitochondrial Ca2+ ([Ca2+]out) from panel B were fitted using exponential functions to obtain time constants during the Pi-independent and Pi-dependent Ca2+ uptake phases. MgATP and AMP-PNP accelerated Pi-independent Ca2+ uptake rates, while MgCl2 slightly prolonged Ca2+ uptake only in the presence of Pi. For phase (i), Pi-independent uptake, a double exponential was employed: [Ca2+]out(t) = Afast[exp(-t/τfast] + Aslow[exp(-t/τslow)]. Afast and Aslow are the maximum amplitude; τfast and τslow are the time constants of the fast and slow components of exponential decay, respectively. Only the slow decay time constants are plotted for different treatments. In phase (ii), the [Ca2+]out was fitted by a single exponential function [Ca2+]out(t) = A[exp(−t/τ] + C.
FIGURE 8.

ATP effects were not altered by inhibition of ANT or the mitochondrial ATP synthase. A, oligomycin (ATP synthase inhibitor) pretreatment did not alter MgATP facilitation of Pi-independent Ca2+ uptake. B, ADP did not accelerate Pi-independent Ca2+ uptake. C, bongkrekic acid (BKA; ANT inhibitor) did not alter the ATP response.
Finally, the non-hydrolyzable ATP analog, AMP-PNP, was shown to have a similar effect on mitochondrial Ca2+ dynamics as MgATP (Fig. 7), i.e. increased Ca2+ uptake rate and decreased [Ca2+]mito. These data indicate that ATP facilitates mitochondrial Ca2+ uptake through a novel direct effect of the molecule itself, presumably requiring nucleoside triphosphate uptake by a non-canonical transporter, since matrix Ca2+ buffering was also increased by ATP.
Discussion
The present study quantitatively investigated the fundamental role of Pi transport on mitochondrial Ca2+ uptake and buffering by monitoring extra- and intra-mitochondrial Ca2+ and ΔΨm. The major findings were that Pi directly facilitates mitochondrial Ca2+ uptake in a concentration-dependent manner while simultaneously buffering [Ca2+]mito to a fixed setpoint. Using Pi-depleted mitochondria, a novel regulatory mechanism involving MgATP was also revealed, whereby it increased mitochondrial Ca2+ uptake. The MgATP effect did not require ATP hydrolysis. The results demonstrate that there is a strict dependence of maximal MCU, as well as [Ca2+]mito, on anion flux rate, highlighting the regulatory complexity of cardiac mitochondrial Ca2+ dynamics.
The ability of mitochondria to accumulate massive amount of Ca2+ has been known for more than fifty years (3, 14, 25). While previous work established that Pi increases mitochondrial Ca2+ loading (11, 26) and participates in mitochondrial Ca2+ buffering to clamp matrix Ca2+ at a limiting setpoint (21, 27), the dual role of Pi has not been studied quantitatively in energized cardiac mitochondria while measuring changes in total Ca2+ uptake, matrix Ca2+, and ΔΨm in parallel. The protocol used here uniquely permitted the study of both the Pi-dependent and Pi-independent components of mitochondrial Ca2+uptake and defined the efficacy of the anion for activation of uptake versus buffering. Pi and Vi were approximately equally effective in supporting both functions, while acetate facilitated uptake but had little effect on matrix buffering.
ATP has been shown to participate in regulating mitochondrial Ca2+ accumulation. It was first reported by Vasington and Murphy (3), and DeLuca and Engstrom (25) that ATP was necessary for the uptake of Ca2+. In the former study, Pi or ATP enhanced, and respiratory inhibitors or uncouplers inhibited, “active” Ca2+ uptake, while in the latter study, it was concluded that neither Pi nor coupled respiration was required for Ca2+ uptake/binding (25). However, these studies did not take into account possible effects of Mg2+, ATP, Pi, or even substrate availability on the electrochemical driving force for Ca2+ uptake (indeed, this work took place prior to wide acceptance of the chemiosmotic hypothesis). Later, Carafoli and Lehninger showed that ATP (or ADP) is taken up along with Ca2+ and Pi to form hydroxyapatite-like precipitates in the mitochondrial matrix. Without the uptake of ATP (or ADP), no granules were formed (5). Weinbach and Von Brandt then found that the granules contained adenine nucleotides (28). The possible co-precipitation of Ca-Pi and nucleotide was, therefore, a potential explanation of why ATP accelerated mitochondrial Ca2+ uptake. Our findings show that, independent of Pi, ATP, but not ADP, can regulate mitochondrial Ca2+ uptake and buffering, and that hydrolysis is not required. The transporter mediating this effect, and whether ATP buffers by forming a high affinity complex with Ca2+, remains to be determined.
Recent studies have confirmed that MCU is a selective Ca2+ channel (29) comprised of a defined pore subunit (MCUa) (30, 31). In most studies, it is assumed that maximal MCU rates are a function of the pore's properties, regulated by accessory proteins in the Ca2+ uptake complex (e.g. MCUb (32), MICU1 and MICU2 (33–36), MCUR (37), EMRE (38)). However, our findings in intact mitochondria demonstrate that the maximal Ca2+ uptake rate is limited by the anion transport rate. This effect is masked under physiological conditions because Pi is usually at saturating concentrations with respect to effect on Ca2+ uptake (EC50 of Pi = 87.5 μm). Interestingly, the [Ca2+]mito level was buffered and reached a steady state near 20–30 μm when the phosphate concentration was > 0.01 mm (Fig. 1 B) or when [Ca2+]out level was > 2 μm (Fig. 2). This calcium-phosphate buffering effect allows estimation of the buffering threshold; however, even though the extramitochondrial phosphate concentration required for calcium buffering was ∼0.01 mm, [Pi]mito may not necessarily be equal to [Pi]out. In this regard, employing phosphorous NMR spectroscopy, Garlick et al. (39, 40) reported that mitochondrial Pi concentration was 2.5× the cytoplasmic level in intact hearts (∼6 mm). In contrast, in solution, the calcium-phosphate complex only forms when calcium is in the millimolar range (Ksp = 3 × 10−30 for Ca3(PO4)2,) (20), a Ca2+ concentration at least an order of magnitude higher than we measured either outside or inside the mitochondria.
The method described here also provides an indirect way of measuring phosphate influx since the mitochondrial Ca2+ uptake rate is proportional to the phosphate concentration and sensitive to the phosphate carrier inhibitor. By assuming 1:1 or 1:1.5 Pi:Ca transport stoichiometry (27), we could approximate the phosphate flux to be around 0.5–1 nmol/mg/sec at 1 mm [Pi], and this rate is close to the range of the reported phosphate transport rate in the rat liver mitochondria at 2 mm, which was determined by the inhibitor stop method with 32P at 0 °C (41). In keeping with the overall hypothesis that anion flux limits Ca2+ uptake, this maximum Pi transport rate corresponded to the maximum MCU rates previously reported for guinea pig cardiac mitochondria (21).
Phosphate is known to play a role in regulating mitochondrial Ca2+ and function. It has previously been reported that Pi is required for bulk mitochondrial Ca2+ uptake (42). Possible mechanisms of phosphate facilitation of Ca2+ uptake could be explained by (i) precipitation of mitochondrial Ca2+ by forming a Ca-Pi complex to maintain the electrochemical driving force for Ca2+, (ii) the requirement for co-transport of an anion via PiC with Ca2+ to maintain charge balance (supported by the acetate result), or (iii) direct interaction with Ca2+ transporters. In the current study, we confirmed that phosphate participates in clamping mitochondrial [Ca2+]mito level to maintain a Ca2+ gradient for Ca2+ uptake. Dissipation of the proton gradient by phosphate influx was unlikely to be the main reason for enhancing Ca2+ uptake, since Vi could equally increase uptake without restoring ΔΨm (Fig. 5C).
Mitochondrial matrix or extramitochondrial Pi, by themselves, were probably not direct allosteric regulators of the mitochondrial calcium uniporter because concomitant Pi influx was absolutely required to maintain fast Ca2+ uptake. The PiC has also been implicated in the regulation of mPTP opening to modulate cell death (13, 43); however, whether the mechanism is through a direct PiC-mPTP interaction or merely the result of a loss of Pi influx to regulate matrix Ca2+, or the Pi-Ca2+ complex, is unclear. To verify the importance of Pi in regulating mitochondrial Ca2+ dynamics, further testing on PiC knockdown cells or animals (13, 16) would be helpful to understand the molecular basis of the Pi-dependent process described herein.
Nucleotide effects on mitochondrial Ca2+ uptake have been reported previously (44); however, ATP or AMP-PNP were shown to inhibit the uniporter from a site on the outside of the inner membrane. Thus, the effect of ATP to increase Pi-independent Ca2+ uptake shown here has not been observed before. The nature of the ATP transporter involved remains to be determined, as we ruled out adenine nucleotide translocase or the ATP synthase as contributors to the mechanism. An ATP-Mg/Pi carrier (SLC25A23) has also been described in cardiac mitochondria, representing another possible anion influx pathway. Knockdown of this exchanger was recently shown to impact mitochondrial Ca2+ dynamics (16), but it is unclear how this ATP/Pi antiporter would be able to function in the Pi-depleted mitochondria used in the present study.
In conclusion, these findings support a major role for anion influx in determining the maximal Ca2+ influx rate and matrix Ca2+ concentration in cardiac mitochondria. Pi uptake is required for maximal enhancement of MCU rate, as well as maximal buffering capacity. ATP is not strictly required for Ca2+ uptake, but facilitates Pi-independent uptake (ADP does not substitute). The Ca-Pi buffer system clamps the free Ca2+ at an upper limit, allowing massive accumulation of Ca2+ without increasing the efflux rate or diminishing the electrochemical driving force for entry. The mitochondria are therefore able to regulate metabolism via Ca2+ sensing and also can act as a cellular Ca2+ sink when there is a chronic, potentially pathological, increase in extramitochondrial Ca2+. Notably, we show that the first role is modulated by the Pi effect on Ca2+ uptake, effectively incorporating Pi, which also rises with energy demand, into the sensing process. Considering that Pi, ATP, and Ca2+ dynamics are altered under metabolic stress, it will be important to define how the two effects of Pi contribute to the pathophysiology of cardiac disease.
This work was supported by National Institutes of Health Grants R01HL101235 and R01HL108917. The authors declare that they have no conflicts of interest with the contents of this article.
- Pi
- inorganic phosphate
- Vi
- inorganic vanadate
- mPTP
- mitochondrial permeability transition pore
- RaM
- rapid uptake mode
- MCU
- mitochondrial Ca2+ uniporter
- PiC
- phosphate carrier
- ANT
- adenine nucleotide translocase
- AMP-PNP
- 5'-adenylyl imidodiphosphate
- TMRM
- tetramethylrhodamine methyl ester.
References
- 1. Rizzuto R., De Stefani D., Raffaello A., Mammucari C. (2012) Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566–578 [DOI] [PubMed] [Google Scholar]
- 2. Liu T., O'Rourke B. (2009) Regulation of mitochondrial Ca2+ and its effects on energetics and redox balance in normal and failing heart. J. Bioenerg. Biomembr. 41, 127–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vasington F. D., Murphy J. V. (1962) Ca ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J. Biol. Chem. 237, 2670–2677 [PubMed] [Google Scholar]
- 4. Chance B. (1965) The Energy-Linked Reaction of Calcium with Mitochondria. J. Biol. Chem. 240, 2729–2748 [PubMed] [Google Scholar]
- 5. Carafoli E., Rossi C. S., Lehninger A. L. (1965) Uptake of Adenine Nucleotides by Respiring Mitochondria during Active Accumulation of Ca++ and Phosphate. J. Biol. Chem. 240, 2254–2261 [PubMed] [Google Scholar]
- 6. Nicholls D. G., Ferguson S. (2013) Bioenergetics 4th Ed., Academic Press [Google Scholar]
- 7. Balaban R. S., Bose S., French S. A., Territo P. R. (2003) Role of calcium in metabolic signaling between cardiac sarcoplasmic reticulum and mitochondria in vitro. Am. J. Physiol. Cell Physiol. 284, C285–C293 [DOI] [PubMed] [Google Scholar]
- 8. Wei A. C., Liu T., Winslow R. L., O'Rourke B. (2012) Dynamics of matrix-free Ca2+ in cardiac mitochondria: two components of Ca2+ uptake and role of phosphate buffering. J. Gen. Physiol. 139, 465–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wohlrab H. (1986) Molecular aspects of inorganic phosphate transport in mitochondria. Biochim. Biophys. Acta 853, 115–134 [DOI] [PubMed] [Google Scholar]
- 10. Krämer R. (1996) Structural and functional aspects of the phosphate carrier from mitochondria. Kidney Int. 49, 947–952 [DOI] [PubMed] [Google Scholar]
- 11. Lehninger A. L. (1974) Role of phosphate and other proton-donating anions in respiration-coupled transport of Ca2+ by mitochondria. Proc. Natl. Acad. Sci. U.S.A. 71, 1520–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zoccarato F., Nicholls D. (1982) The role of phosphate in the regulation of the independent calcium-efflux pathway of liver mitochondria. Eur. J. Biochem. 127, 333–338 [DOI] [PubMed] [Google Scholar]
- 13. Kwong J. Q., Davis J., Baines C. P., Sargent M. A., Karch J., Wang X., Huang T., Molkentin J. D. (2014) Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 21, 1209–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carafoli E. (2010) The fateful encounter of mitochondria with calcium: How did it happen? Biochim. Biophys. Acta 1797, 595–606 [DOI] [PubMed] [Google Scholar]
- 15. Fiermonte G., De Leonardis F., Todisco S., Palmieri L., Lasorsa F. M., Palmieri F. (2004) Identification of the mitochondrial ATP-Mg/Pi transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution. J. Biol. Chem. 279, 30722–30730 [DOI] [PubMed] [Google Scholar]
- 16. Hoffman N. E., Chandramoorthy H. C., Shanmughapriya S., Zhang X. Q., Vallem S., Doonan P. J., Malliankaraman K., Guo S., Rajan S., Elrod J. W., Koch W. J., Cheung J. Y., Madesh M. (2014) SLC25A23 augments mitochondrial Ca(2)(+) uptake, interacts with MCU, and induces oxidative stress-mediated cell death. Mol. Biol. Cell 25, 936–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sparagna G. C., Gunter K. K., Sheu S. S., Gunter T. E. (1995) Mitochondrial calcium uptake from physiological-type pulses of calcium. A description of the rapid uptake mode. J. Biol. Chem. 270, 27510–27515 [DOI] [PubMed] [Google Scholar]
- 18. Buntinas L., Gunter K. K., Sparagna G. C., Gunter T. E. (2001) The rapid mode of calcium uptake into heart mitochondria (RaM): comparison to RaM in liver mitochondria. Biochim. Biophys. Acta 1504, 248–261 [DOI] [PubMed] [Google Scholar]
- 19. Wei A. C., Aon M. A., O'Rourke B., Winslow R. L., Cortassa S. (2011) Mitochondrial energetics, pH regulation, and ion dynamics: a computational-experimental approach. Biophys. J. 100, 2894–2903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chalmers S., Nicholls D. G. (2003) The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J. Biol. Chem. 278, 19062–19070 [DOI] [PubMed] [Google Scholar]
- 21. Wei A. C., Liu T., Cortassa S., Winslow R. L., O'Rourke B. (2011) Mitochondrial Ca2+ influx and efflux rates in guinea pig cardiac mitochondria: low and high affinity effects of cyclosporine A. Biochim. Biophys. Acta 1813, 1373–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tyler D. D. (1969) Evidence of a phosphate-transporter system in the inner membrane of isolated mitochondria. Biochem. J. 111, 665–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Papa S., Kanduc D., Lofrumento N. E. (1973) Phosphate transport in mitochondria action of mersalyl on the binding and transport of inorganic phosphate. FEBS Lett. 36, 9–11 [DOI] [PubMed] [Google Scholar]
- 24. DeMaster E. G., Mitchell A. (1973) A comparison of arsenate and vanadate as inhibitors or uncouplers of mitochondrial and glycolytic energy metabolism. Biochemistry 12, 3616–3621 [DOI] [PubMed] [Google Scholar]
- 25. Deluca H. F., Engstrom G. W. (1961) Calcium uptake by rat kidney mitochondria. Proc. Natl. Acad. Sci. U.S.A. 47, 1744–1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Langer G. A., Nudd L. M. (1980) Addition and kinetic characterization of mitochondrial calcium in myocardial tissue culture. Am. J. Physiol. 239, H769–H774 [DOI] [PubMed] [Google Scholar]
- 27. Nicholls D. G., Chalmers S. (2004) The integration of mitochondrial calcium transport and storage. J. Bioenerg. Biomembr. 36, 277–281 [DOI] [PubMed] [Google Scholar]
- 28. Weinbach E. C., Von Brand T. (1967) Formation, isolation and composition of dense granules from mitochondria. Biochim. Biophys. Acta 148, 256–266 [DOI] [PubMed] [Google Scholar]
- 29. Kirichok Y., Krapivinsky G., Clapham D. E. (2004) The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364 [DOI] [PubMed] [Google Scholar]
- 30. Chaudhuri D., Sancak Y., Mootha V. K., Clapham D. E. (2013) MCU encodes the pore conducting mitochondrial calcium currents. Elife 2, e00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. De Stefani D., Raffaello A., Teardo E., Szabò I., Rizzuto R. (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Raffaello A., De Stefani D., Sabbadin D., Teardo E., Merli G., Picard A., Checchetto V., Moro S., Szabò I., Rizzuto R. (2013) The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 32, 2362–2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Patron M., Checchetto V., Raffaello A., Teardo E., Vecellio Reane D., Mantoan M., Granatiero V., Szabò I., De Stefani D., Rizzuto R. (2014) MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 53, 726–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Plovanich M., Bogorad R. L., Sancak Y., Kamer K. J., Strittmatter L., Li A. A., Girgis H. S., Kuchimanchi S., De Groot J., Speciner L., Taneja N., Oshea J., Koteliansky V., Mootha V. K. (2013) MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One 8, e55785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Csordás G., Golenár T., Seifert E. L., Kamer K. J., Sancak Y., Perocchi F., Moffat C., Weaver D., de la Fuente Perez S., Bogorad R., Koteliansky V., Adijanto J., Mootha V. K., Hajnóczky G. (2013) MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab. 17, 976–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baughman J. M., Perocchi F., Girgis H. S., Plovanich M., Belcher-Timme C. A., Sancak Y., Bao X. R., Strittmatter L., Goldberger O., Bogorad R. L., Koteliansky V., Mootha V. K. (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mallilankaraman K., Cárdenas C., Doonan P. J., Chandramoorthy H. C., Irrinki K. M., Golenár T., Csordás G., Madireddi P., Yang J., Müller M., Miller R., Kolesar J. E., Molgó J., Kaufman B., Hajnóczky G., Foskett J. K., Madesh M. (2012) MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 14, 1336–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sancak Y., Markhard A. L., Kitami T., Kovács-Bogdan E., Kamer K. J., Udeshi N. D., Carr S. A., Chaudhuri D., Clapham D. E., Li A. A., Calvo S. E., Goldberger O., Mootha V. K. (2013) EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342, 1379–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Garlick P. B., Soboll S., Bullock G. R. (1992) Evidence that mitochondrial phosphate is visible in 31P NMR spectra of isolated, perfused rat hearts. NMR Biomed. 5, 29–36 [DOI] [PubMed] [Google Scholar]
- 40. Garlick P. B., Brown T. R., Sullivan R. H., Ugurbil K. (1983) Observation of a second phosphate pool in the perfused heart by 31P NMR; is this the mitochondrial phosphate? J. Mol. Cell Cardiol. 15, 855–858 [DOI] [PubMed] [Google Scholar]
- 41. Paradies G., Ruggiero F. M. (1991) Effect of aging on the activity of the phosphate carrier and on the lipid composition in rat liver mitochondria. Arch. Biochem. Biophys. 284, 332–337 [DOI] [PubMed] [Google Scholar]
- 42. Harris E. J., Zaba B. (1977) The phosphate requirement for Ca2+-uptake by heart and liver mitochondria. FEBS Lett. 79, 284–290 [DOI] [PubMed] [Google Scholar]
- 43. Leung A. W., Varanyuwatana P., Halestrap A. P. (2008) The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J. Biol. Chem. 283, 26312–26323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Litsky M. L., Pfeiffer D. R. (1997) Regulation of the mitochondrial Ca2+ uniporter by external adenine nucleotides: the uniporter behaves like a gated channel which is regulated by nucleotides and divalent cations. Biochemistry 36, 7071–7080 [DOI] [PubMed] [Google Scholar]