

Background: Lys-377 mutants of the E. coli melibiose permease are defective in the cotransport of substrates.
Results: Lys-377 mutants do not bind substrates, whereas Ile-22 mutants bind Na+ but not sugars. Both observations are explained by molecular dynamics simulations.
Conclusion: Lys-377 is crucial for the organization of the binding site. Ile-22 may participate in sugar binding.
Significance: Uncovering the architecture of the substrate binding site is essential for understanding the transport mechanism.
Keywords: FRET, IR spectroscopy, membrane biophysics, molecular dynamics, sugar transport, secondary transporter, sodium ligands
Abstract
We examine the role of Lys-377, the only charged residue in helix XI, on the functional mechanism of the Na+-sugar melibiose symporter from Escherichia coli. Intrinsic fluorescence, FRET, and Fourier transform infrared difference spectroscopy reveal that replacement of Lys-377 with either Cys, Val, Arg, or Asp disables both Na+ and melibiose binding. On the other hand, molecular dynamics simulations extending up to 200–330 ns reveal that Lys-377 (helix XI) interacts with the anionic side chains of two of the three putative ligands for cation binding (Asp-55 and Asp-59 in helix II). When Asp-59 is protonated during the simulations, Lys-377 preferentially interacts with Asp-55. Interestingly, when a Na+ ion is positioned in the Asp-55-Asp-59 environment, Asp-124 in helix IV (a residue essential for melibiose binding) reorients and approximates the Asp-55-Asp-59 pair, and all three acidic side chains act as Na+ ligands. Under these conditions, the side chain of Lys-377 interacts with the carboxylic moiety of these three Asp residues. These data highlight the crucial role of the Lys-377 residue in the spatial organization of the Na+ binding site. Finally, the analysis of the second-site revertants of K377C reveals that mutation of Ile-22 (in helix I) preserves Na+ binding, whereas that of melibiose is largely abolished according to spectroscopic measurements. This amino acid is located in the border of the sugar-binding site and might participate in sugar binding through apolar interactions.
Introduction
The melibiose transporter from Escherichia coli (MelB) catalyzes the symport of the sugar melibiose and H+, Na+, or Li+ by using the cation free energy to drive the uphill transport of the sugar (1, 2). A variety of biochemical and biophysical approaches have resulted in a transport model comprising several intermediate states. Cotransport with Na+ or Li+ involves first cation binding, which increases the melibiose affinity. This is followed by the binding of sugar, conformational changes leading to an inward-open conformation, release of sugar, and, finally, release of the cation (1, 3, 4). Some important residues for the transport mechanism, especially for the binding step, have been identified. Therefore, it is believed that the cation binding site comprises at least Asp-55, Asp-59 (helix II), and Asp-124 (helix IV), whereas the sugar most likely interacts at least with Asp-19 and Asp-124 (helices I and IV) (2, 5, 6).
A recent important achievement has been the resolution of the crystal structure of the melibiose transporter from Salmonella thyphimurium (MelBST) at 3.35 Å, confirming the structural model of 12 transmembrane α-helices (6). The structure, in a predominantly outward-facing, partially occluded conformation, validates the alternating access mechanism for transport by showing inverted topology repeats (7). On the other hand, the structure confirms an adequate location of the side chains proposed as ligands for the substrates. Because MelBST and MelB have more than 85% sequence identity and, most importantly, all essential side chains for transport are conserved, we can use this structure to derive conformational insights for the transport mechanism in E. coli and as a model to perform molecular dynamics on MelB.
In this work, we investigated the role of Lys-377, the sole charged side chain of helix XI, by analyzing the effects of replacing this lysine with either Cys, Val, Arg, or Asp. We also studied second-site revertants from Lys-377 variants with spontaneous mutations at position Ile-22: K377C/I22S, K377R/I22S, and K377V/I22S (8). According to the crystallographic structure (6), Lys-377 is located in the inner space of the protein near Asp-55 and Asp-59 (Fig. 1), side chains that have been shown to be important for substrate binding (5). Furthermore, Lys-377 has been proposed to interact with Asp-59 (8). On the other hand, mutations of Lys-377 have been shown previously to be deleterious for the transport activity (8–10). Therefore, it is indispensable to determine the structural and functional role of this important side chain. With respect to Ile-22, it is located in the other side of the central cavity, at the same level as Lys-377 (Fig. 1), and mutants of Ile-22 present a decreased transport capability (11). Therefore, both Lys-377 and Ile-22 could be involved in substrate binding.
FIGURE 1.
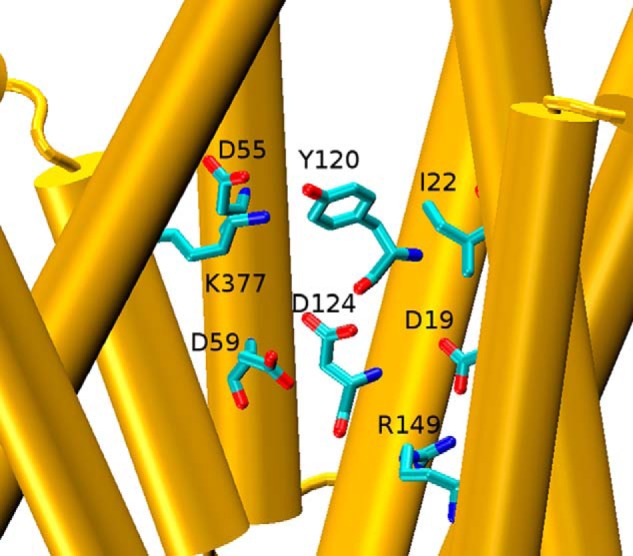
Disposition of key side chains in the MelBST crystal structure (molecule A of PDB code 4M64 (6). The central space containing Lys-377 and Ile-22 is shown along with the four internally located acidic side chains, Tyr-120, and Arg-149. Helices VIII and X were removed to facilitate the visualization. Shown is a side view with the periplasmic side on top.
Here we analyzed the effects on the binding of cosubstrates to MelB reconstituted into proteoliposomes or embedded in vesicles upon the exchange of Lys-377 to polar, apolar, anionic, and cationic amino acids: K377C, K377V, K377R, and K377D, respectively. To this end, we used intrinsic fluorescence, FRET, and Fourier transform infrared spectroscopy (12–14). We show that Cys, Val, Arg, or Asp mutants of Lys-377 lack cation and melibiose binding. Furthermore, we found that the I22S and I22A mutants preserve cation binding but no longer bind melibiose. We explain and complement the experimental findings with the aid of molecular dynamics simulations of MelB embedded in a hydrated lipid bilayer.
Experimental Procedures
Materials
E. coli total lipid extract was from Avanti Polar Lipids, Inc. Synthesis of the fluorescent sugar analog 2′-(N-dansyl)-aminoethyl-1-thio-d-galactopyranoside (D2G)6 was carried out by Dr. B. Rousseau and Y. Ambroise (Institut de Biologie et Technologies-Saclay, CEA, France). All other materials were of reagent grade and obtained from commercial sources.
MelB Expression, Purification, and Reconstitution
MelB and the mutants were cloned, expressed, and purified as reported previously (15–17). Briefly, a recombinant pK95ΔAHB plasmid with a cassette containing the melB gene encoding a permease with a His6 tag at its C-terminal end and devoid of its four native cysteines (Cys-less MelB) was used as a background for further permease engineering and as a control. The proteins were overexpressed in E. coli DW2-R (ΔmelB, ΔlacZY) transformed with the appropriate plasmid, grown at 30 °C in M9 medium supplemented with 0.5% glycerol, 0.2% (w/v) casamino acids, 10 mm thiamine, and 0.1 mm ampicillin until an A600 of 1.6–1.8 was reached. For protein purification, cells were homogenized in medium containing 50 mm Tris-HCl (pH 8.0), 50 mm NaCl, and 5 mm 2-mercaptoethanol and disrupted using a microfluidizer (model 110S, Microfluidics) with three passes at 20,000 p.s.i. Cell debris was removed by low-speed centrifugation for 10 min. The supernatant was collected and ultracentrifuged at 310,000 × g for 30 min. The membrane fraction was incubated with 1% (w/v) 3-(laurylamido)-N,N′-dimethylaminopropylamine oxide (Anatrace) for 30 min at 4 °C. After another ultracentrifugation step at 310,000 × g for 15 min, the supernatant was collected and loaded to a nickel-nitrilotriacetic acid affinity resin (Sigma-Aldrich) and washed with 20 mm Tris, 100 mm NaCl, 10% Glycerol, 10 mm melibiose, 10 mm imidazole, 0.1% (w/v) dodecyl-β-d-maltopyranoside (Anatrace), and 5 mm 2-mercaptoethanol (pH 8.0). The protein was eluted at pH 8.0 with 300 mm imidazole and 0.1% dodecyl-β-d-maltopyranoside. MelB reconstitution into liposomes of E. coli total lipids (protein/lipid ratio 1/2, w/w) was performed by removing the detergent with Bio-Beads SM-2 (Bio-Rad). MelB content was assayed by a Lowry procedure including 0.2% (w/v) sodium dodecyl sulfate and using bovine serum albumin as the standard (18).
Preparation of Membrane Vesicles
Right-side-out (RSO) and inside-out (ISO) membrane vesicles were prepared from the same culture following established methods (19). RSO vesicles were prepared from cells washed twice with 50 mm Tris-HCl (pH 8.0) and pelleted at 4 °C by centrifugation at 10,000 × g for 10 min. Upon resuspension of the pellet with 80 ml/1 g of cells in a 50 mm Tris-HCl buffer (pH 8.0) and 30% (w/v) sucrose, the cells were incubated at room temperature under vigorous stirring for 1 h. 0.5 mg/ml lysozyme and 10 mm potassium-EDTA (pH 7.0) were added simultaneously, followed by centrifugation at 13,000 × g. The pellet was resuspended using a glass syringe in the smallest possible volume (3–5 ml) of KPi buffer (pH 6.6) containing 30% (w/v) sucrose and 20 mm MgSO4, and 1 mg/ml of DNase and RNase were added. The small volume was released in 0.5 liter of KPi buffer (pH 6.6) pre-equilibrated at 37 °C. After 15 min of incubation under vigorous stirring, the protoplast generation was enhanced by addition of 10 mm K-EDTA (pH 8.0), followed by 15 mm MgSO4 after 15 min. The unbroken cells were precipitated by centrifuging the lysate at 800 × g (4 °C) for 1 h, leaving only RSO vesicles in the supernatant. Further centrifugation at a higher speed (16,000 × g, 4 °C) resulted in sedimentation of the RSO vesicles. Finally, the vesicles were washed at least three times using KPi buffer (pH 7.0) harboring 20 mm MgSO4 and resuspended at a protein concentration of 10–20 mg/ml in 100 mm KPi buffer (pH 6.6), frozen in liquid nitrogen, and stored at −80 °C until use.
ISO membrane vesicles were prepared by microfluidizer pressure. Frozen cells were thawed rapidly at 25 °C and washed twice with 10 mm Tris-HCl (pH 8.0). EDTA (15 mm), lysozyme (1 mg/ml), DNase (20 μg/ml), RNase (20 μg/ml), and MgSO4 (15 mm) were added and incubated at 4 °C for 30 min. The cell debris and unbroken cells were removed by low-speed centrifugation for 30 min, and the supernatant was collected and centrifuged at 146,000 × g for 30 min. The ISO vesicles were washed three to four times with 100 mm KPi buffer (pH 6.6), resuspended in the same buffer at about 10–20 mg of protein/ml, frozen in liquid nitrogen, and stored at −80 °C until use.
The MelB content of membrane vesicles was measured using the histidine tag-specific reagent His-ProbeTM-HRP (Thermo Scientific) directed against the His6 introduced at the C terminus of the transporter sequence, following the protocol of the manufacturer. In short, membrane samples (10 μg protein) were incubated with SDS-PAGE loading buffer containing 2% SDS for 5 min at room temperature and loaded on a 12%-SDS-PAGE gel. After electrophoresis, the proteins were transferred to nitrocellulose membranes, blocked with Tris-buffered saline containing 25 mg/ml bovine serum albumin for 1 h, and washed twice with Tris-buffered saline. The nitrocellulose membranes were then incubated with the His-ProbeTM-HRP reagent for 1 h at room temperature and washed three times with buffer. After this, the nitrocellulose membranes were developed by incubation with SuperSignalWorking solution containing luminol and peroxide and quantified with ImageJ software.
FTIR Difference Spectra
The experimental setup was the same as that described in a previous study (14, 20). In summary, about 20 μl of MelB containing proteoliposome suspension (about 100 μg of protein) in 100 mm KCl, 20 mm MES (pH 6.6) was spread homogeneously on a germanium Attenuated Total Reflectance crystal (Harrick, Ossining, NY; 50 × 10 × 2 mm, yielding 12 internal reflections at the sample side) and dried under a stream of nitrogen. The substrate-containing buffer and the reference buffer were alternatively perfused over the proteoliposome film at a rate of ∼1.5 ml/min. The film was exposed to substrate-containing buffer for 4 min and washed with reference buffer for 10 min (or 30 min for the buffer containing 50 mm melibiose). For each cycle, 1000 scans at a resolution of 4 cm−1 were recorded, and a minimum of 25 spectra were taken and averaged to increase the signal-to-noise ratio (i.e. a total of ≥25,000 scans for every difference spectrum). Spectra were recorded with an FTS6000 Bio-Rad spectrometer equipped with a mercury-cadmium-telluride detector. Data corrections for the difference spectra were carried out following a protocol developed previously (20).
Quantitative Comparison of Intensity and Similarity of FTIR Difference Spectra
Quantitative comparison of the intensity and similarity of IRdiff spectra were performed as described previously (5). In brief, the difference spectra were normalized for the amount of protein contributing to the IR signal in the 1700–1500 cm−1 interval. Quantitative comparison of two normalized difference spectra (an input and a reference spectrum) was performed by a linear regression in the 1710–1500 cm−1 region on the first derivative of the difference spectra and in the 1700–1500 cm−1 region on the second derivative of the absorbance spectra. The correlation analysis provides two significant parameters: the correlation coefficient (R2), which discloses the spectral similarity of the input with respect to the reference spectrum, and the slope, which measures the relative intensity of common features in the input spectrum with respect to the reference spectrum.
Fluorescence Spectra
Fluorescence measurements were done at 20 °C with a QuantaMasterTM spectrofluorometer. The data were processed with Felix 32 software (Photon Technology International). Trp fluorescence spectra were acquired by setting the excitation wavelength at 290 nm (half-bandwidth of 5 nm) and collecting the emission spectrum in 100 mm KPi buffer (pH 7.0). Na+-dependent FRET signals (λex, 290 nm; half-bandwidth, 5 nm) of RSO or ISO membrane vesicles (100 μg of protein/ml) or proteoliposomes (30 μg of protein/ml) were obtained by incubation with the sugar fluorescent analog D2G at a final concentration of 10 μm in 100 mm KPi buffer (pH 7.0) containing 100 mm KCl.
Molecular Dynamics Simulations
The starting coordinates where those of molecule A of MelBST (6) (PDB code 4M64). The CHARMM-GUI server (21–23) was used to mutate the required amino acids to those of MelB. A short energy minimization was performed in vacuo using the Nanoscale Molecular Dynamics program (24) to remove any possible steric clash. This E. coli MelB was inserted into a bilayer of 90 × 90 Å2 produced using the CHARMM-GUI Membrane Builder (25) consisting of 154 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) molecules by the replacement method. In this step, the protein orientation was calculated from the Orientations of Proteins in Membranes database by minimizing its transfer energy from water to the membrane (26). The coordinates of this protein-lipid complex were transferred to the Visual Molecular Dynamics (VMD) program (1.9.2 version (27)) and was complemented with a water layer of 18 Å on top and at the bottom of the bilayer, with water also filling the free space in the protein interior (a total of 13,709 water molecules). NaCl at a concentration of 0.3 m was finally added randomly with the “Add Ions” extension of VMD (131 Na+ and 135 Cl− per MelB). The system had a total of 75,800 atoms.
The system was energy-minimized, followed by equilibration for 200 ps using an integration step of 1 fs and NVT conditions (constant number of particles, volume, and temperature) using the Nanoscale Molecular Dynamics program. Equilibration was done by slowly heating the system from 0 to 298 K in steps of 2 K. Molecular dynamics were subsequently produced using the ACEMD program (28) running on a personal computer with a Graphic Processing Unit graphic card. For protein and lipid atoms, the CHARMM force field parameters (29, 30) were used, and, for water, the TIP3P model (31) was used. Two different replicas were produced for a minimum of 200 ns at constant pressure and temperature using particle mesh Ewald method full-system periodic electrostatics (32, 33). A time step of 4.0 fs, rigid bonds on all hydrogen-heavy atom bonds, and a hydrogen scale parameter of 4 were used to speed up the calculations (34). When indicated, protonation of Asp-59 was done by using the corresponding patch in the VMD extension “Auto PSF Builder.” Root-mean-square deviation (RMSD) was calculated using the VMD extension “RMSD Visualizer Tool” after alignment of the backbone Cα atoms.
Results
Characterization of Mutants
The effect of replacement of Lys-377 in the functionality of MelB was first assessed on MacConkey indicator plates containing 10 mm melibiose, which give red colonies linked to melibiose entry into the cytoplasm, subsequent metabolism, and acidification of the surrounding outer medium. The wild-type form of MelB devoided of any cysteine, C-less, gave red colonies, as expected, whereas cells expressing the K377C, K377V, and K377D mutants showed a white phenotype, indicating the absence of inward translocation of melibiose. K377R showed a slightly pink color, indicative of the presence of small melibiose entry, either passive or active.
As an additional characterization, the structure-sensitive infrared amide I and II protein backbone vibrations of the liposome-reconstituted MelB were used to assess whether significant modifications of the overall MelB structure could be introduced by the mutations at Lys-377. Only two slight spectral changes were observed in the amide I and II region of either the absorbance or the second derivative spectra (Fig. 2). The amide I maximum downshifted from 1658 cm−1 in WT and C-less to about 1656 cm−1 in all variants, indicating the presence of minor modifications in the environment of transmembrane α helices upon the exchange of Lys-377. A band at around 1630 cm−1 slightly incremented its absorbance in the variants. On the basis of its wave number, we can argue about the formation of β sheets or about H-bonding/protonation changes of the side chains of Lys, Arg, or Asn residues (35–37). However, it is evident that no gross structural alteration was introduced by any of the mutations on Lys-377, indicating that the overall native MelB structure was retained in these mutants. Additionally, no aggregation signals were noted in the infrared spectra around 1631 cm−1 (38).
FIGURE 2.
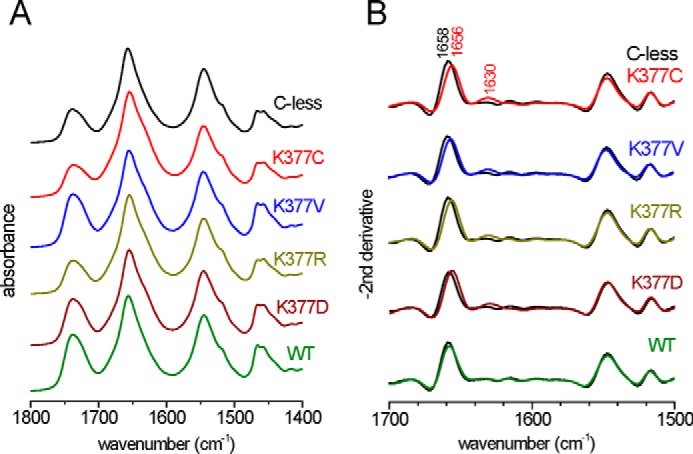
Spectral comparison of Lys-377 mutants to WT and C-less MelB using the structure-sensitive amide I and II vibrations. A, FTIR absorbance spectra of hydrated samples after correcting for water contributions. B, the second derivative in the amide I/II region provides details about structural changes induced by the mutation. The second derivative spectrum of each mutant and of WT is compared with that of C-less (black).
Defective Binding of Substrates to Lys-377 Mutants: Infrared Difference Spectra
The substrate-induced infrared difference spectra (IRdiff) of purified MelB reconstituted into liposomes provide a means to detect substrate binding to the transporter and to analyze the triggered conformational changes (20). First, Fig. 3A compares the Na+-induced IRdiff spectrum of C-less with those of some Lys-377 mutants. In each case, the difference spectrum consists, in comparison with C-less, of a featureless line indicative of a lack of interaction between the added Na+ and the mutated transporter, even at 50 mm NaCl (about 15 times above the affinity constant for C-less (4)). Second, a comparison of 50 mm melibiose-induced IRdiff spectra in the presence of Na+ of the Lys-377 mutants with those of C-less is shown in Fig. 3B. Again, a featureless signal is observed, indicating the absence of an interaction between the sugar and the mutated MelB. Quantification of the similarity and the intensity of these difference spectra was carried out by a spectral correlation analysis (5) and is presented in Fig. 3, right panels. It is clear that none of the Lys-377 mutants produce any quantifiable signal upon substrate incubation, corroborating the visual inspection of the difference spectra.
FIGURE 3.
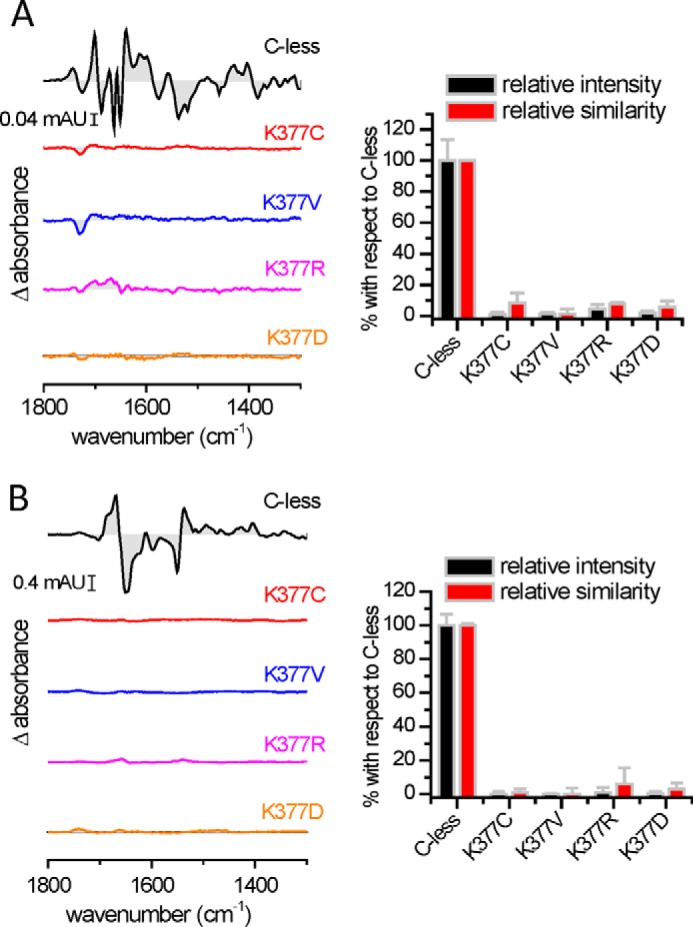
Substrate-induced infrared difference changes of MelB reconstituted into proteoliposomes. Substrate-induced IRdiff spectra at 4-cm−1 resolution of MelB C-less, K377C, K377V, K377R, and K377D at 25 °C (20 mm MES and 100 mm KCl (pH 6.6)) normalized to the amount of probed protein. A, 50 mm Na+-induced IRdiff spectra. B, 50 mm melibiose-induced IRdiff spectra in the presence of 10 mm Na+. The spectral similarity and intensity of the substrate-induced IRdiff spectra are shown in the right panels. The error bars correspond to the mean ± S.E. of three independent spectra.
Fluorescence Experiments
Fig. 4A shows the changes in fluorescence intensity of Trp residues of MelB reconstituted into proteoliposomes upon substrate binding. As for the IRdiff spectra, no response was obtained upon consecutive addition of melibiose and Na+ from any of the Lys-377 mutants, even at high substrate concentration. The only exception was the conservative K377R variant, for which a small signal was observed upon Na+ addition after incubation with melibiose.
FIGURE 4.
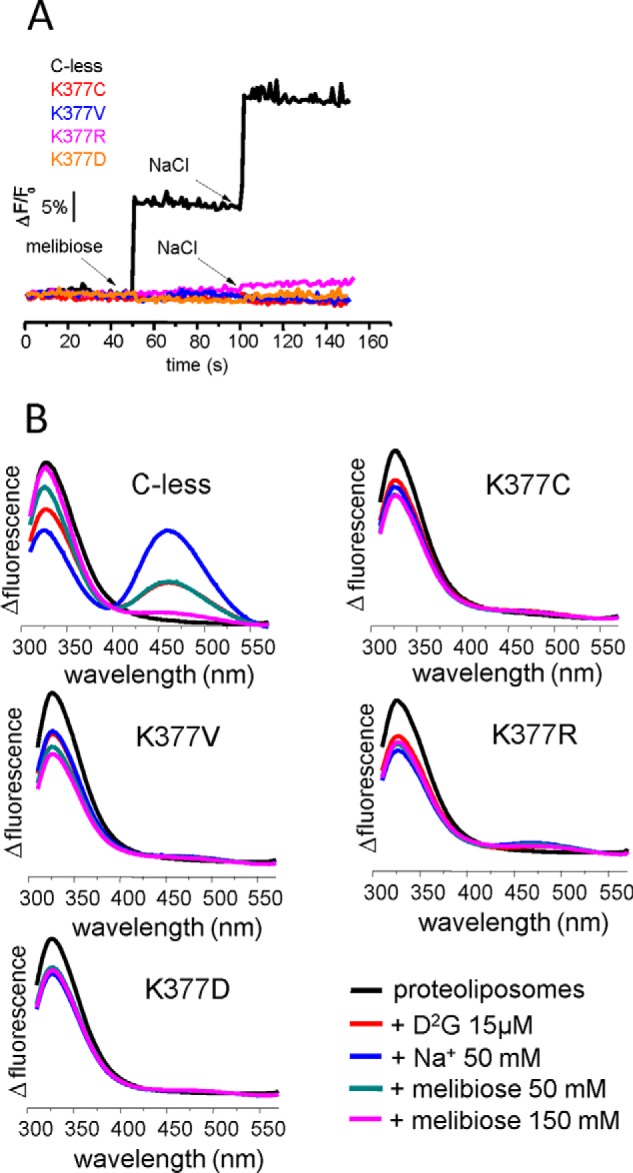
Substrate-induced changes in protein fluorescence emission and FRET of MelB in proteoliposomes. A, tryptophan fluorescence intensity changes in proteoliposomes (λex = 290 nm; half-bandwidth = 5 nm, and λem = 325 nm; half-bandwidth = 5 nm) containing purified C-less, K377C, K377V K377R and K377D at 20 μg/ml in 100 mm KPi after the consecutive addition of sugar and Na+ to a final concentration of 50 mm (arrows). B, FRET from Trp side chains to D2G (λex = 290 nm) in nominally Na+-free 100 mm potassium phosphate and 100 mm KCl. Emission fluorescence was recorded before and after the consecutive additions of 15 μm D2G, 50 mm NaCl, 50 mm melibiose, and 150 mm melibiose as indicated. Each spectrum is the mean of three scans.
The fluorescent sugar analog D2G allows a more sensitive approach to detect substrate interaction. It has been demonstrated previously that FRET from Trp side chains to D2G occurs when D2G binds to MelB (13), which further increases upon Na+ binding and decreases upon D2G displacement by addition of melibiose in excess (Fig. 4B, C-less). As shown in Fig. 4, no FRET was observed upon incubation of proteoliposomes with the fluorescent sugar analog D2G, in contrast to C-less. Again, the only exception is K377R. It shows a modest response to D2G addition, weakly sensitivity to the addition of Na+ and melibiose (Fig. 4B). In summary, the lack of substrate-induced changes in Trp fluorescence and in the FRET between Trp and D2G is in keeping with the absence of an interaction between the substrates and the Lys-377 mutants, in agreement with the results from IRdiff spectroscopy.
The previous spectroscopic experiments were carried out on proteoliposomes in which MelB has an orientation opposite to that in the cell (4, 39). The absence of binding of the mutated protein when reconstituted in proteoliposomes could be due to a conformation locked open toward the inward-facing space, as reported previously for R149C (39), or to an irreversible denaturizing effect of detergents during protein purification (although the spectral comparison in Fig. 2 argues against this last possibility). To fully discard the above possibilities, we analyzed the binding of substrates directly in RSO and ISO vesicles (39). We quantified the amount of MelB in the vesicles using the His probe HRP and found a MelB content comparable with that of control vesicles containing C-less. FRET experiments with membrane vesicles confirmed the results gathered from studying proteoliposomes, i.e. no FRET was observed upon D2G incubation, and there was no FRET enhancement upon addition of Na+ for any Lys-377 variant (data not shown). The only exception was K377R, for which a small effect was observed in both RSO and ISO vesicles.
In summary, the spectroscopic experiments strongly suggest that a Lys at position 377 is required for the binding of substrates. The requirement of Lys-377 for cation binding is especially intriguing given its positive charge, which makes Lys-377 an unlikely ligand for Na+. To better understand the role of Lys-377 in cation binding, we performed all-atom molecular dynamics simulations (see below).
Spontaneous Second-site Revertants of Lys-377 Mutants Partially Restore the Substrate-binding Capacity
Second-site revertants of defective single-site mutants have been described that recover the transport ability to some extent. For K377C, it has been reported that the additional mutation I22S restores the red color of colonies grown on MacConkey plates (8). Surprisingly, no active transport was detected for K377C/I22S when using radioactive melibiose (8). A possible explanation of this discrepancy was that the [3H]melibiose concentration used in the transport experiments was about 300 times lower than that used in the MacConkey plates (29 mm), reflecting a lower affinity for the substrates in the revertants (8). Therefore, it was of interest to examine substrate binding to these revertants by using a similar concentration as that used for the native MelB. We obtained the same spontaneous revertants, K377C/I22S, K377R/I22S, and K377V/I22S, described previously in the transport studies on MacConkey plates (8).
To identify the molecular origin of the revertant effect, we analyzed the behavior of the second-site revertants involving Ile-22 with respect to their capacity to bind substrates. To this end, we used the site-directed single mutant I22S as a reference. Although I22S shows a Na+-induced IRdiff spectrum very similar to C-less, the IRdiff spectra of the double mutants indicate strongly reduced Na+ binding (Fig. 5).
FIGURE 5.
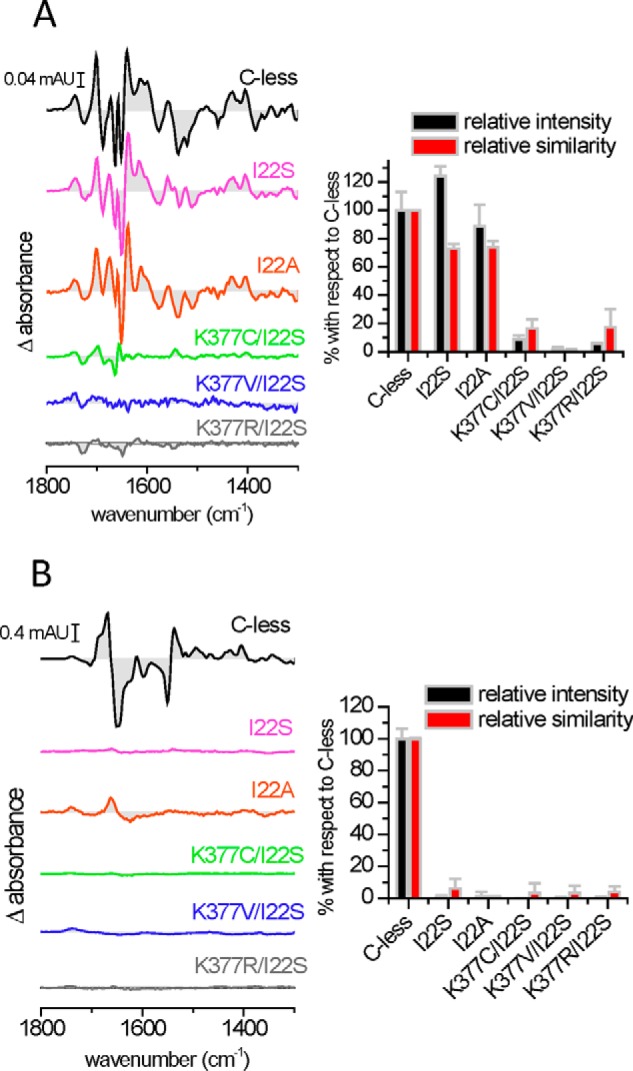
Substrate-induced infrared difference changes of MelB I22 mutants and Lys-377 double mutants reconstituted into proteoliposomes. Substrate-induced IRdiff spectra at 4-cm−1 resolution of MelB C-less, Ile-22 mutants, and Lys-377 mutants. A, 50 mm Na+-induced spectra. B, 50 mm melibiose-induced spectra in the presence of 10 mm Na+. Right panels, the respective intensities and similarities are plotted with respect to C-less. The experimental conditions were as indicated in Fig. 3.
Surprisingly, the I22S single mutant elicited a near complete absence of melibiose-induced IRdiff spectrum (Fig. 5B), much like the Lys-377 single mutants (Fig. 3B). To see whether the non-polar to polar change in the I22S mutant could be deleterious, we also examined the I22A mutant and observed the same absence of melibiose binding activity (Fig. 5B). Consistent with these results, none of these single or double mutants displayed any substrate-induced intrinsic fluorescence signal or D2G FRET in proteoliposomes (Fig. 6). The binding of substrates was also investigated by measuring FRET directly in RSO and ISO membrane vesicles, which does not involve any solubilization or purification process. Although the MelB protein content of these vesicles detected with the His tag-specific reagent HRP was comparable with C-less, no signal of the sugar analog D2G could be detected, a result consistent with the lack of sugar binding concluded from the IRdiff and intrinsic fluorescence data (data not shown).
FIGURE 6.
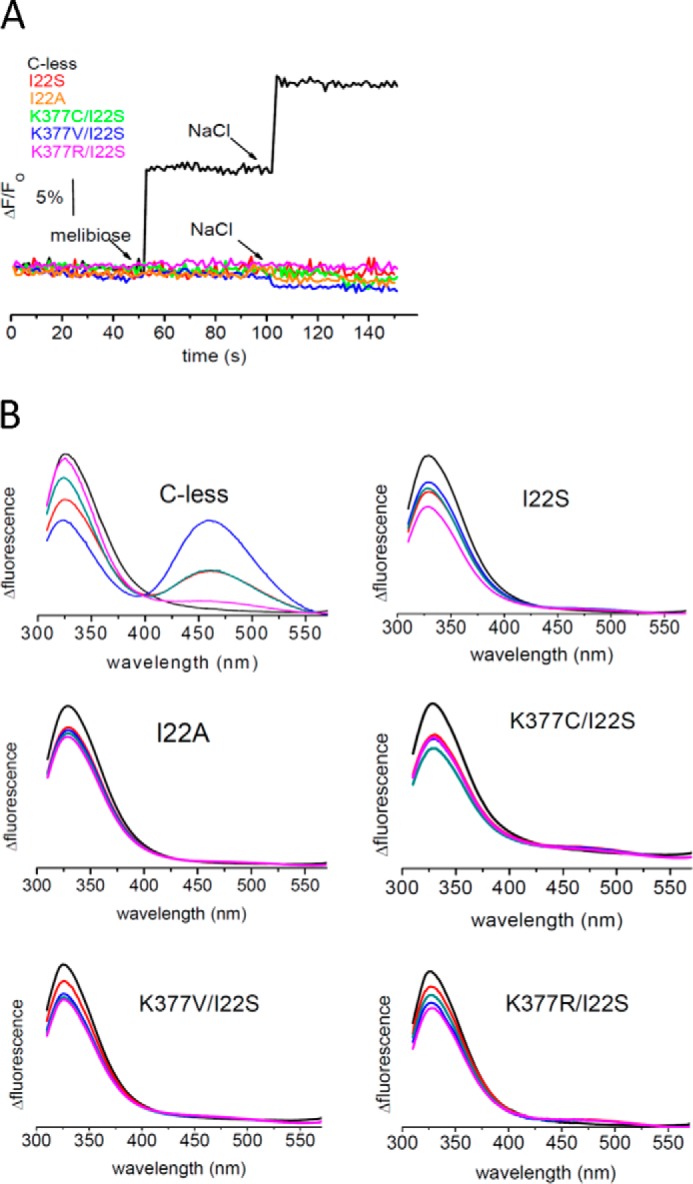
Substrate-induced changes in protein fluorescence emission and FRET of MelB in proteoliposomes. A, time course of Trp fluorescence intensity upon substrates incubation in proteoliposomes (λex = 290 nm, λem = 340 nm) for the indicated MelB mutants. B, FRET signal between MelB Trp and D2G (λex = 290 nm) in proteoliposomes. The experimental conditions were as indicated in Fig. 4.
Molecular Dynamics Simulations
The MelBST-A crystal structure (6) was used as a template for E. coli MelB by mutating all side chains of MelBST that differ from those of MelB (a total of 60 replacements). After a short energy minimization in vacuo to remove steric clashes, the MelB structure was included in a bilayer of POPE because the major head group component of the E. coli membrane lipids is phosphatidylethanolamine (40). Layers of waters including accessible areas of the protein interior and 0.3 m NaCl were added to achieve a neutral system (Fig. 7A). The system was first energy-minimized, followed by an equilibration step to warm up slowly from 0 to 298 K, and then two replicas of molecular dynamics were performed. The plot of the RMSD of the Cα backbone atoms shows that the protein drift from the initial structure stabilizes at about 20 ns to a value of around 4 Å (Fig. 7B). At 2 ns, the RMSD is 2.2 Å, providing evidence for good dynamic stability of the simulation (41). No entry of Na+ was detected in these simulations. From the very beginning of the simulation, Lys-377NZ (HXI) moved from interacting with Tyr-120OH (HIV) to the dyad Asp-59/Asp-55 (HII) (Fig. 7A), forming a bidentate ionic bond (about 3.7 Å between the NZ atom of Lys-377 (HIV) and the CG atom of either Asp-59 or Asp-55 in helix II). The stabilized distance between Lys-377NZ (HXI) and Tyr-120OH (HIV) was around 10 Å (Fig. 7C). On the other hand, Asp-124 (HIV) moved to form another cluster with Arg-149 (HV), Lys-18, and Asp-19 (HI), with a stabilized distance of 2.7 Å between Asp-124OD1 (HIV) and Lys-18NZ (HI). The two cationic clusters appear to be separated by Tyr-120 in helix IV (Fig. 7A).
FIGURE 7.

Molecular dynamics of MelB. A, the simulated system after equilibration for 200 ps. The expanded figure shows a snapshot of the proposed cation binding site environment after 200 ns of simulation. The protein is shown as a schematic (blue), with the cytoplasmic side at the bottom. The POPE bilayer (light blue) and the waters (red) are drawn in lines. Na+ (blue) and Cl− (red) are drawn in a van der Waals representation. The MelBST-A crystal structure (PDB code 4M64) (6) was mutated in all amino acids corresponding to those of MelB, minimized briefly, and included in a bilayer of POPE with layers of water molecules and NaCl 0.3 m finally added (see “Experimental Procedures”). The figure was generated with VMD software (25). B, drift from the initial structure as measured by the RMSD of backbone Cα atoms. C, time course of the specified distances, indicative of the location of the NZ atom of Lys-377 interacting with the CG atom of Asp-55 and Asp-59 but distant from the OH atom of Tyr-120 or the CG atom of Asp-124.
On the other hand, Asp-59 (HII) is located in the crystal structure in a non-polar environment that could raise its pKa and, therefore, remain protonated at neutral pH according to calculations (6). To check for the effect of this protonation state, we repeated the same MD simulations by keeping Asp-59 protonated. In this case, Lys-377NZ (HXI) moved from interacting with Tyr-120OH (HIV) toward Asp-55 (HII) with a stabilized distance of 3.5 Å between Asp-55CG and Lys-377NZ (Fig. 8, A and B). The distance between Lys-377NZ (HXI) and Tyr-120OH (HIV) was around 8 Å (Fig. 8B). Therefore, regardless of the protonated state of Asp-59, Lys-377 moved away from Tyr-120OH and formed ionic interactions with the Asp-55 side chain and also with the deprotonated form of the Asp-59 side chain during the simulations. The behavior of Asp-124 (HIV) was similar to that of deprotonated Asp-59: it moved toward the cluster formed by Lys-18, Asp-19, and Arg-149 with a stabilized distance of 2.7 Å between Asp-124OD1 and Lys-18NZ.
FIGURE 8.
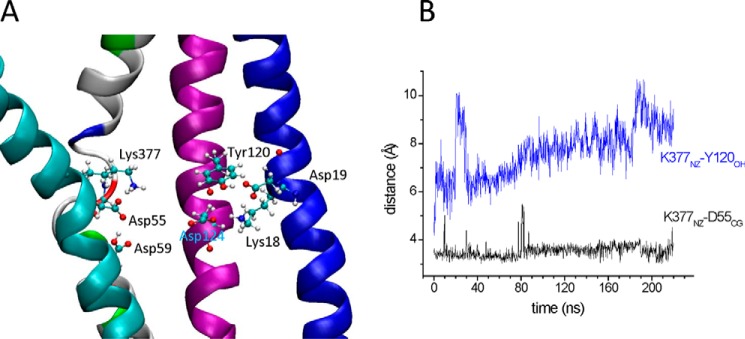
Simulation of MelB with Asp-59 protonated. A, snapshot of the substrate binding sites region of MelB with Asp-59 protonated after 220 ns of simulation. B, time course of the distances between the NZ atom of Lys-377 and the CG atom of Asp-55 or the OH atom of Tyr-120.
Asp-124 Moves toward the Cationic Site upon Na+ Binding
It has been hypothesized that Asp-55 and Asp-59 are ligands to Na+. Furthermore, a conformational change involving Asp-124 has been suggested to follow Na+ binding (5, 6). To find out the effect of Na+ binding on the Lys-377 (HXI) and the Asp-55/Asp-59 dyad (HII), we manually inserted an Na+ in place of a water molecule located between the two Asp side chains and performed MD simulations as above. The cation remained close to both side chains during the length of the simulations, whereas Asp-124 (HVI) moved toward the Na+ (Fig. 9A). Therefore, when Na+ binds to its putative binding site, Asp-124 points toward the Asp-55/Asp-59/Lys-377/Na+ cluster, leaving the Lys-18/Asp-19/Arg-149 region. It is apparent that the three acidic side chains become Na+ ligands and that the complex is stabilized by Lys-377 to compensate for the excess of negative charge (Fig. 9B).
FIGURE 9.
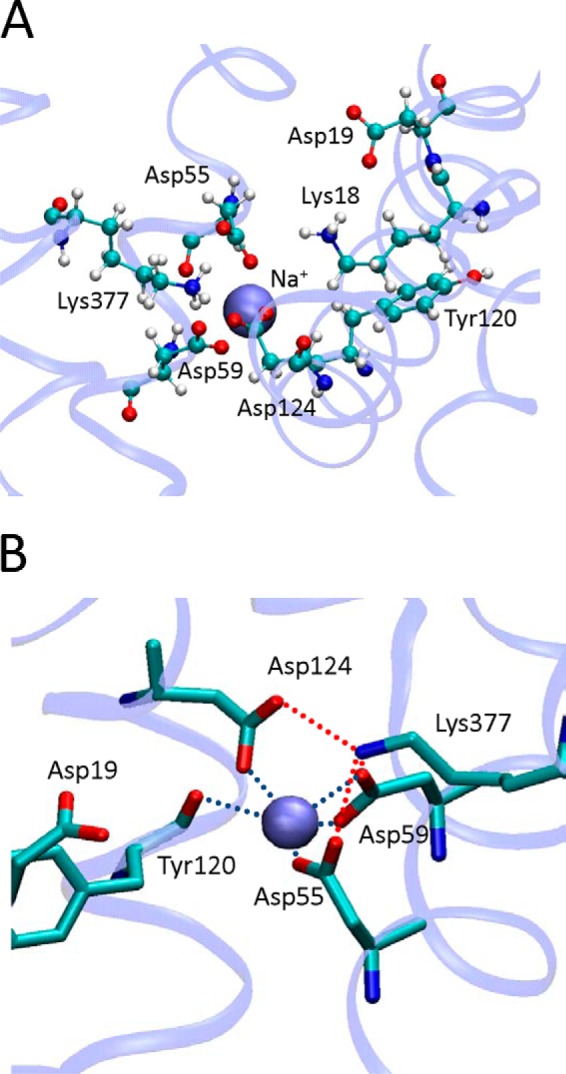
Organization of the Na+ binding site. A, snapshot of the proposed binding site environment of MelB with a Na+ positioned initially between Asp-55 and Asp-59 after 330 ns of simulation. B, detail of the Na+ binding site showing the ligands from the protein.
Discussion
In this work, the important role of Lys-377 in the MelB structure and transport mechanism is demonstrated. Mutants of Lys-377 that have been found previously to be deficient in sugar transport (8–10) are shown here to be unable to bind Na+ and melibiose, as shown by infrared difference spectroscopy and fluorescence experiments.
As first proposed by Wilson and co-workers (8–10), it is likely that Lys-377 (HXI) interacts with Asp-59 and Asp-55 (HII) to form an ionic complex, at least during certain steps of the transport cycle. However, in the crystal structure of MelBST, Lys-377 interacts with Tyr-120 and not with Asp-55 or Asp-59 (6). Furthermore, the apparent pKa of Asp-59 may attain values of around 9, according to empirical calculations (6), implying a protonated neutral side chain at neutral pH not very well suited for being a ligand for Na+. However, it is not clear whether the interaction of Lys-377 with Tyr-120, or the apparent high pKa of Asp-59 inferred in the crystal structure corresponds to the real state of the empty MelB. The crystal lattice might provide an environment differing from the lipidic membrane, exacerbated by the artificially low temperatures commonly used to protect the crystal from x-ray radiation damage. In addition, the relative low resolution of the electron density provided by the diffraction pattern (3.35 Å) might be insufficient for an unambiguous location of amino acid side chains and for the resolution of internal water molecules.
MD simulations of MelB embedded in a POPE bilayer show that Lys-377 leaves the interaction with Tyr-120 (as observed in the outward-facing, partially occluded conformation of MelBST in the crystal structure) and points toward Asp-55 and Asp-59. In this manner, in the empty transporter, the negative cluster formed by Asp-55 and Asp-59 is stabilized by Lys-377 and by water molecules. The ionic bond Lys-377-Asp-59 and the surrounding water molecules increase the polarity of the carboxylate group, therefore decreasing the apparent pKa of Asp-59 to values slightly below 4, as estimated with PROPKA (42). When Asp-59 is set as protonated during the MD simulations, Lys-377 points toward Asp-55. Therefore, a structural role essential for the correct formation of the active site of the cosubstrate is envisaged for Lys-377. Earlier studies have shown an absence of Na+-coupled melibiose transport for mutants of Asp-55, Asp-59, or Asp-124 (43–46). In a previous work, we studied mutants of these side chains and showed that Asp-55 and Asp-59 are essential ligands for Na+ (5). Regarding the role of Asp-124, we showed that it is an essential residue for sugar binding, required as well for normal Na+ binding and Na+-induced conformational changes (5). Most likely, Asp-124 interacts with Na+ when the cation is bound to Asp-55 and Asp-59 (5, 6). Furthermore, a close proximity between Asp-55 and Asp-59 is probably required for Na+ binding because we know that mutants of Asp-55 or Asp-59 alone lack Na+ binding (5, 43–46). We therefore suggest that the main role of Lys-377 is to stabilize the inner region of MelB by compensating repulsive interactions between the anionic side chains of Asp-55 and Asp-59. On the other hand, it is not implausible that, besides its stabilizing role, Lys-377 could also act as ligand to the sugar. In this line, the involvement of Lys side chains in sugar binding has been documented in several sugar transporters, such as the lactose permease LacY (47) or the sodium galactose transporter vSGLT (48). The importance of Lys-377 forming a cluster with Asp-55 and Asp-59 is fully consistent with the total level of conservation of Lys-377 among the members of the glucoside-pentoside-hexuronide:cation symporter family, including the lactose permease from Streptococcus thermophilus and the isoprimeverose permease from Lactobacillus pentosus. Asp-55 and Asp-59 also show high conservation. Even the mammalian MFSD2A transporter shows these three amino acids in an equivalent position (49, 50).
On the other hand, the structure of the empty transporter after MD simulation shows that Ile-22 outlines a second charged cluster formed by Lys-18, Asp-19, Arg-149, and Asp-124 (Fig. 7A). The side chains of Asp-19 and Asp-124 are essential for transport because they are putative ligands for the sugar (5), whereas Arg-149 has a structural role (39). We performed experiments on Ile-22 mutants showing that it is irrelevant in Na+ binding but that it does play a role in melibiose binding. A possible role of Ile-22 as a ligand for melibiose seems contradictory with the reported active transport of Ile-22 mutants (11). However, a situation of low affinity but high transport turnover could account for the observed 40% transport of I22C in ISO vesicles and 87% transport in DW1 cells (11). It is of help to consider other mutations involving Ile-22. Therefore, I22S has been described as partly compensating the loss of the negative charge in E142C (4), and mutations at Ile-22 have been found as second-site revertants for A350C, A383C, L391C, and G395C (8, 51). Given the diversity of locations of these side chains, it seems evident that further experiments are needed to establish an unequivocal role for Ile-22. Therefore, two areas for the binding of the cosubstrates become apparent, connected by Asp-124 (HIV). The cationic site comprises at least the Asp-55 and Asp-59 side chains (HII), with participation of Asp-124 (HIV). The sugar site comprises at least the Asp-19 side chain and, possibly, Lys-18 (HI), with additional participation of Asp-124. Tyr-120 (HIV) appears to separate the two areas. Being located between the two binding sites, Asp-124 most probably can orient indistinctly toward the cationic or the sugar site, giving rise to conformational changes upon cation binding that increase the sugar affinity, as proposed previously (5, 6). The MD simulations run after placing a Na+ in the Asp-55/Asp-59 environment clearly give credit to this presumption because, in this case, Asp-124 moves toward the Asp-55/Asp-59 site.
Interestingly, MD simulations support suggestions claiming a particular nature of Na+ ligands in MelB compared with other transporters (1, 5, 6, 44–46, 52). As depicted in Fig. 9, at the end of 330-ns MD, the Na+ is ligated by three Asp side chains, with distances between the Na+ and the oxygen atoms ranging from 2.2 to 2.7 Å (one from Asp-55, two from Asp-59, and one from Asp-124). A fifth ligand corresponds to the backbone O from Tyr-120 (2.5 Å). In contrast, in other transporters of known structure, the Na+ ligands mostly comprise oxygen carbonyls from the peptide backbone (53–56).
Our approach permits further exploration of the role of Arg-149 (39). On one hand, its location forming an ionic bond with Asp-19 in the crystallographic structure suggests that it could serve as a ligand for the sugar (6). However, at the end of the simulation, either in the presence or the absence of Na+, Arg-149 is 9–10 Å from Asp-19 and is interacting with Trp-342 and Trp-128. Moreover, it has been shown that the R149C mutant in ISO vesicles or reconstituted into liposomes is able to bind melibiose, whereas R149C in RSO vesicles does not (39). Therefore, in this latter work, a role in the inward/outward reorientation mechanism has been proposed for Arg-149. Therefore, mutation of Arg-149 may not much affect the sugar affinity, as observed experimentally (39), but, instead, it may abolish or greatly impede the initiation of conformational changes leading to the inward-open conformation and, therefore, hamper sugar transport.
In conclusion, the most salient information derived from this study of MelB is the implication of a cluster of three Asp residues of the N-terminal domain (HII-HIV) in the coordination of the coupling Na+ and its stabilization by Lys-377 in the C-terminal helix XI. Combining this knowledge with that of the recently solved three-dimensional structure of this permease opens a tantalizing perspective to gain insights not only into the molecular basis of the unique ionic selectivity profile of MelB or that of other members of the GPH family but also into the coupling process governing the ion (or H+)-coupled transport mechanism of these permeases.
Acknowledgments
The Nanoscale Molecular Dynamics program was developed by the Theoretical and Computational Biophysics Group in the Beckman Institute for Advanced Science and Technology at the University of Illinois at Urbana-Champaign. We thank Alex Perálvarez-Marin and Arnau Cordomí for critical reading of the manuscript and Elodia Serrano and Neus Ontiveros for technical assistance.
This work was supported by Ministerio de Ciencia e Innovación and Fondo Europeo de Desarrollo Regional Grant BFU2012-40137-C02--01 and, in part, by a grant from the Commissariat à l'Energie Atomique (CEA-Saclay).
- D2G
- 2′-(N-dansyl)-aminoethyl-1-thio-d-galactopyranoside
- RSO
- right-side out
- ISO
- inside-out
- POPE
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- VMD
- Visual Molecular Dynamics
- RMSD
- root-mean-square deviation
- IRdiff
- infrared difference spectra
- MD
- molecular dynamics.
References
- 1. Pourcher T., Bassilana M., Sarkar H. K., Kaback H. R., Leblanc G. (1990) The melibiose/Na+ symporter of Escherichia coli: kinetic and molecular properties. Philos. Trans. R Soc. Lond. B Biol. Sci. 326, 411–423 [DOI] [PubMed] [Google Scholar]
- 2. Poolman B., Knol J., van der Does C., Henderson P. J., Liang W. J., Leblanc G., Pourcher T., Mus-Veteau I. (1996) Cation and sugar selectivity determinants in a novel family of transport proteins. Mol. Microbiol. 19, 911–922 [DOI] [PubMed] [Google Scholar]
- 3. Bassilana M., Pourcher T., Leblanc G. (1988) Melibiose permease of Escherichia coli: characteristics of co-substrates release during facilitated diffusion reactions. J. Biol. Chem. 263, 9663–9667 [PubMed] [Google Scholar]
- 4. Meyer-Lipp K., Séry N., Ganea C., Basquin C., Fendler K., Leblanc G. (2006) The inner interhelix loop 4–5 of the melibiose permease from Escherichia coli takes part in conformational changes after sugar binding. J. Biol. Chem. 281, 25882–25892 [DOI] [PubMed] [Google Scholar]
- 5. Granell M., León X., Leblanc G., Padrós E., Lórenz-Fonfría V. A. (2010) Structural insights into the activation mechanism of melibiose permease by sodium binding. Proc. Natl. Acad. Sci. U.S.A. 107, 22078–22083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ethayathulla A. S., Yousef M. S., Amin A., Leblanc G., Kaback H. R., Guan L. (2014) Structure-based mechanism for Na+/melibiose symport by MelB. Nat. Commun. 5, 3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Radestock S., Forrest L. R. (2011) The alternating-access mechanism of MFS transporters arises from inverted-topology repeats. J. Mol. Biol. 407, 698–715 [DOI] [PubMed] [Google Scholar]
- 8. Ding P. Z., Wilson T. H. (2000) Physiological evidence for an interaction between helix XI and helices I, II, and V in the melibiose carrier of Escherichia coli. Biochem. Biophys. Res. Commun. 268, 409–413 [DOI] [PubMed] [Google Scholar]
- 9. Ding P. Z., Wilson T. H. (2000) The melibiose carrier of Escherichia coli: cysteine substitutions for individual residues in helix XI. J. Membr. Biol. 174, 135–140 [DOI] [PubMed] [Google Scholar]
- 10. Franco P. J., Jena A. B., Wilson T. H. (2001) Physiological evidence for an interaction between helices II and XI in the melibiose carrier of Escherichia coli. Biochim. Biophys. Acta 1510, 231–242 [DOI] [PubMed] [Google Scholar]
- 11. Ding P. Z., Wilson T. H. (2001) Cysteine mutagenesis of the amino acid residues of transmembrane helix I in the melibiose carrier of Escherichia coli. Biochemistry 40, 5506–5510 [DOI] [PubMed] [Google Scholar]
- 12. Mus-Veteau I., Pourcher T., Leblanc G. (1995) Melibiose permease of Escherichia coli: substrate-induced conformational changes monitored by tryptophan fluorescence spectroscopy. Biochemistry 34, 6775–6783 [DOI] [PubMed] [Google Scholar]
- 13. Maehrel C., Cordat E., Mus-Veteau I., Leblanc G. (1998) Structural studies of the melibiose permease of Escherichia coli by fluorescence resonance energy transfer: I: evidence for ion-induced conformational change. J. Biol. Chem. 273, 33192–33197 [DOI] [PubMed] [Google Scholar]
- 14. León X., Lórenz-Fonfría V. A., Lemonnier R., Leblanc G., Padrós E. (2005) Substrate-induced conformational changes of melibiose permease from Escherichia coli studied by infrared difference spectroscopy. Biochemistry 44, 3506–3514 [DOI] [PubMed] [Google Scholar]
- 15. Botfield M. C., Wilson T. H. (1988) Mutations that simultaneously alter both sugar and cation specificity in the melibiose carrier of Escherichia coli. J. Biol. Chem. 263, 12909–12915 [PubMed] [Google Scholar]
- 16. Pourcher T., Leclercq S., Brandolin G., Leblanc G. (1995) Melibiose permease of Escherichia coli: large scale purification and evidence that H+, Na+, and Li+ sugar symport is catalyzed by a single polypeptide. Biochemistry 34, 4412–4420 [DOI] [PubMed] [Google Scholar]
- 17. Weissborn A. C., Botfield M. C., Kuroda M., Tsuchiya T., Wilson T. H. (1997) The construction of a cysteine-less melibiose carrier from E. coli. Biochim. Biophys. Acta 1329, 237–244 [DOI] [PubMed] [Google Scholar]
- 18. Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 19. Kaback H. R. (1971) Bacterial membranes. Methods Enzymol. XXII, 99–120 [Google Scholar]
- 20. Lórenz-Fonfría V. A., León X., Padrós E. (2012) Studying substrate binding to reconstituted secondary transporters by attenuated total reflection infrared difference spectroscopy. Methods Mol. Biol. 914, 107–126 [DOI] [PubMed] [Google Scholar]
- 21. Woolf T. B., Roux B. (1996) Structure, energetics, and dynamics of lipid-protein interactions: A molecular dynamics study of the gramicidin A channel in a DMPC bilayer. Proteins 24, 92–114 [DOI] [PubMed] [Google Scholar]
- 22. Jo S., Kim T., Im W. (2007) Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE 2, e880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jo S., Lim J. B., Klauda J. B., Im W. (2009) CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J. 97, 50–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu E. L., Cheng X., Jo S., Rui H., Song K. C., Davila-Contreras E. M., Qi Y., Lee J., Monje-Galvan V., Venable R. M., Klauda J. B., Im W. (2014) CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comp. Chem. 35, 1997–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lomize M. A., Lomize A. L., Pogozheva I. D., Mosberg H. I. (2006) OPM: orientations of proteins in membranes database. Bioinformatics 22, 623–625 [DOI] [PubMed] [Google Scholar]
- 27. Humphrey W., Dalke A., Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 27–38 [DOI] [PubMed] [Google Scholar]
- 28. Harvey M. J., Giupponi G., De Fabritiis G. (2009) ACEMD: accelerated molecular dynamics simulations in the microsecond timescale. J. Chem. Theory Comput. 5, 1632–1639 [DOI] [PubMed] [Google Scholar]
- 29. Feller S. E., MacKerell A. D. (2000) An improved empirical potential energy function for molecular simulations of phospholipids. J. Phys. Chem. B 104, 7510–7515 [Google Scholar]
- 30. MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 31. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 32. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., Pedersen L. G. (1995) A smooth Particle Mesh Ewald Method. J. Chem. Phys. 103, 8577–8593 [Google Scholar]
- 33. Harvey M. J., De Fabritiis G. (2009) An implementation of the smooth particle-mesh Ewald (PME) method on GPU hardware. J. Chem. Theory Comput. 5, 2371–2377 [DOI] [PubMed] [Google Scholar]
- 34. Feenstra K. A., Hess B., Berendsen H. J. C. (1999) Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J. Comp. Chem. 20, 786–798 [DOI] [PubMed] [Google Scholar]
- 35. Barth A. (2000) The infrared absorption of amino acid side chains. Prog. Biophys. Mol. Biol. 74, 141–173 [DOI] [PubMed] [Google Scholar]
- 36. Goormaghtigh E., Raussens V., Ruysschaert J. M. (1999) Attenuated total reflection infrared spectroscopy of proteins and lipids in biological membranes. Biochim. Biophys. Acta 1422, 105–185 [DOI] [PubMed] [Google Scholar]
- 37. Barth A., Zscherp C. (2002) What vibrations tell us about proteins. Q Rev. Biophys. 35, 369–430 [DOI] [PubMed] [Google Scholar]
- 38. Li C., Kumar S., Montigny C., le Maire M., Barth A. (2014) Quality assessment of recombinant proteins by infrared spectroscopy. Characterisation of a protein aggregation related band of the Ca2+-ATPase. Analyst 139, 4231–4240 [DOI] [PubMed] [Google Scholar]
- 39. Lin Y., Fuerst O., Granell M., Leblanc G., Lórenz-Fonfría V., Padrós E. (2013) The substitution of Arg149 with Cys fixes the melibiose transporter in an inward-open conformation. Biochim. Biophys. Acta 1828, 1690–1699 [DOI] [PubMed] [Google Scholar]
- 40. Dowhan W. (1997) Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu. Rev. Biochem. 66, 199–232 [DOI] [PubMed] [Google Scholar]
- 41. Law R. J., Capener C., Baaden M., Bond P. J., Campbell J., Patargias G., Arinaminpathy Y., Sansom M. S. (2005) Membrane protein structure quality in molecular dynamics simulation. J. Mol. Graph. Model. 24, 157–165 [DOI] [PubMed] [Google Scholar]
- 42. Rostkowski M., Olsson M. H., Søndergaard C. R., Jensen J. H. (2011) Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Struct. Biol. 11, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pourcher T., Deckert M., Bassilana M., Leblanc G. (1991) Melibiose permease of Escherichia coli: mutation of aspartic acid 55 in putative helix II abolishes activation of sugar binding by Na+ ions. Biochem. Biophys. Res. Commun. 178, 1176–1181 [DOI] [PubMed] [Google Scholar]
- 44. Pourcher T., Zani M. L., Leblanc G. (1993) Mutagenesis of acidic residues in putative membrane-spanning segments of the melibiose permease of Escherichia coli: I: effect on Na+-dependent transport and binding properties. J. Biol. Chem. 268, 3209–3215 [PubMed] [Google Scholar]
- 45. Zani M. L., Pourcher T., Leblanc G. (1993) Mutagenesis of acidic residues in putative membrane-spanning segments of the melibiose permease of Escherichia coli: II: effect on cationic selectivity and coupling properties. J. Biol. Chem. 268, 3216–3221 [PubMed] [Google Scholar]
- 46. Wilson D. M., Wilson T. H. (1992) Asp-51 and Asp-120 are important for the transport function of the Escherichia coli melibiose carrier. J. Bacteriol. 174, 3083–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Abramson J., Smirnova I., Kasho V., Verner G., Iwata S., Kaback H. R. (2003) The lactose permease of Escherichia coli: overall structure, the sugar-binding site and the alternating access model for transport. FEBS Lett. 555, 96–101 [DOI] [PubMed] [Google Scholar]
- 48. Faham S., Watanabe A., Besserer G. M., Cascio D., Specht A., Hirayama B. A., Wright E. M., Abramson J. (2008) The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science 321, 810–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Angers M., Uldry M., Kong D., Gimble J. M., Jetten A. M. (2008) Mfsd2a encodes a novel major facilitator superfamily domain-containing protein highly induced in brown adipose tissue during fasting and adaptive thermogenesis. Biochem. J. 416, 347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nguyen L. N., Ma D., Shui G., Wong P., Cazenave-Gassiot A., Zhang X., Wenk M. R., Goh E. L., Silver D. L. (2014) Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 509, 503–506 [DOI] [PubMed] [Google Scholar]
- 51. Ding P. Z. (2004) Loop X/XI, the largest cytoplasmic loop in the membrane-bound melibiose carrier of Escherichia coli, is a functional re-entrant loop. Biochim. Biophys. Acta 1660, 106–117 [DOI] [PubMed] [Google Scholar]
- 52. Yousef M. S., Guan L. (2009) A 3D structure model of the melibiose permease of Escherichia coli represents a distinctive fold for Na+ symporters. Proc. Natl. Acad. Sci. U.S.A. 106, 15291–15296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yamashita A., Singh S. K., Kawate T., Jin Y., Gouaux E. (2005) Crystal structure of a bacterial homologue of Na+/Cl-dependent neurotransmitter transporters. Nature 437, 215–223 [DOI] [PubMed] [Google Scholar]
- 54. Boudker O., Ryan R. M., Yernool D., Shimamoto K., Gouaux E. (2007) Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature 445, 387–393 [DOI] [PubMed] [Google Scholar]
- 55. Ressl S., Terwisscha van Scheltinga A. C., Vonrhein C., Ott V., Ziegler C. (2009) Molecular basis of transport and regulation in the Na+/betaine symporter BetP. Nature 458, 47–52 [DOI] [PubMed] [Google Scholar]
- 56. Khafizov K., Perez C., Koshy C., Quick M., Fendler K., Ziegler C., Forrest L. R. (2012) Investigation of the sodium-binding sites in the sodium-coupled betaine transporter BetP. Proc. Natl. Acad. Sci. U.S.A. 109, E3035–E3044 [DOI] [PMC free article] [PubMed] [Google Scholar]