

Background: Cellulosomal cohesin-dockerin types are reversed in Bacteroides cellulosolvens.
Results: Combined crystallographic and computational approaches of a lone cohesin yielded a structural model of the cohesin-dockerin complex that was verified experimentally.
Conclusion: The dockerin dual-binding mode is not exclusive to enzyme integration into cellulosomes; it also characterizes cell-surface attachment.
Significance: This combined approach provides a platform for generating testable hypotheses of the high affinity cohesin-dockerin interaction.
Keywords: cellulase, cellulose, cellulosome, computational biology, computer modeling, protein-protein interaction
Abstract
Cohesin-dockerin interactions orchestrate the assembly of one of nature's most elaborate multienzyme complexes, the cellulosome. Cellulosomes are produced exclusively by anaerobic microbes and mediate highly efficient hydrolysis of plant structural polysaccharides, such as cellulose and hemicellulose. In the canonical model of cellulosome assembly, type I dockerin modules of the enzymes bind to reiterated type I cohesin modules of a primary scaffoldin. Each type I dockerin contains two highly conserved cohesin-binding sites, which confer quaternary flexibility to the multienzyme complex. The scaffoldin also bears a type II dockerin that anchors the entire complex to the cell surface by binding type II cohesins of anchoring scaffoldins. In Bacteroides cellulosolvens, however, the organization of the cohesin-dockerin types is reversed, whereby type II cohesin-dockerin pairs integrate the enzymes into the primary scaffoldin, and type I modules mediate cellulosome attachment to an anchoring scaffoldin. Here, we report the crystal structure of a type I cohesin from B. cellulosolvens anchoring scaffoldin ScaB to 1.84-Å resolution. The structure resembles other type I cohesins, and the putative dockerin-binding site, centered at β-strands 3, 5, and 6, is likely to be conserved in other B. cellulosolvens type I cohesins. Combined computational modeling, mutagenesis, and affinity-based binding studies revealed similar hydrogen-bonding networks between putative Ser/Asp recognition residues in the dockerin at positions 11/12 and 45/46, suggesting that a dual-binding mode is not exclusive to the integration of enzymes into primary cellulosomes but can also characterize polycellulosome assembly and cell-surface attachment. This general approach may provide valuable structural information of the cohesin-dockerin interface, in lieu of a definitive crystal structure.
Introduction
Plant cell wall carbohydrates are among the most abundant sources of organic carbon on the planet with an estimated 1011 tons recycled annually (1). Degradation of this composite structure by an extensive consortium of microbial cellulases and hemicellulases is an important biological phenomenon of significant environmental and industrial interest.
Some anaerobic micro-organisms organize plant cell wall-degrading enzymes in a highly intricate, extracellular, and multienzyme complex termed the cellulosome. The molecular basis for the integration of proteins into the cellulosome was first described in Clostridium thermocellum (2, 3). Early studies defined two major types of cohesin-dockerin interactions, based on sequence and structural diversity of the two protein modules. Type I dockerins, located in the cellulases and hemicellulases, and reiterated type I cohesins of a macromolecular scaffold, termed CipA or primary scaffoldin, mediate the integration of the various enzymes into the complex (4). The cohesins in the primary scaffoldins carry multiple copies of cohesin modules that do not usually discriminate between different dockerin modules, and thus the composition of these complexes is generally dictated by the level to which the ∼70 catalytic components are expressed. Thus, although cellulosome-integrating primary scaffoldin, CipA, possesses only nine type I cohesins, the genome of C. thermocellum encodes over 70 dockerin-containing open reading frames. Consistent with the model based on the C. thermocellum system, in the majority of bacterial cellulosomes type I cohesin-dockerin interactions mediate assembly of enzymes into these complexes. A restricted sub-set of bacteria, such as C. thermocellum, Acetivibrio cellulolyticus, and Clostridium clariflavum (5–7), also anchor cellulosomes onto the cell surface. In these bacteria primary scaffoldins contain a divergent C-terminal type II dockerin that specifically recognizes type II cohesins of anchoring scaffoldins appended to the cell envelope of the bacterium (8, 9). Lack of cross-specificity between types I and II cohesin-dockerin pairs ensures the correct assembly of the multienzyme cellulosome complex and its attachment to the cell surface.
Bacteroides cellulosolvens, which has been recently reclassified as Pseudobacteroides cellulosolvens (10), is a mesophilic anaerobic bacterium, capable of actively degrading plant structural carbohydrates, in particular crystalline cellulose and hemicellulose (11, 12). B. cellulosolvens cellulosolvens has a particularly complex cellulosome with multicomponent scaffoldins similar to those of C. thermocellum and A. cellulolyticus (Fig. 1). However, cohesins carried by the primary and anchoring scaffoldins are of a reversed type to what has been described in C. thermocellum and A. cellulolyticus (14). Thus, the cellulosome of B. cellulosolvens comprises a primary scaffoldin ScaA that contains 11 type II cohesins, which bind cellulosomal enzymes containing type II dockerins (13). In addition, ScaA comprises a C-terminal type I dockerin that interacts with type I cohesins located in the ScaB-anchoring scaffoldin. ScaB bears 10 type I cohesins and an S-layer homology module that tethers the entire cellulosome to the bacterial cell surface (14). The functional implications of this reversed specificity in B. cellulosolvens cellulosome architecture remain unclear.
FIGURE 1.

Schematic representation of the B. cellulosolvens cellulosome. The primary scaffoldin, ScaA, binds up to 11 cellulosomal enzymes through type II cohesin dockerin interactions (in green). ScaA contains a C-terminal type I dockerin (in yellow) that binds to one of the 10 type I cohesins of anchoring scaffoldin ScaB. ScaB binds to the cell surface through a C-terminal S-layer homology (SLH) domain (in violet).
Structure-function studies on types I and II cohesin-dockerin complexes have provided insights into the mechanism of cellulosome assembly and cell-surface attachment, respectively (4, 8, 15, 16). Within the dockerin sequence, there is a tandem duplication of a 22-residue segment that displays remarkable structural conservation. Thus, the structure of the first duplicated segment, containing the N-terminal helix, is usually structurally superimposable with the C-terminal segment. This symmetry, coupled with three-dimensional crystal structures and mutagenesis studies (15, 16), indicates that C. thermocellum and Clostridium cellulolyticum type I dockerins contain two equivalent ligand-binding sites that support a dual mode of dockerin binding to cohesins. In contrast, the structure of type II cohesin-dockerin complexes indicates a single binding mode of action (8). Although it has been suggested that a dual-binding mode enhances conformational flexibility of the quaternary structure of a highly populated multienzyme complex, it remains unclear why such a property seems to be restricted to type I complexes.
Elucidation of the molecular determinants of cohesin-dockerin recognition is crucial to understand the mechanism of cellulosome assembly and cell-surface attachment. The crystal structures of several types I and II cohesins have been determined (4, 17–19). These protein modules display similar folds comprising nine β-strands that form a flattened β-barrel with a typical jelly roll topology (4, 20). Analysis of the dockerin-binding platforms suggests that subtle differences in the topology of the binding sites and a lack of sequence identity in the β-strands that include the core of the ligand-binding site explains why types I and II cohesins display distinct specificities for their target dockerins. Considering that in B. cellulosolvens the cohesin-dockerin types are reversed in comparison with most of the characterized cellulosomes, it remains unknown whether the structural determinants of specificity between types I and II cohesins are also reversed in comparison with C. thermocellum and A. cellulolyticus. This report provides insight into the mechanism of type I cohesin-dockerin interactions that mediate cellulosome attachment to the bacterial cell surface.
Experimental Procedures
Cloning and Expression
DNA encoding the seventh cohesin module of ScaB (residues 1039–1186), designated BcCohI,2 was synthesized in vitro (NZYTech Ltd., Lisboa, Portugal) with a codon usage optimized for expression in Escherichia coli. The synthesized gene contained engineered NcoI and XhoI restriction sites at the 5′ and 3′ ends, respectively. After sequence verification, the gene was subcloned into the prokaryotic expression vector pET28a (Novagen). Recombinant BcCohI contained a C-terminal His6 tag. Tuner (DE3) cells, transformed with the pET28a derivative, were grown at 37 °C to an A600 0.5, and recombinant protein expression was induced by adding 0.2 mm isopropyl β-d-thiogalactopyranoside. Bacterial cells were further incubated at 19 °C for 16 h.
CBM-Coh and Xyn-Doc fusion proteins were cloned and expressed as described previously (24). The CBM in each case included the C. thermocellum scaffoldin-borne CBM3a, and Xyn represents a PCR construct of Geobacillus stearothermophilus xylanase T-6 (25). The type I ScaA dockerin (residues 2238–2319; DocScaA) and its mutant derivatives (DocS23A/D24A, DocS60A/D61A, and DocS23A/D24A/S60A/D61A) were designed and synthesized (NZYTech Ltd.) with codon usage optimized for expression in E. coli. The genes contained engineered EcoRI and XhoI recognition sequences at the 5′ and 3′ ends, respectively, and were subcloned into pET32a vector (Novagen). DocScaA and mutant derivatives (as above) were expressed in E. coli Origami cells. Growth was performed at 37 °C to mid-exponential phase (A600 = 0.5) in Luria broth. Recombinant protein expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside and incubated for 16 h at 19 °C.
Protein Purification
BcCohI and other recombinant cohesins and dockerins produced in this study were purified by immobilized metal ion affinity chromatography (IMAC) using Sepharose columns charged with nickel (HisTrapTM, GE Healthcare, Uppsala, Sweden) following conventional protocols (26). For crystallography, BcCohI was further purified by size exclusion chromatography as follows. BcCohI was buffer-exchanged in PD-10 Sephadex G-25 M gel filtration columns (GE Healthcare) into 20 mm Na-Hepes buffer, pH 7.5, containing 200 mm NaCl and 2 mm CaCl2. The protein was loaded into a HiLoad 16/60 Superdex 75 column (GE Healthcare) at a flow rate of 1 ml min−1, previously equilibrated with 20 mm Na-Hepes buffer, pH 7.5, containing 200 mm NaCl and 2 mm CaCl2. This step enabled separation of BcCohI from eventual minor E. coli contaminants. Fractions containing the purified cohesin were then concentrated with Amicon® Ultra-15 centrifugal devices with a 10-kDa cutoff membrane (MilliporeTM) and washed three times with 0.5 mm CaCl2. The final protein concentration was measured using a NanoDrop 2000C UV-visible spectrophotometer (Thermo Scientific) with a molar extinction coefficient (ϵ) 33,350 m−1 cm−1 and was adjusted to 15 and 30 mg/ml in 0.5 mm CaCl2.
CBM-Coh fusion proteins were purified using amorphous (phosphoric acid-treated) cellulose as an affinity matrix, and His-tagged Xyn-Docs were isolated using IMAC. Precise details of the purification scheme in each case can be obtained in earlier reports (24, 25). DocScaA and its mutant derivatives were purified through IMAC as described above.
Crystallization
The crystallization conditions were screened by the sitting-drop vapor-phase diffusion method using the commercial kits Crystal Screen, Crystal Screen 2, PEG Ion Screen I and II from Hampton Research (Aliso Viejo, CA), and JCSG-plus HT96 screens (Molecular Dimensions, Suffolk, UK) by the robotic nanodrop dispensing system Oryx8 (Douglas Instruments). Drops of 0.7 μl of BcCohI concentrated to 7 or 15 mg/ml, and 0.7 μl of reservoir solution per condition were prepared at 292 K. Diffracting crystals were obtained at 15 mg/ml from three different conditions as follows: 0.05 m ammonium sulfate, 0.1 m sodium acetate, and 20% PEG 4K; 0.2 m KCl and 20% PEG 3350; and 0.2 m sodium chloride and 20% w/v PEG 3350. These crystals were cryo-cooled in liquid nitrogen, after passing through cryo-protectant solution of the crystallization buffer containing 30% glycerol.
Data Collection, Processing, and Refinement
Data were collected on beamline ID14-4 at the European Synchrotron Radiation Facility (ESRF, Grenoble, France), using a Q315r CCD detector (ADSC), with the crystals cooled at 100 K using a Cryostream (Oxford Cryosystems Ltd.). EDNA and Mosflm (27) were used for strategy calculation during data collection. All data were processed with the programs iMOSFLM (28) and AIMLESS (29) from the CCP4 suite (Collaborative Computational Project, No. 4, 1994 (30)). BcCohI crystals belong to the orthorhombic space group (P212121) with unit cell dimensions a = 30.38 Å, b = 52.77 Å, and c = 90.46 Å. Final data and structure-quality statistics are shown in Table 1, and the best crystal was diffracted to a resolution of 1.84 Å. Data collection statistics are given in Table 1. The Matthews coefficient indicated the presence of just a single cohesin module (Vm = 2.13 Å3 Da−1; solvent content ∼42%) (31). BALBES was used for carrying out molecular replacement (32). The best solution was found using the cohesin from Carvalho et al. (15) (PDB code 2ccl, sequence identity 39%). This gave a single BcCohI module in the space group P212121, with initial Rfactor/Rfree of 34.6/44.2% using REFMAC5 (33) and a Q-factor of 0.688. The output from BALBES was submitted to ARP/wARP (34) which gave a PDB model in two chains (133 residues) with a sequence coverage of 87% and an Rfactor of 32.1%. This model was further adjusted and refined using REFMAC5 (33) interspersed with model improvement in COOT (35) to give the final model (Table 1). A TLS model for grouped atom movement with one TLS group per chain was used in the final round of refinement.
TABLE 1.
X-ray data and structure quality statistics for the final refined models of BcCohI
Protein was purified by immobilized metal affinity chromatography followed by gel filtration. Crystals were grown in 0.2 m sodium chloride and 20% w/v PEG 3350 (protein concentration of 15 g/liter). The structure was solved by molecular replacement using PDB code 2ccl as a search model (15). CC½ is the half-data set correlation coefficient. Values in parentheses are for the highest resolution shells.
| Data quality | |
|---|---|
| X-ray source | ESRF, beamline ID14-4 |
| Wavelength (Å) | 0.9393 |
| Unit cell parameters | a = 30.38 Å |
| b = 52.77 Å | |
| c = 90.46 Å | |
| Space group | P212121 |
| Resolution of data (Å) | 42.93-1.84 |
| Total reflections | 93,268 (3119) |
| Unique reflections | 13,158 (1293) |
| Multiplicity | 7.0 (3.8) |
| Completeness (%) | 99.46 (98.40) |
| Mean I/σ(I) | 8.9 (1.3) |
| Wilson B-factor | 23.64 |
| Rmergea | 0.161 (1.014) |
| Rpimb | 0.057 (0.567) |
| CC1/2 | 0.995 (0.549) |
| Mosaicity | 0.34 |
| Rwork | 0.1842 (0.2962) |
| Rfree | 0.2566 (0.3301) |
| No. of non-hydrogen atoms | 1389 |
| Macromolecules | 1222 |
| Water | 167 |
| Protein residues | 154 |
| Root mean square (bonds) | 0.015 |
| Root mean square (angles) | 1.76 |
| Ramachandran favored (%) | 93 |
| Ramachandran outliers (%) | 0 |
| Clash score | 8.21 |
| Average B-factor | 31.1 |
| Macromolecules | 29.9 |
| Solvent | 40 |
| PDB code | 4ums |
a Rmerge = Σh Σi|I(h,i) − 〈I(j)〉|/Σh Σi I(h,i), where I(h,i) is the intensity of the measurement of reflection h and 〈I(h)〉 is the mean value of I(h,i) for all i measurements.
b Rpim= (Σhkl√1/(n − 1)Σj = 1n|Ihkl,j − 〈Ihkl〉|)/Σhkl Σj Ihkl,j), where 〈Ihkl〉 is the average of symmetry-related observations of a unique reflection.
Data Deposition
Coordinates and observed structure factor amplitudes have been deposited in the Protein Data Bank under the accession codes 4ums (supplemental File 1) and r4umssf (supplemental File 2); PDB validation report, (supplemental File 3).
Interface Modeling and Analysis
Because an experimental structure of the B. cellulosolvens dockerin is unknown, we used computational modeling and docking to predict its structure and dock it against the B. cellulosolvens cohesin. Briefly, for each dockerin structure in the PDB (PDB codes 1ohz, 2b59, 2ccl, 2vn5, 2vn6, 2ozn, 2y3n, 3kcp, 3ul4, 4dh2, 4fl4, and 4iu2), we used Rosetta Design (36, 37) to thread the sequence of the B. cellulosolvens dockerin and minimize the side chain and backbone conformation (complete execution instructions are provided in the supplemental File 4). We then docked each modeled dockerin against the cohesin using the rigid-body orientations observed in experimentally determined dockerin-cohesin structures (PDB codes as mentioned above). Each resulting binary model was refined using RosettaDock in all-atom refinement mode generating 750 models. The nine top-scoring models (by computed binding energy, buried surface area, and packing statistics) and the two models shown under “Results” were selected based on visual inspection. All backbones used for modeling were pre-refined using Rosetta energy minimization procedures (complete execution instructions are provided in thesupplement material). The all-atom energy function used in all Rosetta simulations was talaris2014, which is dominated by contributions from van der Waals packing, hydrogen bonding, and solvation.
Isothermal Titration Calorimetry
Isothermal titration calorimetry was deployed to measure the affinity of BcCohI for its dockerin partners essentially as described previously (15). Briefly, the wild type and mutant forms of the dockerin (20 μm), fused to thioredoxin, were stirred at 307 rpm in the reaction cell, which was injected with 10-μl aliquots of a 100 μm solution of cohesin at 220-s intervals. The buffer consisted of 50 mm Na-Hepes buffer, pH 7.5, containing 2 mm CaCl2 and 0.5 mm tris(2-carboxyethyl)phosphine, and titrations were carried out at 35 °C unless otherwise stated. Integrated heat effects, after correction for heats of dilution, were analyzed by nonlinear regressing using a single-site model (Microcal ORIGIN version 7.0, Microcal Software, Northampton, MA).
Results and Discussion
Structure of the B. cellulosolvens Type I Cohesin
The structure of the B. cellulosolvens type I cohesin, BcCohI, the 7th cohesin of the B. cellulosolvens anchoring scaffoldin ScaB, was solved by molecular replacement. BcCohI displays an elliptical structure with nine β-strands, which form two β-sheets aligned in an elongated β-barrel that displays a classical “jelly roll fold” (Fig. 2). The two sheets comprise β-strands 9, 1, 2, 7, and 4 on one face and β-strands 8, 3, 6, and 5 on the other face (Table 2). Strands 1 and 9 align parallel to each other, thus completing the jelly roll topology, although the other β-strands are antiparallel. No crystals of BcCohI in complex with its cognate dockerin could be obtained.
FIGURE 2.
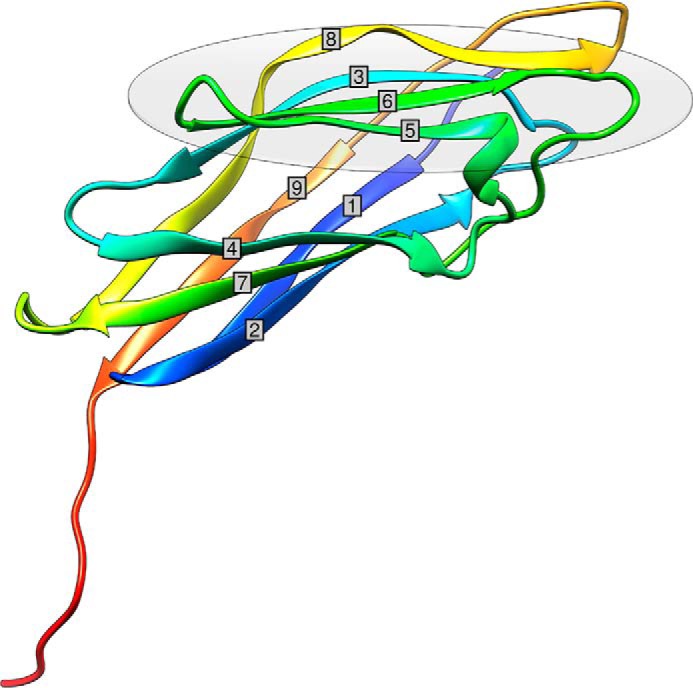
Three-dimensional structure of BcCohI color-ramped from N (blue) to C (red) terminus. BcCohI has a β-sandwich fold, and the nine cohesin β-strands are labeled. The gray oval represents the putative dockerin-interacting cohesin face.
TABLE 2.
The sequences that comprise the β-strands in BcCohI
| β-Strand | Residues in the cohesin |
|---|---|
| 1 | Asn-1043 to Glu-1052 |
| 2 | Gly-1055 to Asn-1065 |
| 3 | Ile-1071 to Tyr-1080 |
| 4 | Leu-1085 to Gly-1093 |
| 5 | Phe-1103 to Asn-1106 |
| 6 | Lys-1112 to Asp-1119 |
| 7 | Ala-1134 to Lys-1141 |
| 8 | Gly-1147 to Lys-1163 |
| 9 | Phe-1174 to Ala-1182 |
Structural similarity search using SSM (21) revealed that the closest and functionally relevant structural homologs of BcCohI are the type I cohesins from C. thermocellum (PDB codes 3ul4 for CtCoh-OlpA; 4dh2 for CtCoh-OlpC; 1aoh for CtCoh-CipA, and 2ccl for CtCoh-CipA complexed with a mutated dockerin) and C. cellulolyticum (PDB codes 1g1k for Coh alone and 2vn5/6 for the cohesin complexed with dockerins), with a z score of 6.6, root mean square deviation of 1.0 Å over 117 aligned residues out of a possible 120 amino acids and a total sequence identity of 38%. BcCohI also shows strong homology with the type I cohesin from C. perfringens (root mean square deviation >2.3 Å and a sequence identity of <30%; PDB codes 2vo8 and 2jh2) and a more distant homology to carbohydrate-binding modules (CBMs) presenting a β-sandwich fold, which also comprises two β-sheets that adopt a β-jelly roll fold (root mean square deviation >3 Å, and sequence identity of <20%) (22). This observation suggests a common evolutionary origin for CBMs and cohesins with ligand-interacting platforms having evolved for the recognition of carbohydrates or proteins, respectively.
Comparison with Type I Cohesins
As shown in Fig. 3, the B. cellulosolvens ScaA type I dockerin (Bc-DocA) specifically recognizes BcCohI (Bc-B3). The capacity of BcCohI to interact with dockerins of different origins was evaluated. The data, presented in Fig. 4, revealed that BcCohI (Bc-B3) is unable to bind to type I dockerins from C. cellulolyticum, A. cellulolyticus, Archaeoglobus fulgidus, and C. thermocellum and type III dockerins from Ruminococcus flavefaciens. In addition, BcCohI does not recognize any of the type II dockerins tested, including those found in B. cellulosolvens enzymes. The data confirm the strict species specificity in type I cohesin-dockerin interactions. The biological significance of this lack of cross-species specificity in cohesin-dockerin complexes is not entirely clear. It is likely that the grafting of enzymes with different origins in the same complex may adversely affect the catalytic efficiency of the cellulosome.
FIGURE 3.

Interaction of the type I Bc-DocA with a cohesin microarray. CBM-Coh fusion proteins, as listed in the key, were applied in triplicate in successive 2-fold dilution, starting from a maximum concentration of 8 μm (∼5 nl). The XynDoc fusion protein (10 ml per slide) was then applied at a final concentration of 2 nm, and the amount of dockerin bound to the cohesin samples was visualized by immunofluorescence. The triplet of spots at the base of each sample area denotes a marker, composed of a Xyn-CBM conjugate, which indicates the location of the samples on the cellulose slide. GenBankTM accession numbers for sequences described in this figure are as follows: A. cellulolyticus ScaA (AF155197), ScaB (AY221112), ScaC (AY221113), and ScaD (AY221114); A. fulgidus Orfs 2375 and 2376 (AE000782); B. cellulosolvens ScaA and ScaB (AF224509) and Cel48A (AAR23324); C. acetobutylicum CipA (AE001437); C. cellulolyticum CipC (U40345) and OrfX (Cc-OX, AF081458); C. thermocellum primary scaffoldin, CipA (Q06851), and anchoring scaffoldins, OlpA (Ct-OA, Q06848), SdbA (U49980), OlpB (Q06852), and Orf2p (Q06853); R. flavefaciens ScaA, ScaB, ScaC, and ScaE (AJ278969).
FIGURE 4.
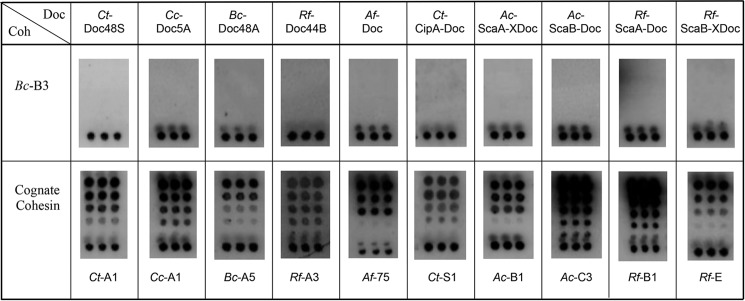
Interaction of dockerins from various sources with a type I cohesin from B. cellulosolvens ScaB (Bc-B3). Results are shown for four enzyme-borne dockerins (from C. thermocellum, C. cellulolyticum, B. cellulosolvens, and R. flavefaciens), the lone dockerin from A. fulgidus, and five scaffoldin-borne dockerins from C. thermocellum, A. cellulolyticus, and R. flavefaciens. None of these dockerins interacted with Bc-B3, but all of them interacted selectively with the target cohesins from their own species. Experiments were carried out as described in the legend to Fig. 3, with the designated XynDoc fusion proteins. See key and legend in Fig. 3 for the nomenclature of the different cohesins identified in this figure. GenBankTM accession numbers for dockerin sequences described in this figure are as follows: Ct-Doc48A (WP_003516749), Cc-Doc5A (WP_015924614), Bc-Doc48A (AAR23324), Rf-Doc44B (WP_026053020), Af-Doc (WP_010879862), Ct-CipA-Doc (Q06851), Ac-ScaA-XDoc (AF155197), Ac-ScaB-Doc (AY221112), Rf-ScaA-Doc, and Rf-ScaB-XDoc (AJ278969).
In a previous study, Nakar et al. (23) used a gene-swapping approach to identify a three-residue substitution in the C. cellulolyticum type I cohesin that conferred recognition of C. thermocellum dockerins with an affinity that was only 5-fold lower than C. thermocellum cohesins. This suggests that in type I cohesins only a small number of residues modulate dockerin specificity. Despite low sequence similarity with type I cohesins from C. thermocellum and C. cellulolyticum (<39% identity), previously determined three-dimensional structures of their cohesin-dockerin complexes are remarkably similar. The dockerin-binding sites of these cohesins consist essentially of a planar surface comprising β-strands 8, 3, 5, and 6 (4, 20). Given the near-identical fold displayed by type I cohesins, it is likely that the location of the ligand-binding site in BcCohI is also conserved. By overlaying the structure of B. cellulosolvens type I cohesin with the type I cohesin-dockerin complexes of C. thermocellum (Fig. 5) and C. cellulolyticum (data not shown), the major difference in the overall fold comprises the longer length of β-strand 8 in BcCohI. However, the position of this strand is identical to the other type I modules and different from that of type II cohesins where β-strand 8 is tilted toward the other strands in the dockerin-contacting sheet, thus promoting a larger contribution of this strand to dockerin recognition. In addition, the loop connecting β-strands 7 and 8 is longer in BcCohI but is diverted away from the putative dockerin-binding site.
FIGURE 5.

Superposition of BcCohI with the type I cohesin-dockerin complexes from C. thermocellum. Structural overlay of C. thermocellum type I cohesin-dockerin complexes in two binding orientations (PDB codes 1ohz and 2ccl) superposed with BcCohI. Residues involved in C. thermocellum cohesin-dockerin binding are highlighted in stick representation. Dockerin residue numbers are boxed and indicate equivalent residue positions on CtCoh-1ohz (gray colored structure) and CtCoh-2ccl (brown color), respectively. The cohesin residue numbers refer to structurally equivalent positions on BcCohI (green) and CtCoh-1ohz-A (salmon), respectively. Calcium ions are represented as spheres colored in purple. The top inset is a magnified view from the opposite side, around the blue spherical surface that highlights the canonical C. thermocellum Ser/Thr interface residue pair. Figures were prepared using UCSF Chimera (38).
The internal symmetry of the dockerins, coupled with several mutagenesis studies (15, 16), revealed that C. thermocellum and C. cellulolyticum type I dockerins contain two equivalent cohesin-binding sites, which have been maintained during evolution. In C. thermocellum, two virtually identical cohesin-binding faces are observed, whereby a Ser/Thr pair, located at positions 11/12 and 45/46, respectively, of each binding face, dominates the hydrogen-bonding network with the cohesin counterpart. These two dockerin-binding modes have been demonstrated through the crystal structures of several complexes. In one binding mode, helix-1 of the dockerin dominates cohesin recognition with residues at positions 11 and 12 of the duplicated segment, thereby playing a central role in the interactions established between the two protein partners (15, 16). In the second binding mode the dockerin is rotated 180° relative to the cohesin, and helix-3, rather than helix-1, plays a central role in complex formation (15, 16). Thus, residues at positions 11 and 12 of the two duplicated segments (11/12 in helix 1 and 45/46 in helix 3) are equivalent and dominate the interactions between the dockerin and its cohesin partner.
The B. cellulosolvens type I dockerin sequence from ScaA (Bc-DocA) also displays remarkable internal symmetry (the functional implications of this property are explored below). However, as in the dockerins from different species, the nature of the cohesin recognition amino acid pairs is essentially different (Ser-Asp in B. cellulosolvens instead of Ser-Thr in C. thermocellum). Indeed, the structure of the C. thermocellum type I cohesin-dockerin complexes overlaid with BcCohI suggests significant differences that may preclude inter-species interactions. Detailed analysis of this overlay revealed that replacement of residue Thr-12/46 of the C. thermocellum dockerin by an Asp-12/46 (as observed in B. cellulosolvens dockerins) would likely clash with Glu-131 of the C. thermocellum type I cohesin. Furthermore, residues that occupy positions 11 and 12 of the C. cellulolyticum dockerin helices 1 and 3 are also highly conserved Ala and Leu/Phe, respectively. The dockerin Leu/Phe make several hydrophobic interactions with the C. cellulolyticum cohesin, most notably with the C-β of Ala-129, with the aliphatic region of the Lys-137 side chain and the α-carbon of Gly-131. In BcCohI, the equivalent residues to Ala-129 and Gly-131 are serines, and thus the hydrophobic nature of the interaction is critically affected. Although other residues may modulate the specificity of type I cohesin-dockerin pairs, the above-mentioned observations may explain why there is a consistent lack of cross-reactivity between B. cellulosolvens, C. thermocellum, and C. cellulolyticum type I cohesin-dockerin complexes.
Structural Model for the Type I Cohesin-Dockerin Interaction of B. cellulosolvens
Because we were unable to obtain a crystal structure for the B. cellulosolvens cohesin in complex with Bc-DocA, we instead used computational modeling to predict bound structures. As described above, dockerin structures have a pseudo 2-fold symmetry axis, and the type I cohesins could associate with their cognate dockerins through two conformations that are related by 180° rotation (4, 15, 16, 18). We therefore modeled the B. cellulosolvens cohesin-dockerin pair in both putative binding modes (Fig. 6). The buried surface area upon binding in the two models (one for each mode) is ∼1,100 Å2, which is lower than the average for experimental structures of cohesin-dockerin interactions (1,659 ± 119 Å2); the relatively small, buried surface area in the model potentially reflects inaccuracies in modeling. Notwithstanding this uncertainty, the binding modes of natural cohesin-dockerin interactions are highly conserved (2, 6), thus providing confidence in the modeled binding modes. Both models present an intricate hydrogen-bonding network involving cohesin residues Asn-34, Asn-35, Asp-37, Ser-120, and Ser-122 and either Ser-11 and Asp-12 or Ser-45 and Asp-46 on the dockerin. Similar to C. thermocellum type I cohesins, Asn-35 and Asp-37 create an optimal environment to accommodate the hydroxyl side chain of the Ser residue located at position 11 or 45 of the B. cellulosolvens dockerin. In addition, Asn-35, Ser-120, and Ser-122 coordinate the recognition of the acidic side chain of Asp located at the critical position 12 or 46 of the dockerin, which is one the most important modulators of B. cellulosolvens dockerin specificity. The high similarity between the hydrogen bond networks in the two models is a consequence of the pseudo 2-fold symmetry of the dockerin and the conservation of Asp at positions 12 and 46 and Ser at positions 11 and 45. Inspection of the primary sequence of the 10 B. cellulosolvens type I cohesin modules present in ScaB reveals that Asn-35 and Ser-122 are invariant in the 10 ScaB cohesins (data not shown). In addition, Asp-37 and Ser-120 are conserved in the majority of the proteins and when substituted are usually replaced with amino acids with similar biochemical properties. These observations suggest that the specificity of B. cellulosolvens type I ScaB cohesins for ScaA dockerin is invariable.
FIGURE 6.
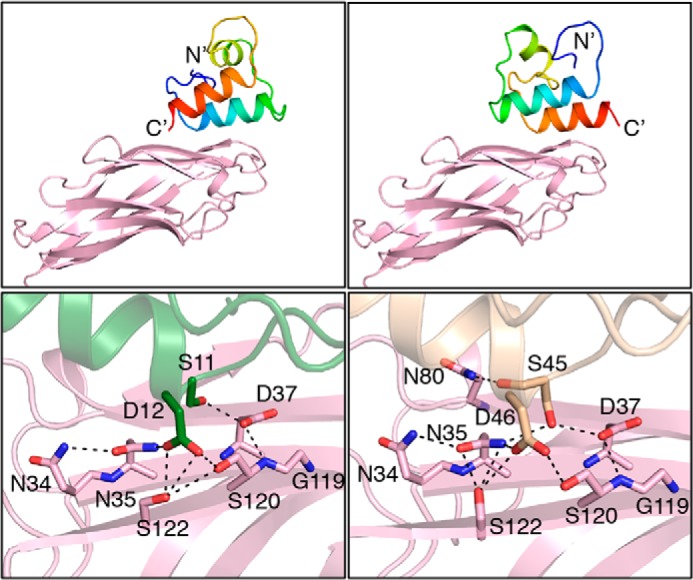
Computational model of the type I B. cellulosolvens cohesin-dockerin complex. Left and right panels represent two different model structures related to one another by a pseudo 2-fold symmetry axis about the interaction between the monomers. Hydrogen bonds mediating the interactions are represented by dashed lines. Panels were prepared using PyMOL (39). Top panel shows the dockerin in rainbow hues (blue to red) from the N to C terminus, respectively. The cohesin structure is colored lavender. In the bottom panels, the interface is magnified, and the major interacting residues are shown.
B. cellulosolvens Dockerins Present Two Functionally Similar Cohesin-binding Faces
The biological significance of the dual-binding mode displayed by C. cellulolyticum and C. thermocellum dockerins is an intriguing question, although it has been speculated that it might confer the required flexibility for cellulosome assembly. In contrast, clostridial type II dockerins only present a single cohesin-binding surface. This suggests that cellulosome assembly (type I interactions) requires cohesin-dockerin flexibility, although cell-surface attachment and poly-cellulosome formation (type II interactions) may be sustained by a single-binding mode. The observation that in B. cellulosolvens the cohesin-dockerin interactions are reversed in comparison with other cellulosomes offers the possibility of evaluating whether flexibility of a type I interaction is involved in cell-surface attachment.
Inspection of the primary sequence of the ScaA dockerin reveals a highly symmetrical protein (Table 3). Alignment of its two duplicated segments reveals complete conservation of the putative residues that participate in cohesin recognition, thus strongly suggesting that this dockerin contains two cohesin-binding interfaces presenting identical ligand specificities (Table 3). These observations were further supported by the modeling studies (Fig. 6), which indicated extensive involvement of the Ser-11/45 and Asp-12/46 residues in the respective binding modes. To probe this experimentally, the relevant Ser-Asp pairs of the two duplicated segments in the dockerin (DocScaA) of the primary scaffolding ScaA were mutated to an Ala pair, individually or in combination (Table 3). The affinity of the three dockerin mutant derivatives and the wild type dockerin (DocScaA) BcCohI was probed by isothermal titration calorimetry. Titrations of the wild type dockerin, DocScaA, and the mutants DocS23A/D24A and DocS60A/D61A into the ScaB cohesin BcCohI revealed ΔH values ranging between 25 and 35 kcal·m−1. Because of the very high affinity of the interactions, accurate Ka values could not be determined, although a qualitative value of ≥109 m−1 could be assigned. In contrast, the quadruple mutant, dockerin DocS23A/D24A/S60A/D61A, in which all four residues of the two cohesin-binding faces were mutated to alanine, displays an affinity that is below the detection limit of the technique (Fig. 7).
TABLE 3.
Protein sequences of B. cellulosolvens type I dockerin (DocScaA) and ScaA mutant dockerin derivatives
Near-perfect internal symmetry (duplicated segment) of the dockerin sequence is shown in bold font. Amino acid changes in the mutants are highlighted in gray. Positions of Ser-11/45 and Asp-12/46 are shown (i.e. 23/24 and 60/61, respectively, in the dockerin derivatives used in these studies).

FIGURE 7.

Binding of ScaA dockerin and its mutant derivatives to BcCohI evaluated by isothermal titration calorimetry. ScaA dockerin is highly symmetric and potentially contains two identical cohesin-binding surfaces. The key residues that participate in cohesin recognition at each cohesin-binding surface were changed to alanine. ScaA dockerins DocS23A/D24A and DocS60A/D61A only have one functional binding face, whereas in the quadruple mutant, DocS23A/D24A/S60A/D61A, the two cohesin-binding faces were inactivated. The upper part of each panel shows the raw heats of binding, and the lower parts comprise the integrated heats after correction for heat dilution. The curve represents the best fit to a single-site binding model. BcCohI-DocScaA; B, BcCohI-DocS23A/D24A; C, BcCohI-Doc/S60A/D61A; and D, BcCohI-DocS23A/D24A/S60A/D61A.
Collectively, these observations suggest that abrogation of cohesin recognition requires the mutation of the two putative cohesin-binding faces of the ScaA dockerin. The inactivation of only one of the faces does not affect the capacity of the dockerin to interact with its cognate protein module, because the alternative face is available for binding. Therefore, the data confirmed the hypothesis that the type I dockerin from ScaA displays a dual-binding mode against B. cellulosolvens type I cohesins. So far, the dual-binding mode was shown to be an important property of type I dockerins that are part of cellulosomal enzymes, suggesting that it may increase the internal flexibility of a high molecular mass multienzyme complex. These data suggest that the dual-binding mode is not restricted to enzyme-borne dockerins and support an important role for flexibility of cellulosome anchoring onto the cell surface in B. cellulosolvens.
Crystallization and structure determination of isolated cohesin modules are relatively straightforward, compared with crystallization of dockerins, both alone or in complex with their cohesin counterpart. Such was the case with BcCohI. Although ultimate proof for dual recognition in cohesin/dockerin pairs requires experimentally determined molecular structures, combined sequence analysis and all-atom structural modeling can generate hypotheses for experimental testing. The mutation and binding studies based on these hypotheses support the role of conserved residues on cohesin and dockerin pairs in mediating a dual-binding mode of interaction in BcCohI.
Supplementary Material
Acknowledgments
We acknowledge ESRF, Grenoble, France (beamline 1D14-4), for provision of synchrotron-radiation facilities. We thank Dr. Teresa Santos Silva for help with data collection and Cecília Bonifácio for help with the crystallization. We are grateful for the assistance and contributions of Rachel Haimovitz and Yoav Barak (Chemical Research Support, The Weizmann Institute of Science).
This work was supported in part by Fundação para a Ciência e a Tecnologia (Lisbon, Portugal) Grants PTDC/BIA-PRO/103980/2008 and EXPL/BIA-MIC/1176/2012, European Union Seventh Framework Programme FP7 2007–2013 under the WallTraC Project Grant 263916 and under BioStruct-X Grant 283570; Israel Science Foundation and a European Research Council Starter's Grant (to the Fleishman laboratory); Israel Science Foundation Grant 1349/13, European Union, Area NMP.2013.1.1-2 “Self-assembly of Naturally Occuring Nanosystems,” CellulosomePlus Project 604530, and an ERA-IB Consortium Grant EIB.12.022 (acronym FiberFuel) (to the Bayer laboratory). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Files 1–4.
The atomic coordinates and structure factors (code 4ums) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- BcCohI
- type I cohesin module from B. cellulosolvens scaffoldin ScaB
- CBM
- carbohydrate-binding module
- Bc-DocA
- type I dockerin from B. cellulosolvens scaffoldin ScaA
- PDB
- Protein Data Bank
- CtCoh
- type I cohesin from C. thermocellum
- IMAC
- immobilized metal ion affinity chromatography.
References
- 1. Gilbert H. J. (2010) The biochemistry and structural biology of plant cell wall deconstruction. Plant Physiol. 153, 444–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bayer E. A., Belaich J.-P., Shoham Y., Lamed R. (2004) The cellulosomes: multienzyme machines for degradation of plant cell wall polysaccharides. Annu. Rev. Microbiol. 58, 521–554 [DOI] [PubMed] [Google Scholar]
- 3. Fontes C. M., Gilbert H. J. (2010) Cellulosomes: highly efficient nanomachines designed to deconstruct plant cell wall complex carbohydrates. Annu. Rev. Biochem. 79, 655–681 [DOI] [PubMed] [Google Scholar]
- 4. Carvalho A. L., Dias F. M., Prates J. A., Nagy T., Gilbert H. J., Davies G. J., Ferreira L. M., Romão M. J., Fontes C. M. (2003) Cellulosome assembly revealed by the crystal structure of the cohesin-dockerin complex. Proc. Natl. Acad. Sci. U.S.A. 100, 13809–13814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Artzi L., Dassa B., Borovok I., Shamshoum M., Lamed R., Bayer E. A. (2014) Cellulosomics of the cellulolytic thermophile Clostridium clariflavum. Biotechnol. Biofuels 7, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leibovitz E., Béguin P. (1996) A new type of cohesin domain that specifically binds the dockerin domain of the Clostridium thermocellum cellulosome-integrating protein CipA. J. Bacteriol. 178, 3077–3084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xu Q., Gao W., Ding S.-Y., Kenig R., Shoham Y., Bayer E. A., Lamed R. (2003) The cellulosome system of Acetivibrio cellulolyticus includes a novel type of adaptor protein and a cell-surface anchoring protein. J. Bacteriol. 185, 4548–4557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Adams J. J., Pal G., Jia Z., Smith S. P. (2006) Mechanism of bacterial cell-surface attachment revealed by the structure of cellulosomal type II cohesin-dockerin complex. Proc. Natl. Acad. Sci. U.S.A. 103, 305–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pinheiro B. A., Brás J. L., Najmudin S., Carvalho A. L., Ferreira L. M., Prates J. A., Fontes C. M. (2012) Flexibility and specificity of the cohesin–dockerin interaction: implications for cellulosome assembly and functionality. Biocat. Biotransform. 30, 309–315 [Google Scholar]
- 10. Horino H., Fujita T., Tonouchi A. (2014) Description of Anaerobacterium chartisolvens gen. nov., sp. nov., an obligately anaerobic bacterium from Clostridium rRNA cluster III isolated from soil of a Japanese rice field, and reclassification of Bacteroides cellulosolvens Murray et al. (1984) as Pseudobacteroides cellulosolvens gen. nov., comb. nov. Int. J. Syst. Bacteriol. Microbiol. 64, 1296–1303 [DOI] [PubMed] [Google Scholar]
- 11. Giuliano C., Khan A. W. (1984) Cellulase and sugar formation by Bacteroides cellulosolvens, a newly isolated cellulolytic anaerobe. Appl. Environ. Microbiol. 48, 446–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murray W. D., Sowden L. C., Colvin J. R. (1984) Bacteroides cellulosolvens sp. nov., a cellulolytic species from sewage sludge. Int. J. Syst. Bacteriol. 34, 185–187 [Google Scholar]
- 13. Ding S.-Y., Bayer E. A., Steiner D., Shoham Y., Lamed R. (2000) A scaffoldin of the Bacteroides cellulosolvens cellulosome that contains 11 type II cohesins. J. Bacteriol. 182, 4915–4925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu Q., Bayer E. A., Goldman M., Kenig R., Shoham Y., Lamed R. (2004) Architecture of the Bacteroides cellulosolvens cellulosome: description of a cell-surface anchoring scaffoldin and a family-48 cellulase. J. Bacteriol. 186, 968–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carvalho A. L., Dias F. M., Nagy T., Prates J. A., Proctor M. R., Smith N., Bayer E. A., Davies G. J., Ferreira L. M., Romão M. J., Fontes C. M., Gilbert H. J. (2007) Evidence for a dual binding mode of dockerin modules to cohesins. Proc. Natl. Acad. Sci. U.S.A. 104, 3089–3094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pinheiro B. A., Proctor M. R., Martinez-Fleites C., Prates J. A., Money V. A., Davies G. J., Bayer E. A., Fontes C. M., Fierobe H.-P., Gilbert H. J. (2008) The Clostridium cellulolyticum dockerin displays a dual binding mode for its cohesin partner. J. Biol. Chem. 283, 18422–18430 [DOI] [PubMed] [Google Scholar]
- 17. Alber O., Noach I., Rincon M. T., Flint H. J., Shimon L. J., Lamed R., Frolow F., Bayer E. A. (2009) Cohesin diversity revealed by the crystal structure of the anchoring cohesin from Ruminococcus flavefaciens. Proteins 77, 699–709 [DOI] [PubMed] [Google Scholar]
- 18. Carvalho A. L., Pires V. M., Gloster T. M., Turkenburg J. P., Prates J. A., Ferreira L. M., Romão M. J., Davies G. J., Fontes C. M., Gilbert H. J. (2005) Insights into the structural determinants of cohesin-dockerin specificity revealed by the crystal structure of the type II cohesin from Clostridium thermocellum SdbA. J. Mol. Biol. 349, 909–915 [DOI] [PubMed] [Google Scholar]
- 19. Noach I., Frolow F., Jakoby H., Rosenheck S., Shimon L. W., Lamed R., Bayer E. A. (2005) Crystal structure of a type II cohesin module from the Bacteroides cellulosolvens cellulosome reveals novel and distinctive secondary structural elements. J. Mol. Biol. 348, 1–12 [DOI] [PubMed] [Google Scholar]
- 20. Shimon L. J., Bayer E. A., Morag E., Lamed R., Yaron S., Shoham Y., Frolow F. (1997) A cohesin domain from Clostridium thermocellum: the crystal structure provides new insights into cellulosome assembly. Structure 5, 381–390 [DOI] [PubMed] [Google Scholar]
- 21. Krissinel E., Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 22. Boraston A. B., Bolam D. N., Gilbert H. J., Davies G. J. (2004) Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem. J. 382, 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakar D., Handelsman T., Shoham Y., Fierobe H.-P., Belaich J. P., Morag E., Lamed R., Bayer E. A. (2004) Pinpoint mapping of recognition residues on the cohesin surface by progressive homologue swapping. J. Biol. Chem. 279, 42881–42888 [DOI] [PubMed] [Google Scholar]
- 24. Haimovitz R., Barak Y., Morag E., Voronov-Goldman M., Shoham Y., Lamed R., Bayer E. A. (2008) Cohesin-dockerin microarray: Diverse specificities between two complementary families of interacting protein modules. Proteomics 8, 968–979 [DOI] [PubMed] [Google Scholar]
- 25. Barak Y., Handelsman T., Nakar D., Mechaly A., Lamed R., Shoham Y., Bayer E. A. (2005) Matching fusion-protein systems for affinity analysis of two interacting families of proteins: The cohesin-dockerin interaction. J. Mol. Recognit. 18, 491–501 [DOI] [PubMed] [Google Scholar]
- 26. Najmudin S., Pinheiro B. A., Prates J. A., Gilbert H. J., Romão M. J., Fontes C. M. (2010) Putting an N-terminal end to the Clostridium thermocellum xylanase Xyn10B story: Crystal structure of the CBM22–1–GH10 modules complexed with xylohexaose. J. Struct. Biol. 172, 353–362 [DOI] [PubMed] [Google Scholar]
- 27. Leslie A. G., Powell H. R. (2007) in Evolving Methods for Macromolecular Crystallography (Read R., J., Sussman J. L., eds) pp. 41–51, Springer, Dordrecht, Netherlands [Google Scholar]
- 28. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Evans P. R. (2011) An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 67, 282–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP 4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Matthews B. W. (1968) Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 32. Long F., Vagin A. A., Young P., Murshudov G. N. (2008) BALBES: a molecular-replacement pipeline. Acta Crystallogr. D Biol. Crystallogr. 64, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., Vagin A. A. (2011) REFMAC 5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for x-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fleishman S. J., Leaver-Fay A., Corn J. E., Strauch E. M., Khare S. D., Koga N., Ashworth J., Murphy P., Richter F., Lemmon G., Meiler J., Baker D. (2011) RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS ONE 6, e20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kuhlman B., Dantas G., Ireton G. C., Varani G., Stoddard B. L., Baker D. (2003) Design of a novel globular protein fold with atomic-level accuracy. Science 302, 1364–1368 [DOI] [PubMed] [Google Scholar]
- 38. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 39. DeLano W. L. (2002) The PyMOL Molecular Graphics System, Version 1.7.2, DeLano Scientific LLC, San Carlos, CA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.