

Background: Phlpp1 is a tumor suppressor that represses Akt2 and other signaling pathways.
Results: Chondrocyte proliferation, matrix production, Akt2 phosphorylation, and Fgf18/Erk1/2 signaling were increased in Phlpp1−/− mice, but levels of the transcription factor FoxO1 were reduced.
Conclusion: Phlpp1 deficiency increases Akt2 activity, which diminishes FoxO1 levels and induces Fgf18 expression to stimulate Erk1/2 activity and chondrocyte proliferation.
Significance: Phlpp1 inhibition may promote cartilage regeneration.
Keywords: Akt PKB, cartilage, fibroblast growth factor (FGF), fibroblast growth factor receptor (FGFR), FOXO, osteoarthritis
Abstract
Endochondral ossification orchestrates formation of the vertebrate skeleton and is often induced during disease and repair processes of the musculoskeletal system. Here we show that the protein phosphatase Phlpp1 regulates endochondral ossification. Phlpp1 null mice exhibit decreased bone mass and notable changes in the growth plate, including increased BrdU incorporation and matrix production. Phosphorylation of known Phlpp1 substrates, Akt2, PKC, and p70 S6 kinase, were enhanced in ex vivo cultured Phlpp1−/− chondrocytes. Furthermore, Phlpp1 deficiency diminished FoxO1 levels leading to increased expression of Fgf18, Mek/Erk activity, and chondrocyte metabolic activity. Phlpp inhibitors also increased matrix content, Fgf18 production and Erk1/2 phosphorylation. Chemical inhibition of Fgfr-signaling abrogated elevated Erk1/2 phosphorylation and metabolic activity in Phlpp1-null cultures. These results demonstrate that Phlpp1 controls chondrogenesis via multiple mechanisms and that Phlpp1 inhibition could be a strategy to promote cartilage regeneration and repair.
Introduction
Skeletal formation is accomplished in part through the process of endochondral ossification. In this developmental program, a cartilage intermediate first arises to serve as a template for de novo bone formation (reviewed in Ref. 1). Strict control of chondrocyte maturation during the cartilaginous phase and subsequent coupling to bone formation ensures acquisition of optimal bone length, density, and shape. Disruption of this process during embryogenesis causes chondrodysplasias and growth plate injuries during adolescence can cause limb shortening (2, 3). Cartilage-specific genetic deficiency of anabolic factors can also result in decreased bone density, highlighting the importance of growth plate cartilage coupling to bone formation (4–6). The endochondral ossification process is reinitiated in adults during fracture healing, heterotopic ossification, and osteoarthritic progression (7).
A number of growth factors and cytokines (e.g. BMPs, Fgfs, Igf1, Hhs, Tgfβ) contribute to the formation and maturation of the cartilaginous phase of endochondral ossification (1). Many of these morphogens directly or indirectly activate Akt (protein kinase B) isoforms that are essential for chondrocyte differentiation (8–16). Akt activity is controlled by phosphorylation and substrate interactions via scaffolding proteins. A number of phosphatases, including PH-domain leucine-rich repeat protein phosphatases (Phlpp)2 1/2, dampen Akt activity to promote apoptosis and inhibit cell proliferation (17–19). Genetic deletion of Phlpp1 (also known as Phlpp, Scop, Plekhe1) slightly reduced snout-to-tail body lengths, but the effects of Phlpp1 on chondrocytes or growth plate structure were not studied (20).
We previously demonstrated that Hdac3 represses Phlpp1 transcription and that Phlpp1 is expressed at higher levels in Hdac3 suppressed chondrocytes (21). Here we examined the effects of Phlpp1 deficiency on skeletal development. Phlpp1−/− mice show increased chondrocyte proliferation and matrix production. Molecular analyses demonstrated Phlpp1 deficiency increased phosphorylation of Akt2, Fgfr3, Mek1/2, and Erk1/2. The elevation in Erk1/2 activity was attributed to increased production of Fgf18, as blocking Fgf receptor (Fgfr)-mediated signaling decreased Erk1/2 phosphorylation. Furthermore, Fgfr inhibition suppressed chondrocyte proliferation induced by Phlpp1 deficiency. We conclude that Phlpp1 deficiency promotes chondrocyte differentiation via multiple pathways.
Materials and Methods
Phlpp1-deficient Mice
Phlpp1−/− (20) and wild type (WT) littermates were maintained in an accredited facility with 12-hour light/dark cycles and supplied water and food (PicoLab® Rodent Diet 20, LabDiet) ad libitum. All animal research was conducted according to National Institutes of Health and the Institute of Laboratory Animal Resources, National Research Council guidelines. The Mayo Clinic Institutional Animal Care and Use Committee preapproved all animal studies. For BrdU labeling studies, mice (n = 5) were injected when they were 5 days old (P5) with 50 μg of 5-bromo-2′-deoxyuridine (BrdU) labeling reagent (Invitrogen) per kg body weight 2 h prior to euthanasia.
Microcomputed Tomography
Bone volume density in the secondary spongiosa of female distal femurs was measured with ex vivo micro-computed tomography (μCT) imaging (μCT35; Scanco Medical AG, Basserdorf, Switzerland). Trabecular bone scans were performed at 7 μm voxels with an energy setting of 70 kVp and a 300 ms integration time on 8-week-old Phlpp1−/− (n = 4) or WT (n = 3) mice.
Immunohistochemical Staining
Tibias from P5 Phlpp1−/− (n = 5) or WT (n = 5) littermates were fixed in 10% neutral buffered formalin, decalcified in 15% EDTA for 7 days, and paraffin embedded. Immunohistochemical (IHC) staining was performed with antibodies directed to BrdU, Ki67 (Cell Signaling Technologies, Boston, MA), or IgG isotype control. Detection was accomplished using the Mouse and Rabbit Specific HRP (ABC) Detection IHC Kit (AbCam, Cambridge, MA) using the substrate 3,3′-diaminobenzidine (Sigma Aldrich). Sections were counterstained with Alcian blue (1% Alcian blue, 3% acetic acid).
Isolation and Culture of Primary Immature Murine Chondrocytes
Primary chondrocytes were collected from P5 Phlpp1−/− or WT mice as previously described (22). Briefly, cartilage from the distal femur and proximal tibia were digested in 3 mg/ml collagenase for 1 h and then overnight in 0.5 mg/ml collagenase. The resulting cell suspension was plated in micromasses consisting of 2 × 105 cells in a 10-μl drop of DMEM. After 1 h, each well was flooded with DMEM supplemented with 5% FBS, 1% antimycotic/antibiotic for the indicated periods of time.
Chondrocyte Metabolic Activity Assays
WT or Phlpp1−/− chondrocytes (8 × 104/well) were cultured in monolayer 96-well plates for 4 days. Cultures were pre-treated with the indicated concentrations of PD173074, U0126 or DMSO for 1 h. CellTiter 96® AQueous One Solution Cell Proliferation Assays (Promega) were performed according to manufacturer's specifications. Briefly, 20 μl of a reagent containing 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) was added to each well and incubated for 2 h at 37 °C. Readings were taken at A490.
Alcian Blue Staining
Micromass cultures were fixed with 10% neutral buffered formalin and stained with Alcian blue for 2 h. Tibias from P5 or 4-week-old mice were fixed in 10% neutral buffered formalin, decalcified in 15% EDTA for 7 days, paraffin embedded, sectioned, and stained with Alcian blue.
ATDC5 Cell Culture
ATDC5 cells were maintained in DMEM supplemented with 10% FBS, 1% antimycotic/antibiotic. For experimental conditions, ATDC5 cells were cultured in DMEM, 5% FBS, 1% antimycotic/antibiotic at a density of 6 × 103cells/cm2. ATDC5 cells were transfected with plasmid mixtures of pcDNA3, pcDNA-Phlpp1ΔC, pcDNA-Phlpp1 (Addgene nos. 22931 and 22404 (28)), pcDNA3-FoxO1A3, or pcDNA3-FoxO1A3HR with Lipofectamine (Invitrogen) using a 1:3 Lipofectamine to DNA ratio as indicated. Cells were also cultured in micromass as described above and treated with the Phlpp inhibitor (NCS 117079) at concentrations shown (23).
Western Blotting
Cell lysates were collected in a buffered SDS solution (0.1% glycerol, 0.01% SDS, 0.1 m Tris, pH 6.8) on ice. Total protein concentrations were obtained with the Bio-Rad DC assay (Bio-Rad). Proteins (20 μg) were then resolved by SDS-PAGE and transferred to a polyvinylidene difluoride membrane. Western blotting was performed with antibodies (1:2000 dilution) for pSer371 p70 S6K, pThr202/Tyr204-Erk1/2, Erk1/2, pSer217/221 Mek1/2, Mek1/2, pSer473-Akt1, pSer474-Akt2, PAN pSer473 Akt, Akt, pSer256 FoxO1, FoxO1, pSer660-PKC, (Cell Signaling Technologies), pTyr724 Fgfr3, Fgfr3 (Abcam, Cambridge, MA), PKCα (BD Biosciences, San Jose, CA), HA (Covance, Princeton, NJ), Flag (Sigma Aldrich), and tubulin (Developmental Studies Hybridoma Bank, Iowa City, IA) and corresponding secondary antibodies conjugated to horseradish peroxidase (HRP) (Santa Cruz Biotechnology, Santa Cruz, CA). Antibody binding was detected with the Supersignal West Femto Chemiluminescent Substrate (Pierce). Blots were stripped using Restore Western blot Stripping Buffer (Pierce) and reprobed when needed. Each experiment was repeated at least three times, and data from a representative experiment are shown.
RNA Isolation and Real-time PCR
Total RNA was extracted from chondrocyte cultures using TRIzol (Invitrogen) and chloroform. Messenger RNA (2 μg) was reverse transcribed using the SuperScript III first-strand synthesis system (Invitrogen). The resulting cDNAs were used to assay gene expression via real-time PCR using the following gene-specific primers: Aggrecan (5′-CCGCTTGCCAGGGGGAGTTG-3′, 5′-GATGATGGGCGCACGCCGTA-3′), Bmp3 (5′-GACCGCGCGACAGGTGGTAG-3′, 5′-CGGCTCTGAAGCTGCGGACC-3′), Bmp4 (5′-AACATCCCAGGGACCAGTGA-3′, 5′-GGATGCTGCTGAGGTTGAAGA-3′), Bmp6 (5′-TAGCAATCTGTGGGTGGTGAC-3′, Col2a1 (5′-ACTGGTAAGTGGGGCAAGAC-3′, 5′-CCACACCAAATTCCTGTTCA-3′), 5′-ACCTCGCTCACCTTGAAGAAG-3′), Fgf1 (5′-TTCAGTCCAGGCACCCTGTG-3′, 5′-TGGTAGGCTCAGCACTGAAGAA-3′), Fgf2 (5′-GGCTGCTGGCTTCTAAGTGT-3′, 5′-GTCCCGTTTTGGATCCGAGT-3′), Fgf18 (5′-TGGGGAAGCCTGATGGTACT-3′, 5′-CCCTTGGGGTAACGCTTCAT-3′), Gapdh (5′-GGGAAGCCCATCACCATCTT-3′, 5′-GCCTCACCCCATTTGATGTT-3′), Ihh (5′-GCTTTCCTGCCGGAGCCCAG-3′, 5′-GGTGGGGGTCCCATCCTCCC-3′), PTHR (5′-XXX-3′, 5′-XXX-3′) PTHrP (5′-CTGGCCCTCGCATCCACGAC-3′, 5′-ACAGCCACTTGCGGCAGAGC-3′), Tgfβ2 (5′-GATAATTGCTGCCTTCGCCC-3′, 5′-GGCTGAGGACTTTGGTGTGT-3′), Tgfβ3 (5′-GATCACCACAACCCACACCT-3′, 5′-CCAGGTTGCGGAAGCAGTAA-3′), Type 2a1 collagen (5′-ACTGGTAAGTGGGGCAAGAC-3′, 5′-CCACACCAAATTCCTGTTCA-3′), Wnt2b (5′-TCCTGGTGGTACATAGGGGC-3′, 5′-GAGCGCATGATGTCTGGGTA-3′), Wnt5a (5′-CCACGCTAAGGGTTCCTATGAG-3′, 5′-ACGGCCTGCTTCATTGTTGT-3′) (24). Fold changes in gene expression for each sample were calculated using the 2−ΔΔCt method relative to control after normalization of gene-specific Ct values to Gapdh Ct values. Each experiment was performed in triplicate and repeated at least three times. The average of these experiments is reported.
Image Quantitation
Images were digitally scanned or collected using phase contrast microscopy. Densitometric values were obtained after converting each image to gray scale and measuring the mean gray values of each condition using Image J software.
Statistical Analysis
Data obtained are the means ± S.E. p values were determined with the Student's t test.
Results
Phlpp1 Deficiency Reduces Bone Length and Density
Phlpp1-deficient mice were previously reported to have slightly decreased snout-to-tail lengths and decreased body weight compared with their WT littermates (20). We confirmed shorter femur lengths (Fig. 1A) and further found a decrease in bone mineral density. Micro-CT analyses revealed that Phlpp1 deficiency decreased bone volume density by 23% at 8 weeks of age (Fig. 1, B, G, H). This was associated with a decrease in trabecular number (Tb.N) (Fig. 1C). Decreased bone mineral density was still apparent in mice at 12 weeks of age (data not shown). Trabecular thickness and trabecular spacing were not altered (Fig. 1, D and E). A trend toward decreased connective density was noted (Fig. 1F). Thus, Phlpp1 deficiency prevents bone lengthening and bone mass accrual.
FIGURE 1.
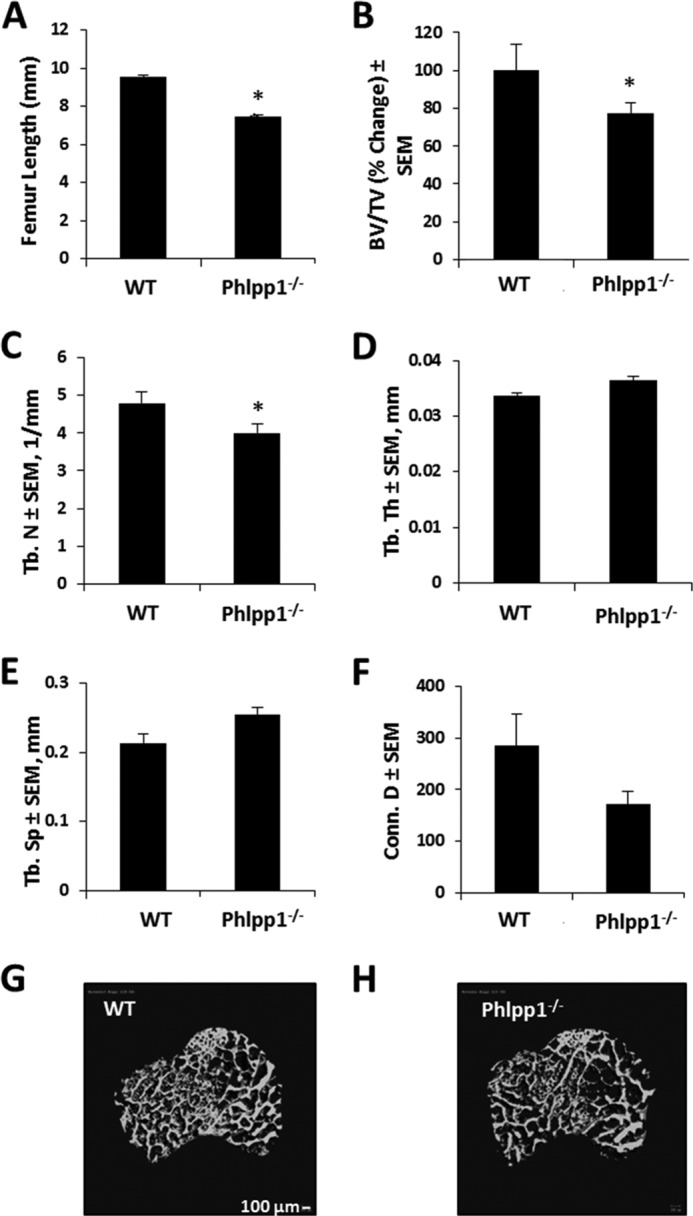
Phlpp1 regulates bone mass. A, femur lengths from 4-week-old animals. B, percent change in bone volume per total volume (BV/TV); (C) trabecular number (Tb. N); (D) trabecular thickness (Tb. Th); (E) trabecular spacing (Tb. Sp); and (F) connective density (Conn. D) of distal femurs from 8-week-old WT (n = 3) and Phlpp1−/− (n = 4) mice were determined by micro-computed tomography. *, p < 0.05 compared with control. G and H, microCT reconstructions are shown of cancellous bone and secondary spongiosa from distal femurs of 8-week-old WT and Phlpp1−/− mice.
Phlpp1 Suppresses Chondrocyte Proliferation and Matrix Production
To better understand how Phlpp1 deficiency might stunt bone lengthening and endochondral bone formation, we examined the growth plates of young mice. Tibial growth plates from Phlpp1−/− mice had 50% more cells per proliferative zone area as well as more intense Alcian blue staining (Fig. 2, A and B). BrdU labeling experiments revealed an increase in the number of BrdU-positive cells in Phlpp1-null mice (Fig. 2C). The number of Ki67-positive cells was also enhanced in Phlpp1−/− tibias (data not shown). Thus, despite shorter lengths and reduced endochondral ossification, growth plate chondrocyte proliferation, and matrix content appear to be increased in Phlpp1-deficient long bones.
FIGURE 2.
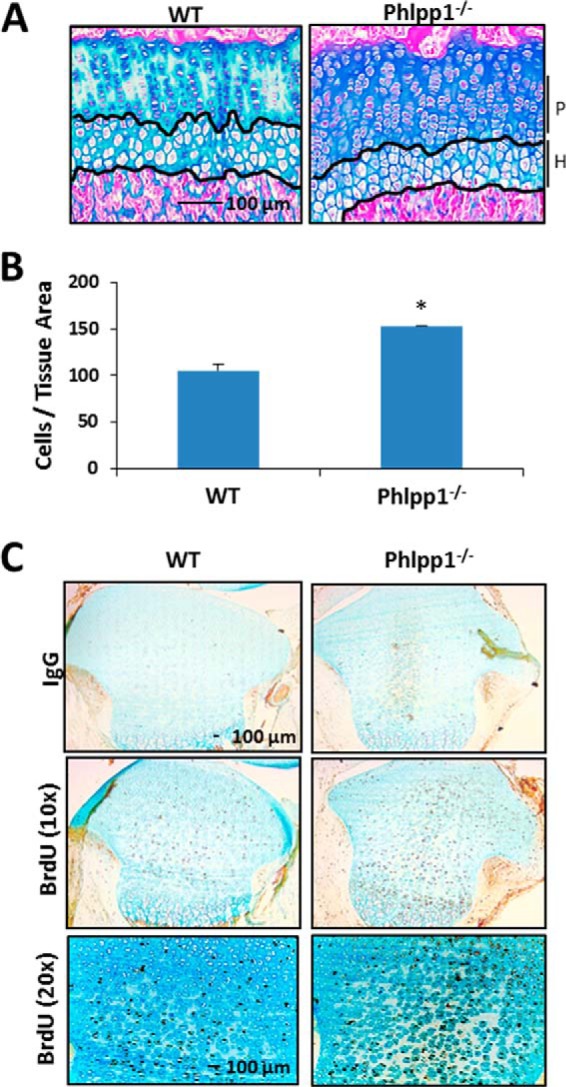
Phlpp1 deficiency increases the number of cells in the proliferative zone. A, sections from paraffin embedded tibias of 4-week-old WT (n = 3) or Phlpp1−/− (n = 4) mice were stained with Alcian blue. Proliferative (P) and hypertrophic (H) zones are noted. B, average number of cells/tissue area within the proliferative zone (P) was determined. C, BrdU staining of tibias from P5 mice, n = 5 WT, n = 5 Phlpp1−/−.
To confirm our in vivo observations, immature murine chondrocytes were cultured ex vivo in micromasses as previously described (21, 22). After 3 days, the chondrocyte micromasses derived from Phlpp1−/− mice stained more intensely with Alcian blue (Fig. 3A). Phlpp1−/− chondrocytes also produced more transcripts for two cartilage matrix genes, type 2a1 collagen (Col2) and aggrecan (Fig. 3, B and C). PTHR expression was slightly increased, but expression of Ihh and PTHrP was unchanged (Fig. 3, D–F). Consistent with data from other cell types (25), the phosphorylation of Akt2, but not Akt1, was increased in Phlpp1 deficient cultures (Fig. 3G). PKC phosphorylation also increased and PKCα was more abundant and stable, as previously observed (26). FoxO1 stability is negatively regulated by Akts, including Akt2 (27–30). In accordance with increased Akt2 activity, phosphorylated and inactive FoxO1 levels increased by 42%, and total levels were reduced by 20% in Phlpp1−/− cultures (Fig. 3G). Notably, we also observed increased activation of Mek1/2 and Erk1/2 in Phlpp1 KO chondrocytes (Fig. 3G).
FIGURE 3.

Phlpp1 deficiency enhances matrix production and Mek/Erk activation in chondrocytes. Chondrocyte micromass cultures from WT or Phlpp1−/− mice were cultured for 3 days. A, cultures were fixed and stained with Alcian blue. Relative expression levels of type 2a1 collagen (B) aggrecan (C), PTHR (D), PRTrP (E), and Ihh (F) were assayed by real time PCR. *, p < 0.05 compared with control. G, Western blotting was performed as indicated. This experiment was repeated three times with data from a representative experiment shown.
Phlpp1 Deficiency Increases Fgf18 Production and Activates Fgfr3, Mek, and Erk1/2 Signaling
To determine how Phlpp1 affects chondrocytes, we surveyed a panel of morphogens known to regulate cartilage development (Fig. 4A). Several cytokines (including Tgfβ2/3 and Wnt2b) were modestly higher in Phlpp1−/− cells, but the most highly induced gene of those tested was Fgf18 (Fig. 4A). Fgf18 binds to Fgfrs. To determine if increased phosphorylation of Mek1/2 and Erk1/2 was due to direct effects of Phlpp1 (19) or due to signaling through Fgfr3 (31–33), we examined Fgfr3 activation. Fgfr3 phosphorylation was increased in Phlpp1−/− chondrocytes (Fig. 3G). An Fgfr inhibitor, PD173074 (34, 35), decreased phosphorylation of Mek1/2 and Erk1/2 in Phlpp1−/− chondrocytes to WT levels (Fig. 4, B and C). Phlpp1−/− chondrocytes showed elevated MTS activity as compared with WT cells (Fig. 4D). The Fgfr inhibitor decreased MTS activity of Phlpp1−/− chondrocytes in a concentration-dependent fashion, but WT chondrocyte MTS activity was unaffected at any concentration (Fig. 4D). A Mek1/2 inhibitor, U0126, also decreased MTS activity of Phlpp1−/− chondrocytes at both concentrations tested (Fig. 4D). WT chondrocyte MTS activity was unaffected at 10 μm, but was suppressed at 50 μm. These results demonstrate that Phlpp1 suppresses chondrocyte metabolic activity by negatively impacting Fgfr/Mek1/2/Erk1/2 signaling.
FIGURE 4.

Phlpp1 deficiency increases Fgf18 expression and Erk1/2 signaling. A–C, Chondrocyte micromass cultures from WT or Phlpp1−/− mice were cultured for 3 days. A, expression levels of indicated morphogens in Phlpp1−/− cells relative to control WT cells were assayed by real-time PCR. The control value was set to 1. *, p < 0.1; **, p < 0.05 compared with control. B, immature chondrocytes from WT or Phlpp1−/− mice were cultured in the presence of 100 nm PD173074. Western blotting was performed with the indicated antibodies and (C) the average gray value of the normalized phospho-Erk1/2 signal was determined (n = 3, normalization against total Erk1/2). D and E, immature chondrocytes from WT or Phlpp1−/− mice were cultured in monolayer in the presence of the indicated concentrations of the Fgfr inhibitor (PD173074), the Mek1/2 inhibitor (U0126) or vehicle control (DMSO) for 4 days. MTS assays were performed. These experiments were repeated three times. Data from a representative experiment are shown. *, p < 0.05 compared with WT vehicle-treated cells; #, p < 0.05 compared with Phlpp1−/− vehicle-treated cells.
FoxO1 Represses Fgf18 Expression in Chondrocytes
FoxO1 represses Fgf18 transcription (36) and therefore may link enhanced Akt2 signaling to Erk1/2 activation in Phlpp1−/− chondrocytes. To test this hypothesis, a Phlpp1 deletion mutant (Phlpp1ΔC) that inhibits interactions between Akt2 and Phlpp1 was introduced in chondrogenic ATDC5 cells (18). Phlpp1ΔC promoted Fgf18 production (Fig. 5). This induction was blocked by expression of a constitutively active form of FoxO1 (FoxO1A3, Fig. 5). Furthermore, DNA binding of FoxO1 was required to repress Fgf18 expression as a FoxO1 mutant construct lacking the DNA-binding domain (FoxO1A3HR) failed to prevent Phlpp1ΔC-induced Fgf18 expression (Fig. 5). WT Phlpp1 also repressed Fgf18 expression (Fig. 5).
FIGURE 5.
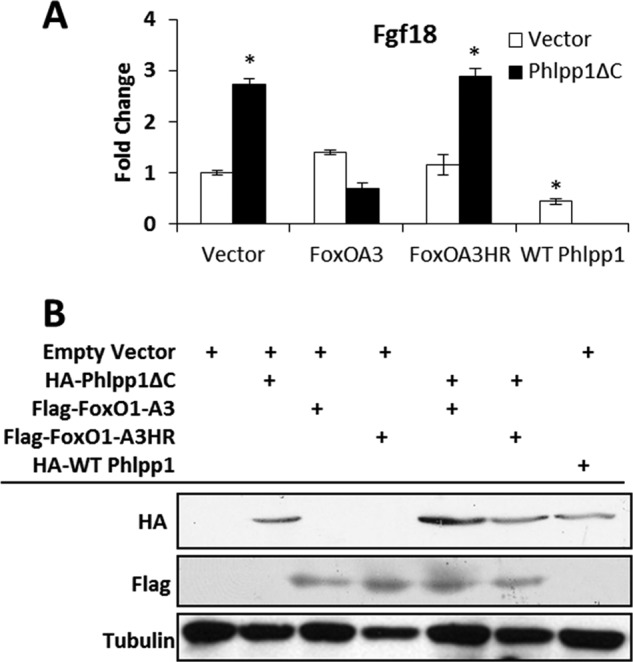
FoxO1 represses Fgf18 expression downstream of Phlpp1. A, ATDC5 cells were transfected with the indicated plasmids and relative expression of Fgf18 was assayed by real time PCR. *, p < 0.05 compared with vector-transfected cells. B, Western blotting was performed to verify expression of each construct. This experiment was repeated three times and a representative experiment is shown.
Chemical Inhibition of Phlpp Induces Fgf18 Expression and Erk1/2 Activation
We next determined if the Phlpp inhibitor, NCS 117079, promotes anabolic signaling in chondrogenic ATDC5 cells (23). This Phlpp inhibitor dose-dependently increased activation of key signaling pathways, including Akt and PKC (Fig. 6A). Erk1/2 and p70 S6 kinase phosphorylation was induced at the 25 μm concentration (Fig. 6A). The Phlpp inhibitor also increased Fgf18 expression and Alcian blue staining of micromass cultures (Fig. 6, B and C). Together these data demonstrate that Phlpp1 inhibition promotes chondrocyte proliferation and Fgf18 expression (Fig. 7).
FIGURE 6.
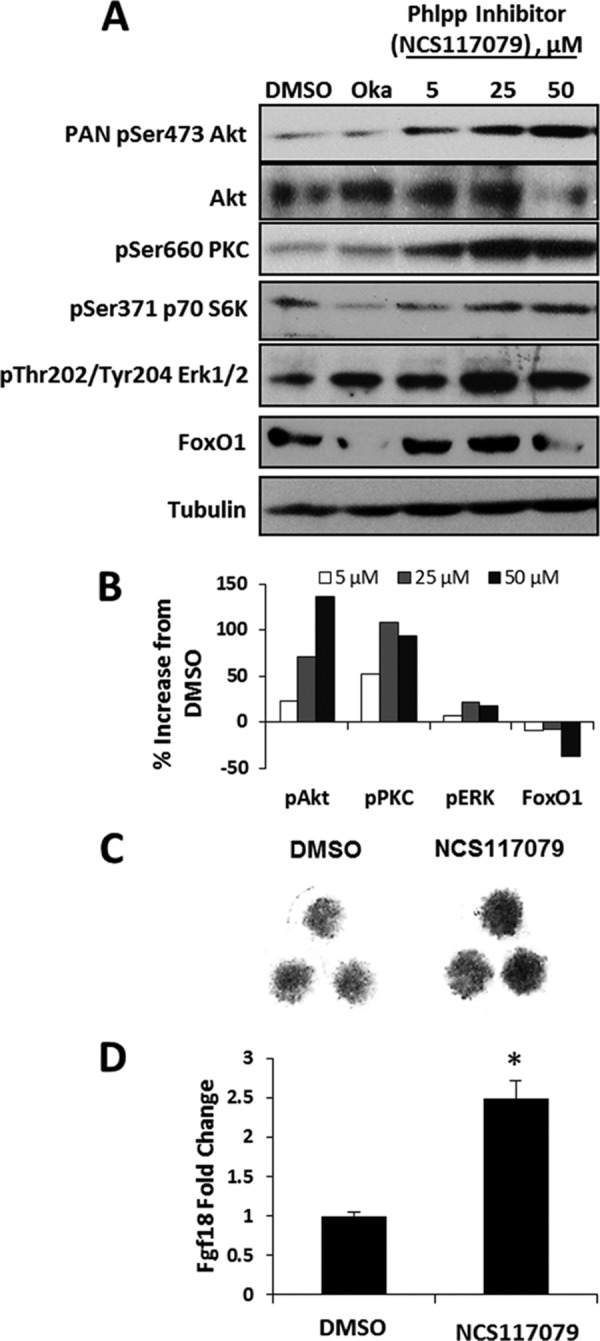
A Phlpp inhibitor increases Fgf18 expression and matrix production in chondrocytes. A, ATDC5 cells were treated with okadaic acid (Oka), increasing concentrations of the Phlpp inhibitor NCS 117079, or vehicle (DMSO) for 24 h. Western blotting was performed as indicated. B, ATDC5 micromass cultures were cultured in the presence of 25 μm of the Phlpp inhibitor NCS 117079 or vehicle (DMSO). Cultures were fixed and stained with Alcian blue. C, ATDC5 cells were treated with 5 μm NCS 117079 or vehicle (DMSO) for 24 h. Relative expression levels of Fgf18 were assayed by real time PCR. *, p < 0.05 compared with control. These experiments were performed three times with data from a representative experiment shown.
FIGURE 7.
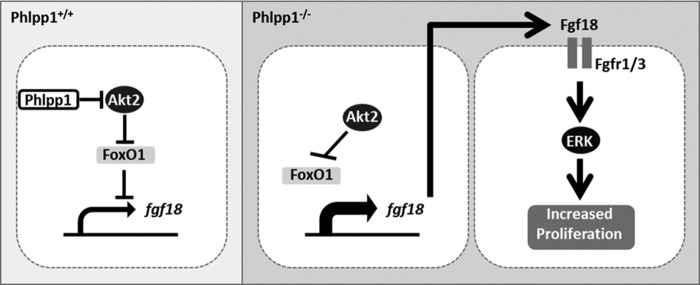
Working Model. Phlpp1 represses Akt2 activity in chondrocytes leading to increased FoxO1-mediated repression of Fgf18 expression. In the absence of Phlpp1, increased Akt activity diminishes FoxO1 levels and increases Fgf18 expression, which stimulates Erk1/2 phosphorylation and chondrocyte proliferation.
Discussion
Because Phlpp1 deficiency causes growth retardation, we sought to determine if Phlpp1 regulates chondrocyte function during endochondral ossification. Our results show that Phlpp1 is a regulator of endochondral bone development, as chondrocyte proliferation and matrix synthesis are enhanced in Phlpp1-deficient mice. This likely occurs due to the ability of Phlpp1 to directly control the activity of anabolic signaling pathways, including Akt2, PKC, and p70 S6 kinase, that are known facilitators of proliferation and matrix production by chondrocytes (21, 37, 38). Activation of these pathways triggers subsequent events, including but not limited to, Fgf18 production and downstream activation of Fgfr and Mek/Erk signaling. Thus, Phlpp1 is active during endochondral bone formation. The effects of Phlpp1 deletion are similar to those observed in mice lacking GDF5 or HIF1α that are characterized by increased chondrocyte proliferation and decreased long bone length (39, 40). The effects of Phlpp1 deficiency on distinct chondrocytes populations, osteoblasts and/or osteoclasts in the developing bone cannot be addressed with this model. Targeted and regulated deletion of Phlpp1 in these and other cell types are needed to fully understand why germline Phlpp1 deficiency produces shorter and thinner bones despite increased BrdU labeling and matrix production.
Phlpp1 preferentially dephosphorylates Akt2, while minimally affecting Akt1 or Akt3 in non-cartilagenous tissues (25). We likewise found that Akt2 phosphorylation was enhanced in Phlpp1−/− chondrocytes. Although little is known about the specific role of Akt2 on chondrocyte differentiation, Akt2−/− mice have reduced body size (41), suggesting a possible growth plate defect. Akt1 phosphorylation was minimally affected in Phlpp1−/− chondrocytes. The substrate preference for Akt2 may explain why both Hdac3 cKO and Phlpp1−/− mice have reduced bone mass. Although Phlpp1 was increased in Hdac3 cKO chondrocytes, the phosphorylation of both Akt1 and Akt2 were reduced (21), suggesting that additional phosphatases, besides Phlpp1, may be deregulated in Hdac3 cKO chondrocytes.
We also noted that Phlpp1 deficiency increases expression of several growth factors, including robust induction of Fgf18. We also demonstrated enhanced Fgf18 production and signaling through Fgfr leading to enhanced Mek/Erk activation and chondrocyte proliferation (Fig. 7). The effects on increased growth factor expression on matrix production, however, were not explored here. Fgf18 is naturally expressed within joints, the perichondrium and growth plates of developing bones (42, 43). Our results are inconsistent with two main observations: 1) that germline Fgf18 deficiency increases chondrocyte proliferation during embryonic development (42, 43), and 2) that Fgfr3 activation decreases chondrocyte proliferation and causes limb shortening in humans (44). However, other studies demonstrate that Fgf18 promotes chondrocyte proliferation and cartilage maintenance, particularly of articular chondrocytes (32, 43, 45–49). Moreover, gain-of-function Fgfr3 mutations expand the pool of proliferative, Sox9-positive chondrocytes and decrease chondrocyte hypertrophy (50, 51). Fgf18 also promotes chondrogenesis, chondrocyte proliferation and cartilage regeneration in surgically induced models of osteoarthritis (31, 32, 43, 52–56). In clinical trials, intra-articular injections of recombinant Fgf18 resulted in dose-dependent increases in total cartilage content, even though thickness of the central medial femorotibial compartment cartilage was unchanged (57). Thus, the effects of Fgf18/Fgfr signaling are complex and may be temporally and spatially controlled. Here we show that increased Fgf18/Fgfr signaling is responsible for some of the effects of Phlpp1 deficiency in micromass cultures. However, the role of Fgf18 within the expanded proliferative zone of the Phlpp1−/− growth plate remains to be determined. Given that Phlpp1 loss of function, either via genetic deficiency or small molecule inhibition, promotes chondrocyte maturation and Fgf18 production in vitro, Phlpp1 is an enticing target for cartilage tissue engineering.
Other groups demonstrated the importance of Akt activity in controlling Fgf18 production and Mek/Erk activation. Conditional deletion of PTEN, a phosphatase that suppresses PI3K/Akt signaling upstream of Phlpp1, in limb progenitor cells enhanced Fgf18 expression (36). The authors attributed this regulation to decreased levels of FoxO1, which represses Fgf18 promoter activity (36). We also found that disrupting the substrate binding ability of Phlpp1 enhances Fgf18 expression, and that this effect is antagonized by expression of an Akt-insensitive FoxO1 mutant.
In summary, we show that germline deletion of Phlpp1 disrupts limb development, but promotes chondrogenesis and increases cartilaginous matrix production, which may be attributed to increases in cartilage anabolic signaling pathways including Akt2, PKC, and p70 S6 kinase, and relief of FoxO1 suppression of Fgf18. Elevated Fgf18 levels signal through Fgfr to activate Mek/Erk signaling and proliferation. Importantly, Phlpp1 chemical inhibition enhances signaling pathway activation, Fgf18 expression and cartilage matrix production. Thus, Phlpp1 may serve as a target to promote cartilage maintenance and/or regeneration and prevent hypertrophic ossification.
Acknowledgments
We thank Xiaodong Li, David Razidlo, and Bridget Stensgard for technical assistance and Dr. Meghan McGee-Lawrence for helpful discussions and μ-CT expertise. The FoxO1A3 and FoxO1A3HR constructs were generous gifts from Dr. Haojie Huang, Mayo Clinic, Rochester, MN. These contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. The authors declare that they have no conflicts of interest with the contents of this article.
This work was supported by National Institutes of Health Grants AR056950, AR61873, AR65397, AR68103, GM055252, and GM067946, the Mayo Clinic, and Mayo Graduate School.
- Phlpp
- PH-domain leucine-rich repeat protein phosphatase
- BMP
- bone morphogenetic protein
- Erk
- extracellular signal-regulated kinase
- Fgf
- fibroblast growth factor
- Fgfr
- fibroblast growth factor receptor
- FoxO
- forkhead box O
- Hdacs
- histone deacetylases
- Hhs
- hedgehogs
- HRP
- horseradish peroxidase
- Igf1
- insulin-like growth factor 1
- PH
- pleckstrin homology
- PKC
- protein kinase C
- Plekhe
- PH domain-containing family E member
- Scop
- suprachiasmatic nucleus circadian oscillatory protein
- Tgfβ
- transforming growth factor β.
References
- 1. Kronenberg H. M. (2003) Developmental regulation of the growth plate. Nature 423, 332–336 [DOI] [PubMed] [Google Scholar]
- 2. Brown J. H., DeLuca S. A. (1992) Growth plate injuries: Salter-Harris classification. Am. Fam. Physician 46, 1180–1184 [PubMed] [Google Scholar]
- 3. Rimoin D. L., Cohn D., Krakow D., Wilcox W., Lachman R. S., Alanay Y. (2007) The skeletal dysplasias: clinical-molecular correlations. Ann. N.Y. Acad. Sci. 1117, 302–309 [DOI] [PubMed] [Google Scholar]
- 4. Panda D. K., Goltzman D., Karaplis A. C. (2012) Defective postnatal endochondral bone development by chondrocyte-specific targeted expression of parathyroid hormone type 2 receptor. Am. J. Physiol. Endocrinol. Metab. 303, E1489–E1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jones D. C., Schweitzer M. N., Wein M., Sigrist K., Takagi T., Ishii S., Glimcher L. H. (2010) Uncoupling of growth plate maturation and bone formation in mice lacking both Schnurri-2 and Schnurri-3. Proc. Natl. Acad. Sci. U. S. A. 107, 8254–8258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Govoni K. E., Lee S. K., Chung Y. S., Behringer R. R., Wergedal J. E., Baylink D. J., Mohan S. (2007) Disruption of insulin-like growth factor-I expression in type IIαI collagen-expressing cells reduces bone length and width in mice. Physiol. Genomics 30, 354–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Staines K. A., Pollard A. S., McGonnell I. M., Farquharson C., Pitsillides A. A. (2013) Cartilage to bone transitions in health and disease. J. Endocrinol. 219, R1–R12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fukai A., Kawamura N., Saito T., Oshima Y., Ikeda T., Kugimiya F., Higashikawa A., Yano F., Ogata N., Nakamura K., Chung U. I., Kawaguchi H. (2010) Akt1 in murine chondrocytes controls cartilage calcification during endochondral ossification under physiologic and pathologic conditions. Arthritis Rheum. 62, 826–836 [DOI] [PubMed] [Google Scholar]
- 9. Ikegami D., Akiyama H., Suzuki A., Nakamura T., Nakano T., Yoshikawa H., Tsumaki N. (2011) Sox9 sustains chondrocyte survival and hypertrophy in part through Pik3ca-Akt pathways. Development 138, 1507–1519 [DOI] [PubMed] [Google Scholar]
- 10. Jin E. J., Park K. S., Bang O. S., Kang S. S. (2007) Akt signaling regulates actin organization via modulation of MMP-2 activity during chondrogenesis of chick wing limb bud mesenchymal cells. J. Cell. Biochem. 102, 252–261 [DOI] [PubMed] [Google Scholar]
- 11. Kita K., Kimura T., Nakamura N., Yoshikawa H., Nakano T. (2008) PI3K/Akt signaling as a key regulatory pathway for chondrocyte terminal differentiation. Genes Cells 13, 839–850 [DOI] [PubMed] [Google Scholar]
- 12. Price J., Zaidi A. K., Bohensky J., Srinivas V., Shapiro I. M., Ali H. (2010) Akt-1 mediates survival of chondrocytes from endoplasmic reticulum-induced stress. J. Cell. Physiol. 222, 502–508 [DOI] [PubMed] [Google Scholar]
- 13. Priore R., Dailey L., Basilico C. (2006) Downregulation of Akt activity contributes to the growth arrest induced by FGF in chondrocytes. J. Cell. Physiol. 207, 800–808 [DOI] [PubMed] [Google Scholar]
- 14. Ulici V., Hoenselaar K. D., Gillespie J. R., Beier F. (2008) The PI3K pathway regulates endochondral bone growth through control of hypertrophic chondrocyte differentiation. BMC Dev. Biol. 8, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rokutanda S., Fujita T., Kanatani N., Yoshida C. A., Komori H., Liu W., Mizuno A., Komori T. (2009) Akt regulates skeletal development through GSK3, mTOR, and FoxOs. Dev. Biol. 328, 78–93 [DOI] [PubMed] [Google Scholar]
- 16. Beier F., Loeser R. F. (2010) Biology and pathology of Rho GTPase, PI-3 kinase-Akt, and MAP kinase signaling pathways in chondrocytes. J. Cell. Biochem. 110, 573–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fayard E., Tintignac L. A., Baudry A., Hemmings B. A. (2005) Protein kinase B/Akt at a glance. J. Cell Sci. 118, 5675–5678 [DOI] [PubMed] [Google Scholar]
- 18. Gao T., Furnari F., Newton A. C. (2005) PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol. Cell 18, 13–24 [DOI] [PubMed] [Google Scholar]
- 19. Brognard J., Newton A. C. (2008) PHLiPPing the switch on Akt and protein kinase C signaling. Trends Endocrinol. Metab. 19, 223–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Masubuchi S., Gao T., O'Neill A., Eckel-Mahan K., Newton A. C., Sassone-Corsi P. (2010) Protein phosphatase PHLPP1 controls the light-induced resetting of the circadian clock. Proc. Natl. Acad. Sci. U. S. A. 107, 1642–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bradley E. W., Carpio L. R., Westendorf J. J. (2013) Histone deacetylase 3 suppression increases PH domain and leucine-rich repeat phosphatase (Phlpp)1 expression in chondrocytes to suppress Akt signaling and matrix secretion. J. Biol. Chem. 288, 9572–9582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gosset M., Berenbaum F., Thirion S., Jacques C. (2008) Primary culture and phenotyping of murine chondrocytes. Nature Protocols 3, 1253–1260 [DOI] [PubMed] [Google Scholar]
- 23. Sierecki E., Sinko W., McCammon J. A., Newton A. C. (2010) Discovery of small molecule inhibitors of the PH domain leucine-rich repeat protein phosphatase (PHLPP) by chemical and virtual screening. J. Med. Chem. 53, 6899–6911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Razidlo D. F., Whitney T. J., Casper M. E., McGee-Lawrence M. E., Stensgard B. A., Li X., Secreto F. J., Knutson S. K., Hiebert S. W., Westendorf J. J. Histone deacetylase 3 depletion in osteo/chondroprogenitor cells decreases bone density and increases marrow fat. PloS one 5, e11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brognard J., Sierecki E., Gao T., Newton A. C. (2007) PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol. Cell 25, 917–931 [DOI] [PubMed] [Google Scholar]
- 26. Gao T., Brognard J., Newton A. C. (2008) The phosphatase PHLPP controls the cellular levels of protein kinase C. J. Biol. Chem. 283, 6300–6311 [DOI] [PubMed] [Google Scholar]
- 27. Guo S., Rena G., Cichy S., He X., Cohen P., Unterman T. (1999) Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J. Biol. Chem. 274, 17184–17192 [DOI] [PubMed] [Google Scholar]
- 28. Nakae J., Park B. C., Accili D. (1999) Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J. Biol. Chem. 274, 15982–15985 [DOI] [PubMed] [Google Scholar]
- 29. Rena G., Guo S., Cichy S. C., Unterman T. G., Cohen P. (1999) Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J. Biol. Chem. 274, 17179–17183 [DOI] [PubMed] [Google Scholar]
- 30. Ericson K., Gan C., Cheong I., Rago C., Samuels Y., Velculescu V. E., Kinzler K. W., Huso D. L., Vogelstein B., Papadopoulos N. (2010) Genetic inactivation of AKT1, AKT2, and PDPK1 in human colorectal cancer cells clarifies their roles in tumor growth regulation. Proc. Natl. Acad. Sci. U. S. A. 107, 2598–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Davidson D., Blanc A., Filion D., Wang H., Plut P., Pfeffer G., Buschmann M. D., Henderson J. E. (2005) Fibroblast growth factor (FGF) 18 signals through FGF receptor 3 to promote chondrogenesis. J. Biol. Chem. 280, 20509–20515 [DOI] [PubMed] [Google Scholar]
- 32. Liu Z., Xu J., Colvin J. S., Ornitz D. M. (2002) Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes Dev. 16, 859–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Haque T., Nakada S., Hamdy R. C. (2007) A review of FGF18: Its expression, signaling pathways and possible functions during embryogenesis and post-natal development. Histol. Histopathol. 22, 97–105 [DOI] [PubMed] [Google Scholar]
- 34. Mohammadi M., Froum S., Hamby J. M., Schroeder M. C., Panek R. L., Lu G. H., Eliseenkova A. V., Green D., Schlessinger J., Hubbard S. R. (1998) Crystal structure of an angiogenesis inhibitor bound to the FGF receptor tyrosine kinase domain. EMBO J. 17, 5896–5904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pardo O. E., Latigo J., Jeffery R. E., Nye E., Poulsom R., Spencer-Dene B., Lemoine N. R., Stamp G. W., Aboagye E. O., Seckl M. J. (2009) The fibroblast growth factor receptor inhibitor PD173074 blocks small cell lung cancer growth in vitro and in vivo. Cancer Res. 69, 8645–8651 [DOI] [PubMed] [Google Scholar]
- 36. Guntur A. R., Reinhold M. I., Cuellar J., Jr., Naski M. C. (2011) Conditional ablation of Pten in osteoprogenitors stimulates FGF signaling. Development 138, 1433–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zeng G., Cui X., Liu Z., Zhao H., Zheng X., Zhang B., Xia C. (2014) Disruption of phosphoinositide-specific phospholipases Cgamma1 contributes to extracellular matrix synthesis of human osteoarthritis chondrocytes. Int. J. Mol. Sci. 15, 13236–13246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhao H., Zhang T., Xia C., Shi L., Wang S., Zheng X., Hu T., Zhang B. (2014) Berberine ameliorates cartilage degeneration in interleukin-1β-stimulated rat chondrocytes and in a rat model of osteoarthritis via Akt signalling. J. Cell Mol. Med. 18, 283–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mikic B., Clark R. T., Battaglia T. C., Gaschen V., Hunziker E. B. (2004) Altered hypertrophic chondrocyte kinetics in GDF-5 deficient murine tibial growth plates. J. Orthop. Res. 22, 552–556 [DOI] [PubMed] [Google Scholar]
- 40. Schipani E., Ryan H. E., Didrickson S., Kobayashi T., Knight M., Johnson R. S. (2001) Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev. 15, 2865–2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Garofalo R. S., Orena S. J., Rafidi K., Torchia A. J., Stock J. L., Hildebrandt A. L., Coskran T., Black S. C., Brees D. J., Wicks J. R., McNeish J. D., Coleman K. G. (2003) Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKBβ. J. Clin. Investig. 112, 197–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lazarus J. E., Hegde A., Andrade A. C., Nilsson O., Baron J. (2007) Fibroblast growth factor expression in the postnatal growth plate. Bone 40, 577–586 [DOI] [PubMed] [Google Scholar]
- 43. Ohbayashi N., Shibayama M., Kurotaki Y., Imanishi M., Fujimori T., Itoh N., Takada S. (2002) FGF18 is required for normal cell proliferation and differentiation during osteogenesis and chondrogenesis. Genes Dev. 16, 870–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rousseau F., Bonaventure J., Legeai-Mallet L., Pelet A., Rozet J. M., Maroteaux P., Le Merrer M., Munnich A. (1994) Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature 371, 252–254 [DOI] [PubMed] [Google Scholar]
- 45. Goldring M. B., Tsuchimochi K., Ijiri K. (2006) The control of chondrogenesis. J. Cell. Biochem. 97, 33–44 [DOI] [PubMed] [Google Scholar]
- 46. Ornitz D. M., Marie P. J. (2002) FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 16, 1446–1465 [DOI] [PubMed] [Google Scholar]
- 47. Iwata T., Chen L., Li C., Ovchinnikov D. A., Behringer R. R., Francomano C. A., Deng C. X. (2000) A neonatal lethal mutation in FGFR3 uncouples proliferation and differentiation of growth plate chondrocytes in embryos. Human Mol. Genet. 9, 1603–1613 [DOI] [PubMed] [Google Scholar]
- 48. Naski M. C., Colvin J. S., Coffin J. D., Ornitz D. M. (1998) Repression of hedgehog signaling and BMP4 expression in growth plate cartilage by fibroblast growth factor receptor 3. Development 125, 4977–4988 [DOI] [PubMed] [Google Scholar]
- 49. Colvin J. S., Bohne B. A., Harding G. W., McEwen D. G., Ornitz D. M. (1996) Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nature Genetics 12, 390–397 [DOI] [PubMed] [Google Scholar]
- 50. Shung C. Y., Ota S., Zhou Z. Q., Keene D. R., Hurlin P. J. (2012) Disruption of a Sox9-beta-catenin circuit by mutant Fgfr3 in thanatophoric dysplasia type II. Human Mol. Genet. 21, 4628–4644 [DOI] [PubMed] [Google Scholar]
- 51. Mugniery E., Dacquin R., Marty C., Benoist-Lasselin C., de Vernejoul M. C., Jurdic P., Munnich A., Geoffroy V., Legeai-Mallet L. (2012) An activating Fgfr3 mutation affects trabecular bone formation via a paracrine mechanism during growth. Human Mol. Genet. 21, 2503–2513 [DOI] [PubMed] [Google Scholar]
- 52. Power J., Hernandez P., Guehring H., Getgood A., Henson F. (2014) Intra-articular injection of rhFGF-18 improves the healing in microfracture treated chondral defects in an ovine model. J. Orthop. Res. 32, 669–676 [DOI] [PubMed] [Google Scholar]
- 53. Yamaoka H., Nishizawa S., Asawa Y., Fujihara Y., Ogasawara T., Yamaoka K., Nagata S., Takato T., Hoshi K. (2010) Involvement of fibroblast growth factor 18 in dedifferentiation of cultured human chondrocytes. Cell Prolif. 43, 67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Valverde-Franco G., Binette J. S., Li W., Wang H., Chai S., Laflamme F., Tran-Khanh N., Quenneville E., Meijers T., Poole A. R., Mort J. S., Buschmann M. D., Henderson J. E. (2006) Defects in articular cartilage metabolism and early arthritis in fibroblast growth factor receptor 3 deficient mice. Human Mol. Genet. 15, 1783–1792 [DOI] [PubMed] [Google Scholar]
- 55. Moore E. E., Bendele A. M., Thompson D. L., Littau A., Waggie K. S., Reardon B., Ellsworth J. L. (2005) Fibroblast growth factor-18 stimulates chondrogenesis and cartilage repair in a rat model of injury-induced osteoarthritis. Osteoarthritis Cartilage 13, 623–631 [DOI] [PubMed] [Google Scholar]
- 56. Ellsworth J. L., Berry J., Bukowski T., Claus J., Feldhaus A., Holderman S., Holdren M. S., Lum K. D., Moore E. E., Raymond F., Ren H., Shea P., Sprecher C., Storey H., Thompson D. L., Waggie K., Yao L., Fernandes R. J., Eyre D. R., Hughes S. D. (2002) Fibroblast growth factor-18 is a trophic factor for mature chondrocytes and their progenitors. Osteoarthritis Cartilage 10, 308–320 [DOI] [PubMed] [Google Scholar]
- 57. Lohmander L. S., Hellot S., Dreher D., Krantz E. F., Kruger D. S., Guermazi A., Eckstein F. (2014) Intra-articular Sprifermin (Recombinant Human Fibroblast Growth Factor 18) in Knee Osteoarthritis: Randomized, Double-blind, Placebo-controlled Trial. Arthritis Rheumatol. 66, 1820–1831 [DOI] [PubMed] [Google Scholar]