

Background: MT-I/II are zinc-binding proteins that are also neuro-protective.
Results: MT-I/II can overcome myelin-mediated inhibition in vitro and in vivo. MT-I/II-deficient mice have reduced spinal axon regeneration.
Conclusion: MT-I/II are required for the conditioning lesion effect and can promote axonal regeneration in the injured CNS.
Significance: MT-I/II have therapeutic potential for the treatment of spinal cord injury.
Keywords: central nervous system (CNS), cyclic AMP (cAMP), myelin, neurite outgrowth, regeneration, axonal regeneration, metallothionein
Abstract
The adult CNS does not spontaneously regenerate after injury, due in large part to myelin-associated inhibitors such as myelin-associated glycoprotein (MAG), Nogo-A, and oligodendrocyte-myelin glycoprotein. All three inhibitors can interact with either the Nogo receptor complex or paired immunoglobulin-like receptor B. A conditioning lesion of the sciatic nerve allows the central processes of dorsal root ganglion (DRG) neurons to spontaneously regenerate in vivo after a dorsal column lesion. After a conditioning lesion, DRG neurons are no longer inhibited by myelin, and this effect is cyclic AMP (cAMP)- and transcription-dependent. Using a microarray analysis, we identified several genes that are up-regulated both in adult DRGs after a conditioning lesion and in DRG neurons treated with cAMP analogues. One gene that was up-regulated under both conditions is metallothionein (MT)-I. We show here that treatment with two closely related isoforms of MT (MT-I/II) can overcome the inhibitory effects of both myelin and MAG for cortical, hippocampal, and DRG neurons. Intrathecal delivery of MT-I/II to adult DRGs also promotes neurite outgrowth in the presence of MAG. Adult DRGs from MT-I/II-deficient mice extend significantly shorter processes on MAG compared with wild-type DRG neurons, and regeneration of dorsal column axons does not occur after a conditioning lesion in MT-I/II-deficient mice. Furthermore, a single intravitreal injection of MT-I/II after optic nerve crush promotes axonal regeneration. Mechanistically, MT-I/II ability to overcome MAG-mediated inhibition is transcription-dependent, and MT-I/II can block the proteolytic activity of α-secretase and the activation of PKC and Rho in response to soluble MAG.
Introduction
Several proteins associated with myelin are well documented inhibitors of axonal regeneration in the adult mammalian central nervous system (CNS). These include myelin-associated glycoprotein (MAG),5 Nogo-A, and oligodendrocyte-myelin glycoprotein (1–6). These proteins mediate inhibition by binding to either the glycosylphosphatidylinositol-linked Nogo receptor or paired immunoglobulin-like receptor B (7, 8). Nogo receptor interacts with Lingo-1 and either p75NTR or TROY to transduce the inhibitory signal and activate the small GTPase Rho, which is protein kinase C (PKC)-dependent (9–14).
Our laboratory has shown that Bt2cAMP can overcome the inhibitory effects of MAG and myelin and promote axonal regeneration (15). When cAMP is elevated in DRG neurons by lesioning the sciatic nerve, the ability of these neurons to extend neurites on MAG and myelin in culture is significantly improved, a phenomenon known as the conditioning lesion effect (16, 17). Injecting Bt2cAMP directly into the DRG cell bodies also induces regeneration of lesioned dorsal column axons, mimicking the conditioning lesion effect in vivo (15, 18). Subsequent work has shown that elevation of intracellular cAMP levels and cAMP-response element-binding protein-mediated transcription are required for the conditioning lesion effect (19). Using microarray analysis to identify genes involved in this pathway, we found that levels of metallothionein (MT)-I RNA are elevated in response to cAMP.
MTs are small cysteine-rich zinc-binding proteins that are found in all CNS tissue; however, their exact physiological role has not yet been elucidated (20, 21). Expression of MT-I and MT-II, an isoform of MT-I, is up-regulated in the CNS after injury and in disease states. Several studies utilizing MT-I/II-deficient mice have shown a neuroprotective role for MTs in ischemia, experimental autoimmune encephalomyelitis, and in response to seizures (22–24). MT-IIA, the major isoform of MT-I and -II found in the CNS, was shown to promote growth on a permissive substrate in rat cortical cultures and to induce a greater number of regenerating sprouts after CNS injury (20, 25). The study described here shows a novel role for MT-I/II in overcoming inhibition by MAG and myelin for a variety of neuronal populations in vitro and promoting in vivo regeneration in the injured optic nerve. The only method of inducing spontaneous axonal regeneration in the injured CNS is by first performing a peripheral nerve lesion, by crushing the sciatic nerve and then subsequently lesioning the dorsal column, referred to as a conditioning lesion (16, 17). We report that mice deficient in MT-I/II do not show regeneration of transected dorsal column axons in response to a conditioning lesion, suggesting that MT-I/II is an important protein for the conditioning lesion effect.
Experimental Procedures
Neuronal Preparations
All animal experiments have received approval from the IACUC at Hunter College and were conducted in accordance with United States Public Health Service's Policy on Humane Care and Use of Laboratory Animals. As described previously, for cortical or hippocampal neurons, cortices and hippocampi were dissected from postnatal day 1 (P1) Long-Evans rat pups of both sexes and incubated twice with 0.5 mg/ml papain in plain Neurobasal-A media (Invitrogen), and papain activity was inhibited by brief incubation in soybean trypsin inhibitor (Sigma) (26). Cell suspensions were layered on an Optiprep density gradient (Sigma) and centrifuged at 1900 × g for 15 min. The purified neurons were then collected and counted. For cerebellar granule neurons, cerebellar cortex was isolated from P5 to P6 rats of both sexes and treated with papain and soybean trypsin inhibitor as described above. After trituration, cells were diluted in Sato's media and counted.
For DRG neurons, DRGs were isolated from P5 to P6 rats of both sexes and treated with 0.015% collagenase in Neurobasal-A media for 45 min at 37 °C. This was followed by a second incubation in collagenase for 30 min at 37 °C, with the addition of 0.1% trypsin and 50 μg/ml DNase I. Trypsin was inactivated with DMEM containing 10% dialyzed fetal bovine serum, and the ganglia were triturated in Sato's media.
Microarray Analysis and Real Time PCR
For the RNA preparations, P21–P23 Long-Evans rats of both sexes were anesthetized with isoflurane, and the right sciatic nerve was transected at the midpoint of the thigh. Animals were killed 18 h later, and the ipsilateral and contralateral L4 and L5 DRGs were collected and snap-frozen. P5 DRG neurons were also prepared as described and incubated for 18 h at 37 °C in the presence or absence of 1.5 mm dibutyryl cAMP (Bt2cAMP). In both cases, the cells were homogenized in TRIzol (Invitrogen), and RNA was purified using the RNeasy RNA isolation kit (Qiagen). Microarray hybridization and quantitative real time PCR were then performed as described previously (27). The full results of the microarray can be viewed on line.
End Point PCR
P28 Long-Evans rats of both sexes received unilateral sciatic nerve lesions and were killed 24 h later. The ipsi- and contralateral lumbar DRGs (L2–L5) were removed and homogenized in TRIzol. RNA was extracted using chloroform, precipitated with isopropyl alcohol in the presence of linear polyacrylamide, and solubilized in RNase-free water. RNA was reverse-transcribed using oligo(dT) and AccuScript High Fidelity RT (Stratagene), and the resulting cDNA was amplified using PfuUltra High Fidelity DNA polymerase (Stratagene). The following primers were used for MT-I/II and glyceraldehyde-3-phosphate dehydrogenase (GAPDH): MT-I/II forward, 5′-ACCAGATCTCGGAATGGAC-3′, and MT-I/II reverse, 5′-TGCACGTGCTGTGCCTGAT-3′; GAPDH forward, 5′-ATGGTGAAGGTCGGTGTGAACG-3′, and GAPDH reverse, 5′-TGGTGAAGACGCCAGTAGACTC-3′. Densitometric measurements were made using ImageJ software (National Institutes of Health).
Intrathecal Delivery of MT-I/II
Osmotic mini pumps with a flow rate of 0.5 μl/h (model 1007D;Alzet) were filled with either sterile saline or MT-I/II (Sigma) at concentrations of 0.25, 0.5, or 1 μg/μl. The osmotic pumps were attached to a cannula and implanted into anesthetized P28 Long Evans rats. A laminectomy was performed between L5 and L6, and the cannula was inserted under the dura mater so that the tip could rest on the dorsal spinal cord approximately between L4 and L5. Animals were sacrificed 24 h later, and the lumbar DRGs (L3 to 5) were processed as described above.
Neurite Outgrowth Assay
Monolayers of control or MAG-expressing Chinese hamster ovary (CHO) cells were plated on eight-well chamber slides as described previously (2). Alternatively, suspensions of purified CNS myelin (1–2 μg/well) were plated in chamber slides and desiccated overnight to create a substrate of myelin. Purified P1 hippocampal, P1 cortical, P5–P6 cerebellar granule neurons, or P5–P6 DRG rat neurons were diluted to 35,000 cells/ml in Sato's media and treated with either 1 mm Bt2cAMP (Calbiochem) or MT-I/II at one of the following concentrations: 5, 10, or 20 μg/ml. For controls, we added an equal volume of PBS corresponding to the volume of Bt2cAMP or MT-I/II that we utilized per well. Neurons from intrathecal delivery experiments received no additional treatment. Neurons were incubated for 14–18 h at 37 °C, fixed with 4% paraformaldehyde (PFA), and immunostained using a monoclonal anti-βIII tubulin antibody (Tuj1;Covance) and Alexa Fluor 568-conjugated anti-mouse IgG (Invitrogen). For quantification, images were taken, and the length of the longest neurite for each neuron was measured using MetaMorph software (Molecular Devices).
Conditioning Peripheral Lesion
Wild-type (WT) 129S1/SvImJ and MT-I/II-deficient mice (The Jackson Laboratory) ∼9–12 weeks old from both sexes were used in our study. For looking at the conditioning peripheral lesion effect of WT and MT-I/II-deficient mice, the right sciatic nerve was isolated in the mid-thigh region and transected. Either 24 h or 7 days later, we sacrificed the animals and isolated the lumbar DRGs (L3–6) from both the lesioned and nonlesioned side of the animal and prepared DRG neuronal preparations according to the protocol listed above. The DRG neurons were plated on monolayers of either MAG-expressing or control CHO cells in SATO media, with or without 50 mm 5,6-dichlorobenzimidazole 1-β-d-ribofuranoside (DRB; Sigma), and allowed to grow for 14 h. The neurons were fixed with 4% PFA and stained with anti-βIII tubulin antibody as listed above, and we the longest neurite for each neuron was measured using MetaMorph software.
Conditioning and Dorsal Column Lesions
The right sciatic nerve of both WT and MT-I/II-deficient mice (n = 6 for both groups) was lesioned. 7 days later under isoflurane anesthesia, a laminectomy was performed at the T8–9 level, and the dorsal column was transected to a depth of 1 mm. Another set of WT and MT-I/II-deficient mice (n = 6 for both groups) received only dorsal column lesions. At 5 weeks after surgery, 2 μl of 1% cholera toxin B (CTB; List Biological) was injected into the right sciatic nerve, and the animals were transcardially perfused with 4% (PFA) 3 days later. Spinal cord sections were immunostained using goat anti-CTB antibody (1:2000; List Biological), biotinylated donkey anti-goat IgG (1:200; List Biological), and avidin-biotin complex (Vector Laboratories). To visualize CTB, the sections were reacted in a solution of 0.05% diaminobenzidine tetrahydrochloride, 0.04% nickel chloride, and 0.015% hydrogen peroxide (27). Individual images of the spinal cord sections were taken under bright field optics and combined into photomontages using Adobe Photoshop (Adobe Systems). The photomontages were then analyzed using ImageJ software. Pixel thresholding was performed to identify CTB-labeled axons, and 10,000-μm2 blocks were drawn at distances 100, 200, and 300 μm rostral and caudal to the lesion site. The area within each block was then measured to determine the area occupied by CTB-labeled axons at that particular point. A minimum of two sections was measured for each animal. The fluorescent CTB staining was carried out as described above except we used secondary antibody coupled to Alexa-488 (Life Technologies, Inc.).
Optic Nerve Crush Experiments
Adult male Fischer rats (200–250 g) were anesthetized with isoflurane and placed in a stereotaxic frame. The right optic nerve was exposed and crushed with fine forceps for 10 s following the protocol from the Benowitz and co-workers (28). Special care was taken to segregate animals that had lens injury (LI) as a result of the intravitreal injection. Experimental groups without lens injury included controls with 5 μl of intravitreal injection of saline (n = 6), recombinant human MT-I/II (5 μg/μl in sterile saline) (n = 6), or intravitreal injections but had gel foam soaked with 5 μg/μl MT-I/II in sterile saline placed over the crush site (n = 4). Another group with lens injury included controls with 5 μl of intravitreal injection of saline (n = 5) or recombinant human MT-I/II (5 μg/μl in sterile saline) (n = 5). Animals were transcardially perfused with 4% PFA after a 14-day postsurgical survival period. Optic nerve sections were immunostained using anti-growth-associated protein-43 (GAP-43) polyclonal antibody (gift from Dr. Larry Benowitz, Harvard University, Cambridge, MA) and FITC-conjugated rabbit anti-sheep IgG (1:500; Jackson ImmunoResearch). Images were taken on a fluorescence microscope using MetaMorph software (Molecular Devices). Quantification of axonal density was performed using ImageJ software. Pixel thresholding was used to identify GAP-43-positive axons within the optic nerve images, and 250,000-μm2 blocks (500 × 500 μm) were drawn on the images. These blocks encompassed the following distances from the lesion site: 0–500, 500–1000, 1000–1500, and 1500–2000 μm. The thresholded pixels in each block were then counted to determine the area occupied by GAP-43-positive axons within that segment of the nerve, and these measurements were reported in square micrometers. To further assess regeneration with MT-I/II, we used a Fischer rat and performed optic nerve crush and then immediately afterward intravitreally injected 5 μl of 5 μg/μl MT-I/II; we took precautions to avoid lens injury. 3 days prior to sacrificing, we intravitreally injected 5 μl of 2 μg/μl CTB coupled to Alexa-488 (Life Technologies, Inc.), and the animals were transcardially perfused with 4% PFA 3 days later and we removed the whole optic nerve. We performed chemical clearing of the whole optic nerve by incubating in a graded series (50, 70, 80, 100, and 100%) with tetrahydrofuran (Sigma) at room temperature on a rocker for 25 min each (29). This was followed by incubating for 20 min with 100% dichloromethane (Sigma) and then clearing with di-benzyl ether (Sigma) overnight; all steps were at room temperature and on a rocker. Placing a Fastwell incubation chamber (Electron Microscopy Sciences) on a microscope slide, we carefully placed our transparent nerve into the Fastwell with 200 μl of dibenzyl ether, coverslipped, and imaged on an Olympus FV1000 MPE multiphoton microscope with a ×25 water immersion lens, scanning ∼250 μm of the whole nerve and then making a two-dimensional flat image of the Z-stacked images.
α-Secretase Activity Assay
Recombinant tumor necrosis factor-α converting enzyme (TACE or ADAM17) is a member of α-secretase class of proteases and is a zinc-dependent enzyme. To monitor its activity with or without 10 μg/ml MT-I/II, we utilized a commercially available α-secretase activity kit and followed the manufacturer's protocol (R&D Systems). We incubated 0.25–1.0 μg TACE with or without 10 μg/ml MT-I/II in a total volume of 50 μl in a 96-well plate. We added 50 μl of the manufacturer's 2× Reaction Buffer and 5 μl of substrate to each well and gently tapped to mix the samples. The samples were incubated in the dark at 37 °C for 60 min, and we read the plate at an absorbance excitation of 355 and emission of 510 nm in a Gemini Fluorimeter with SOFTmax PRO software from Molecular Devices. For measuring α-secretase activity following conditioning lesion, we performed peripheral lesions as described above on 7–9-week-old WT and MT-I/II-deficient mice, and 3 h later we collected both the ipsilateral and contralateral L3–6 DRGs into 100 μl of the 1× Cell Extraction Buffer provided in the kit and then homogenized the samples on ice. We determined the protein concentration for each sample, loaded 50 μg of total protein, adjusted each sample volume to 50 μl with 1× Cell Extraction Buffer, and performed the assay as described above.
PKC Activation
For experiments examining the effect of MAG-Fc and MT-I/II on PKC phosphorylation, P6 rat cerebellar neurons (∼2 million cells per plate) were treated with 10 μg/ml MT-I/II for 1 h prior to adding 20 μg/ml MAG-FC, a soluble form of MAG, or saline as a control for 30 min. Cells were lysed in 50 μl of 1× RIPA Buffer, and Western blotting was performed as described above. Phosphorylated form of PKC was detected with rabbit anti-pan phospho-PKC (1:1000; Cell Signaling Technology) followed by HRP-conjugated anti-rabbit IgG (1:2000; Cell Signaling Technology). Membranes were reacted with Pierce ECL Western blotting substrate or SuperSignal West Femto maximum sensitivity substrate (Thermo Fisher Scientific). Membranes were then stripped using Restore Western blot Stripping Buffer (Thermo Fisher Scientific) and reprobed with rabbit anti-actin (1:1000; Sigma). Densitometric measurements were made using ImageJ software.
Rho Activation Assay
Rat cortical neurons plated in 10-cm2 dishes (∼10 million neurons per dish) were used with a commercially available Rho Activation Assay kit (Millipore). The neurons were placed in plain neurobasal media for 4 h prior to assay. The neurons were treated with 10 μg/ml MT-I/II or sterile saline for 1 h prior to adding 20 μg/ml MAG-FC, a soluble form of MAG, or saline as a control for 30 min. In brief, following the manufacturer's protocol, we lysed the cortical neurons on ice with MLB Buffer supplemented with anti-protease and anti-phosphatase mixtures (Calbiochem) and spun the lysates at 14,000 × g for 5 min. The supernatant was collected and added to 35 μl of agarose beads coupled to rhotekin Rho binding domain and rocked in the cold room for 45 min; a sample of each supernatant was not added to the beads and was used as total Rho loading controls. Beads were washed three times with MLB Buffer, and 20 μl of 2× Laemmli Buffer was added, and samples were prepared for Western blotting. Proteins were separated on pre-cast 4–20% gradient gels (Thermo Fisher Scientific) and transferred to nitrocellulose at 75 V for 1 h. Membranes were probed with rabbit anti-RhoA (1:1000; Cell Signaling Technology) and HRP-conjugated anti-rabbit IgG (1:2000; Cell Signaling Technology). Membranes were visualized with Pierce ECL Western blotting substrate or SuperSignal West Femto maximum sensitivity substrate (Thermo Fisher Scientific). Densitometric measurements were made using ImageJ software.
Statistical Analyses
All analyses were performed using GraphPad Prism software, and data are represented as mean ± S.E. Statistical significance was assessed using paired one-tailed Student's t tests to compare two groups and one-way ANOVAs with Bonferroni's post hoc tests to compare three or more groups.
Results
mt-I/II Expression Is Up-regulated after a Conditioning Lesion
Previously, we and others showed adding Bt2cAMP to primary neurons can overcome both MAG and myelin inhibition, and this effect is transcription-dependent (27, 30). We have similar results when we performed a conditioning lesion and subsequently cultured the DRGs on MAG or myelin; the neurons are not inhibited from putting out long neurites, and this effect is also transcription-dependent (27, 30). Furthermore, a conditioning lesion is known to elevate endogenous levels of cAMP in the DRG cell bodies, and this elevation of cAMP is necessary for the conditioning lesion effect on subsequent regeneration of dorsal column axons (31). To identify cAMP-responsive genes with potential roles in axonal regeneration, RNA was isolated from DRGs treated with 1 mm Bt2cAMP for 18 h or 18 h after a conditioning lesion. These RNAs were hybridized to a custom-made microarray, which corresponded to genes with known roles in axonal growth, cell survival, and inflammation, revealing that 11 increased and four decreased (27, 32). Two genes previously shown to be up-regulated in response to cAMP and a conditioning lesion, arginase I (arg I) and interleukin-6 (il-6), increased in this array (27, 30, 32). Expression of mt-I/II was increased about 3-fold in the microarray. To confirm that the increase in mt-I/II expression was observed in the microarray, we performed RT-PCR analysis on mRNA of DRGs from adult rats that had received a conditioning lesion 1 day prior to collecting the DRGs from both the ipsilateral and contralateral sides. DRGS from the ipsilateral side had more than a 4-fold increase in mT-I/II compared with the contralateral side (Fig. 1). Furthermore, our collaborators in Dr. R. Hart's laboratory performed quantitative RT-PCR where they detected a significant elevation in mt-I levels following 18 h of Bt2cAMP treatment for DRG neurons (1.7-fold greater than control with p < 0.01 t test, data not shown).
FIGURE 1.
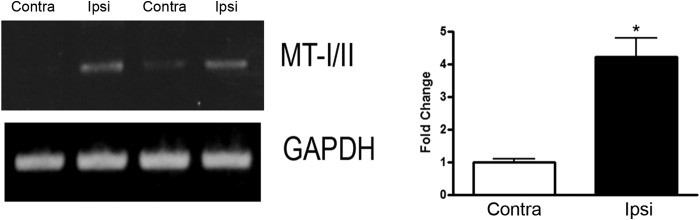
MT-I/II expression is up-regulated 18 h after peripheral conditioning lesion in adult rat DRGs. P28-day-old rats had the peripheral branch of the lumbar (L3–5) DRGs transected 1 day prior to having the DRGs collected from both the ipsilateral (ipsi) and contralateral (contra) sides. RT-PCR analysis of MT-I/II and GAPDH for normalization is performed. The bar graph is the average of DRGs collected from four rats that had peripheral conditioning lesion, where MT-I/II is significantly (*, p < 0.05) elevated on the ipsilateral side compared with the contralateral side.
MT-I/II Overcomes Inhibition of Neurite Outgrowth by MAG and Myelin
Previously, we showed that the proteins from a variety of cAMP-regulated genes can overcome MAG inhibition of neurite outgrowth (27, 30). To assess whether MT-I/II is similar and could also block inhibition of neurite outgrowth by MAG, hippocampal neurons were plated onto either MAG-expressing CHO cells or control cells not expressing MAG, with and without MT-I/II. For all our neurite outgrowths, control wells with no treatment were given an equal volume of PBS corresponding to the volume of MT-I/II that we added. Fig. 2A shows that MT-I/II blocks the inhibition of neurite outgrowth by MAG in a dose-dependent manner. Looking at images of the neurons on CHO cells expressing MAG (Fig. 2C), we see the neurons put out very short neurites compared with neurons on permissive CHO cells (Fig. 2B). Furthermore, MT-I/II is as effective as Bt2cAMP in blocking inhibition, because adding MT-I/II to the culture media at the time of plating overcomes MAG inhibition (Fig. 2E) as robustly as Bt2cAMP (Fig. 2D). DRG neurons are also inhibited by MAG (Fig. 2, G–I), and direct addition of MT-I/II (Fig. 2, K and L), as with hippocampal neurons, blocked the inhibition of neurite outgrowth by MAG once again as potently as Bt2cAMP (Fig. 2, F and J). MT-I/II also blocks inhibition of neurite outgrowth from cortical neurons by myelin as a substrate (Fig. 3). Cortical neurons on PLL put out long neurites (Fig. 3, B and C); however, they are strongly inhibited on myelin (Fig. 3, D–F). Adding MT-I/II to the culture media at the time of plating the cortical neurons overcomes MAG inhibition (Fig. 3, G–I), and these neurons put out significantly longer neurites compared with the control treated with PBS only (Fig. 3A). These results show that MT-I/II cannot only overcome the inhibitory effects of MAG but in all the inhibitors found in myelin and that a variety of neurons are responsive to MT-I/II.
FIGURE 2.
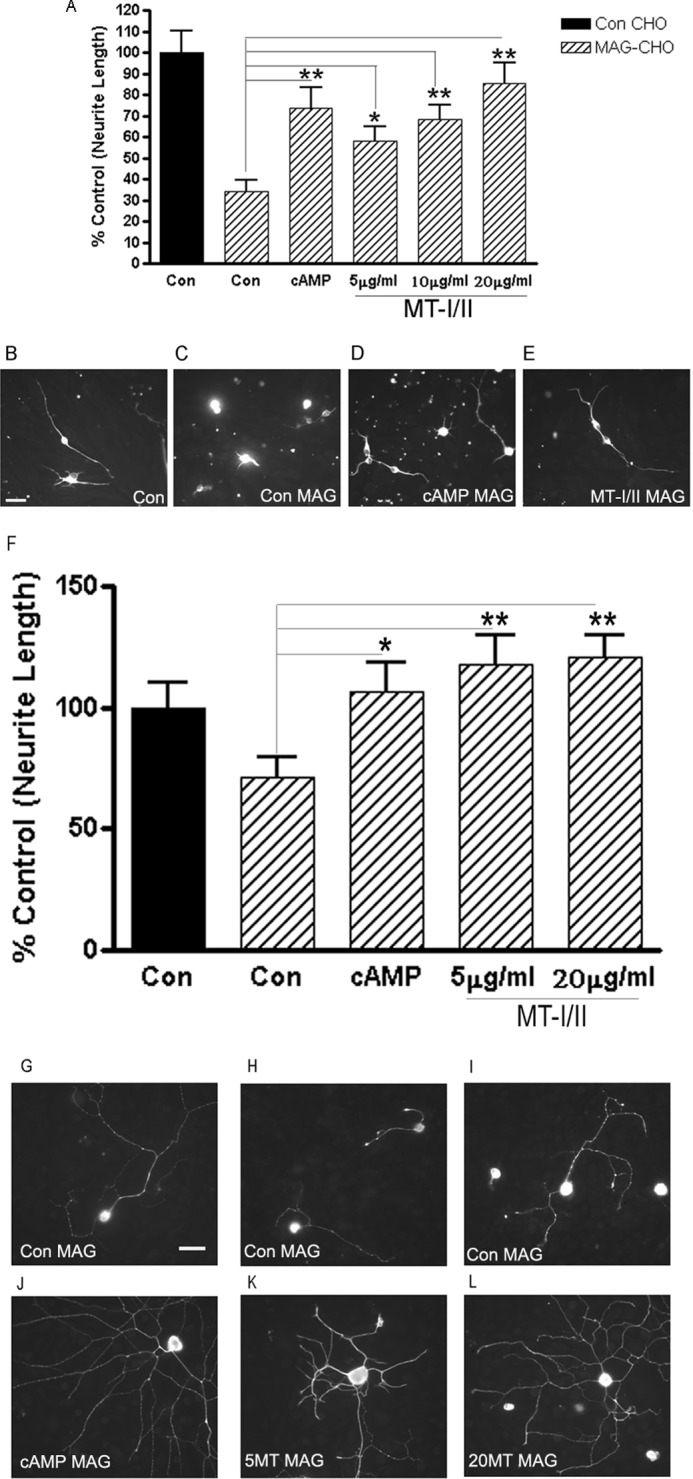
MT-I/II can overcome MAG-mediated inhibition of neurite outgrowth. MT-I/II can overcome MAG inhibition of P1 hippocampal neurons plated on either control (Con) or MAG-expressing CHO cells for 24 h. For control (nontreated) neurons, MAG inhibited neurite outgrowth by greater than 60%; however, MT-I/II directly added to the culture media overcomes the inhibition in a dose-dependent manner (A). MT-I/II is as efficient as cAMP at overcoming MAG-mediated inhibition of neurite outgrowth. Statistics performed are ANOVA, *, p < 0.05, or **, p < 0.01. Representative images of hippocampal neurons shown are plated on control-expressing (B) or MAG-expressing CHO (C) cells and cAMP-treated on MAG (D) and MT-I/II on MAG (E). Similar findings were made for P5 DRG neurons plated on MAG-expressing CHO cells for 18 h, where the neurite outgrowth is inhibited by 35% (F), and with addition of MT-I/II, there was a full reversal of this inhibition. Representative images of DRG neurons plated on MAG display how inhibited they are in G–I; 1 mm cAMP (J), 5 (K), and 20 (L) μg/ml MT-I/II put out significantly longer processes. Scale bar, 20 μm.
FIGURE 3.
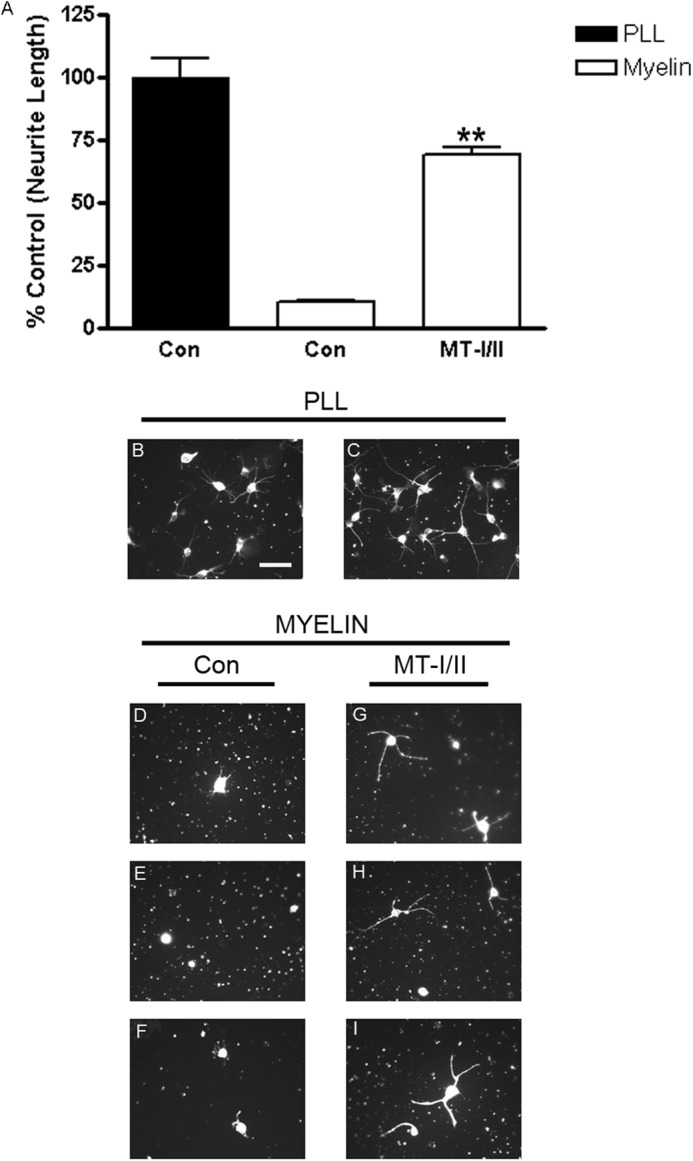
MT-I/II overcomes inhibition by myelin. Primary P1 cortical neurons are isolated and plated directly onto either PLL or 1 μg of myelin in the presence or absence of 20 μg/ml MT-I/II. The bar graph is the average of three independent experiments (A). Control wells with no treatment were given an equal volume of PBS corresponding to the volume of MT-I/II that was added. Neurons plated onto myelin have 80% shorter neurites compared with those on PLL; however, the direct addition of MT-I/II strongly overcomes this inhibitory effect. Representative images of cortical neurons on PLL with (C) and without MT-I/II (B) and plated on myelin without MTs (D–F) and with MT-I/II treatment (G–I) demonstrate that MTs promote neurite outgrowth on myelin. Statistics performed are t test, **, p < 0.01. Scale bar, 20 μm.
Intrathecal Delivery of MT-I/II Blocks Inhibition of DRG Neurons by MAG in Culture
To begin to assess whether MT-I/II could encourage regeneration in vivo, we first performed an experiment to determine whether MT-I/II delivered into the cerebrospinal fluid would overcome inhibition of MAG by adult DRG neurons that are subsequently cultured on MAG-expressing or control CHO cells (Fig. 4). We delivered MT-I/II at different doses intrathecally to adult rats for 24 h using osmotic pumps and then collected the DRGs and grew them on MAG-expressing or control CHO cells, without the addition of any further MT-I/II during the culture period. Control animals received normal saline intrathecally delivered by osmotic pumps. We observed a dose-dependent increase in neurite outgrowth on MAG. Because the MT-I/II is only applied intrathecally suggests that MT-I/II promoted molecular changes in the DRGs neurons in vivo, which enabled them to overcome MAG inhibition once in culture.
FIGURE 4.
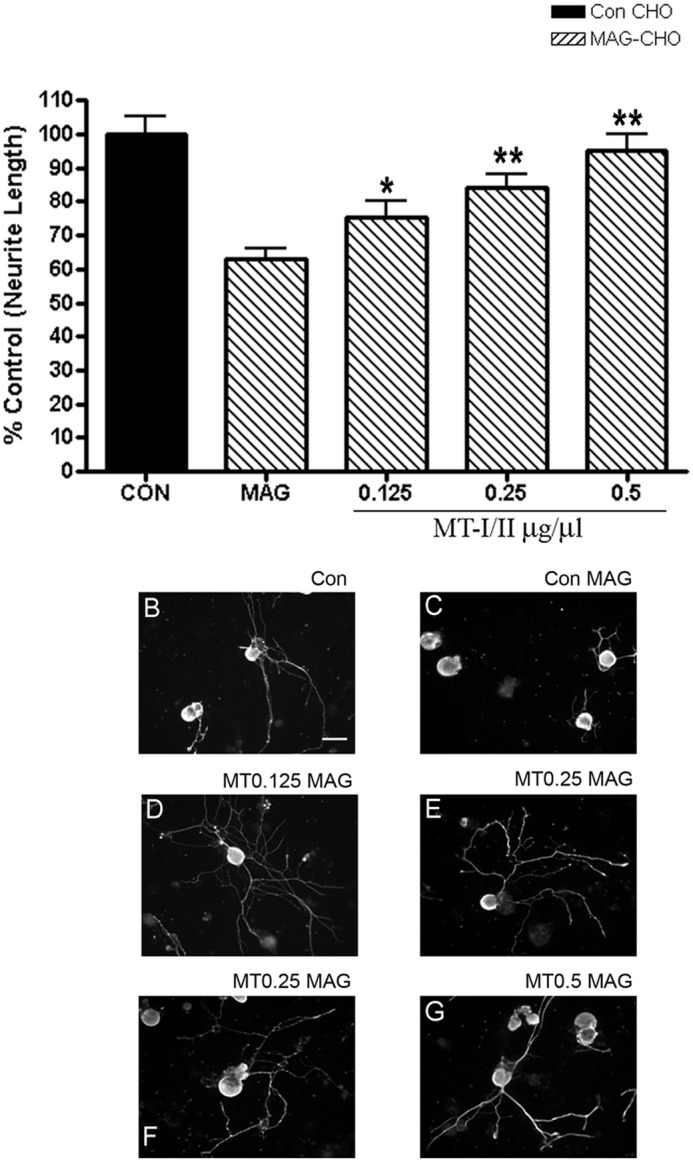
Intrathecal delivery of MT-I/II to lumbar DRGs of P28 rats overcomes MAG-mediated inhibition. P28 rats had pumps inserted intrathecally to deliver vehicle (sterile saline) or MT-I/II (0.125, 0.25, and 0.5 μg/μl) to the lumbar DRGs. 24 h later, DRG neurons from the L3, L4, and L5 DRG were cultured onto monolayers of either control (CON) or MAG-expressing (striped bars) or control CHO cells (black bars) and immunostained for βIII tubulin and quantitated for neurite length (300–500 neurons) (A). The graph is an average of three independent experiments with n = 6 for all conditions. Vehicle control neurons grew long processes on control CHO cells (B) but were inhibited in the presence of MAG by an average of 40% (C). Rats that received MT-I/II had significantly longer neurites on MAG at 0.125 μg/μl (D) and a dose-dependent increase in length with 0.25 (E and F) and 0.5 μg/μl (G). Statistics performed are from one-way ANOVA. *, p < 0.05; **, p < 0.01. Scale bar, 20 μm.
MT-I/II Expression Is Required for the Conditioning Lesion Effect
Because MT-I/II is up-regulated in DRG cell bodies after a conditioning lesion and overcomes inhibition by MAG/myelin, we next wanted to determine whether MT-I/II-deficient mice respond to a conditioning lesion in its ability of DRG neurons to overcome MAG inhibition. To test this possibility, we transected the peripheral branch of DRG neurons in age-matched MT-I/II-deficient and adult wild-type (WT) mice, and at 1 or 7 days post-lesion, the L3–5 DRGs are cultured on MAG-expressing or control CHO cells (Fig. 5, A–E). DRGs from WT mice with a conditioning lesion are able to overcome MAG inhibition at both 1 and 7 days post-lesion, compared with nonlesioned littermate controls. In contrast, the ability of DRGs from lesioned MT-I/II-deficient mice compared with WT mice is strongly reduced in overcoming inhibition by MAG for both the 1- and 7-day time points (**, p < 0.01; *, p < 0.05). This finding suggests that MT-I/II is required for the full conditioning lesion effect. The addition of MT-I/II promotes adult WT DRGs to overcome MAG inhibition (Fig. 5F). Furthermore, Fig. 5F shows that the MT-I/II effect is transcription-dependent, because we directly added the transcriptional inhibitor DRB with MT-I/II, and these DRGs are now inhibited by MAG.
FIGURE 5.
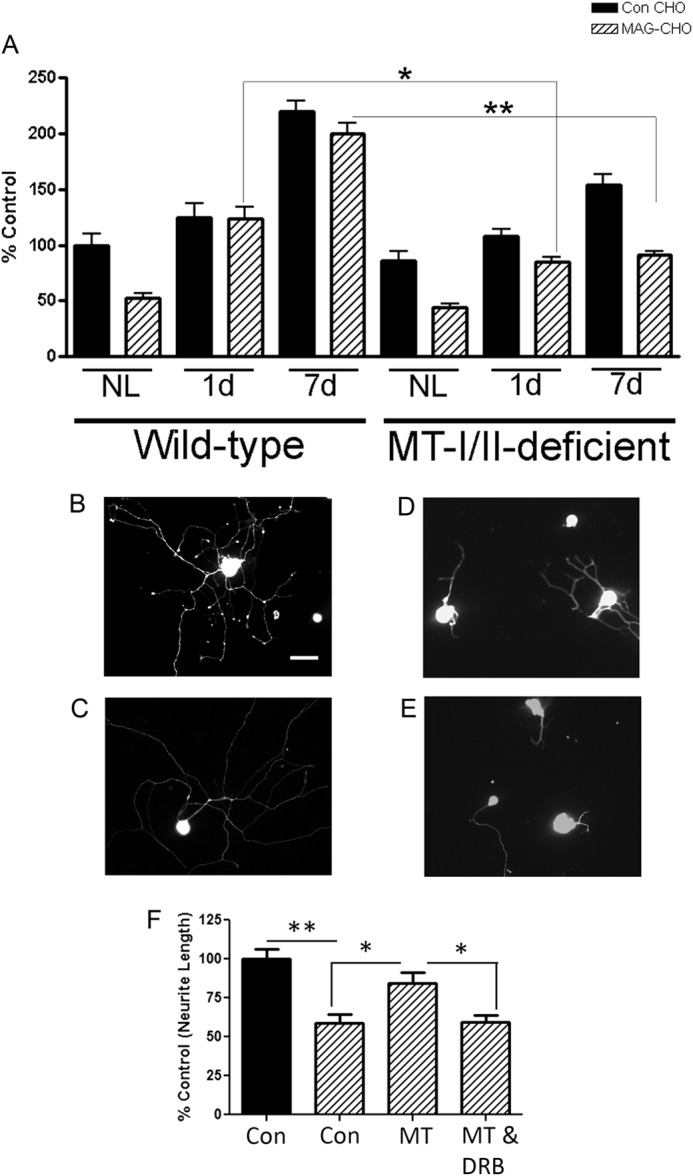
MT-I/II is required for the conditioning peripheral lesion effect and is transcription-dependent. Adult DRGs are isolated from WT and MT-I/II-deficient mice at 1 and 7 days (d) post-conditioning peripheral lesion (CL) (n = 5 for all conditions). The DRG neurons are plated on either control (Con) or MAG-expressing CHO cells for 14 h and stained for β-III tubulin. The longest neurite length for each neuron was quantitated using metamorph (A). DRGs from WT or MT-I/II-deficient mice that did not have a lesion (NL) are inhibited by MAG. WT and MT-I/II-deficient mice with CL overcome MAG inhibition compared with their non-lesion controls, respectively. However, MT-deficient mice 1 or 7 days post-CL put out significantly shorter processes on MAG compared with their WT littermates, where MT-I/II-deficient DRGs had ∼50% shorter processes 7 days post-CL on MAG compared with WT age-matched controls (*, p < 0.05, and **, p < 0.01). B–E, in the representative images below, WT (B and C) and MT-I/II-deficient (D and E) DRGs 7 days post-CL are plated on MAG and stained for β-III tubulin. DRG neurons from adult WT mice are isolated and either added directly to MAG-expressing CHO cells (striped bars) or control CHO cells (black bars) in the presence of MT-I/II (20 μg/ml), with and without DRB (5 μm) as indicated (F), after which they were cultured overnight, fixed, and immunostained for GAP43. In each experiment, the mean length of the longest GAP43-positive neurite for 200–300 neurons was measured (± S.E.). Scale bar, 20 μm.
The conditioning lesion effect on subsequent dorsal column regeneration of MT-I/II-deficient mice was then compared with WT mice. WT and MT-I/II-deficient mice received unilateral transections of the sciatic nerve, and 7 days later, they received a dorsal column lesion at T7–8. CTB subunit labeling of regenerating fibers in the dorsal column axons was performed 6 weeks later and was visualized with either DAB (Fig. 6, A–D) or a secondary antibody coupled to Alexa-488 (Fig. 6, F–H). It is important to note that the conditioning lesion effect in mice is less robust than in rats (17). WT mice had dorsal column axons regenerating to the lesion center (Fig. 6A) with some fibers crossing the lesion; the longest axon crossing the lesion center (*) is demarcated with a white arrow. In contrast, MT-I/II-deficient mice had far fewer fibers coming up the lesion center compared with WT mice (Fig. 6B), and very few of these axons grew across the lesion site. To quantitate these effects, axonal density for sections with DAB staining are quantified at 100, 200, and 300 μm rostral and caudal to the lesion (Fig. 6E). At 100 μm rostral to the lesion, WT mice had almost 87% higher axonal density of CTB-labeled axons (993.3 ± 108.7) compared with MT-I/II-deficient mice (132.8 ± 23.57), suggesting that the conditioning lesion effect is nearly abolished in MT-I/II-deficient mice. Without a conditioning lesion, dorsal column axons fail to regenerate in both WT and MT-I/II-deficient mice (Fig. 6, C and D). We also stained some of the sections from WT and MT-I/II-deficient mice that received a conditioning lesion and a dorsal column with an antibody for CTB and then visualized with secondary antibody coupled to Alexa-488 (Fig. 6, F–H). Similar to what we observed for the DAB-developed sections, WT mice with the conditioning lesion had dorsal column axons regenerating to the lesion center (Fig. 6F) with some fibers crossing the lesion, and the furthest one is demarcated with a white arrow; however, in MT-I/II-deficient mice, the CTB-labeled axons barely made it to the lesion center (Fig. 6G). A more magnified view of an adjacent section of the WT in Fig. 6F displays several axons crossing the lesion center (Fig. 6H).
FIGURE 6.

Lesioned dorsal column axons fail to regenerate after a conditioning peripheral lesion in MT-I/II-deficient mice. WT mice that received a dorsal column lesion after a conditioning peripheral lesion (CL) (A), have CTB-labeled axons (developed with DAB) beyond the lesion site (*). However MT-deficient mice even with CL fail to regenerate even to the lesion site (B). Both WT (C) and MT-I/II-deficient mice (D) fail to regenerate following a dorsal column lesion alone. Measuring average axonal density at ±100, 200, and 300 μm to the lesion site was compared for all conditions in WT and MT-knock-out mice (E). Looking at densities −100 caudal and +100 to +300 rostral, we observed a significantly longer growth of WT with CL compared with MT-I/II-deficient mice with CL (*, p < 0.05, and **, p < 0.01). This suggests that MT-deficient mice fail to regenerate even with CL. Scale bar, 100 μm. We also looked at CTB-labeled axons in WT and MT-deficient mice with CL using a secondary coupled to Alexa-488 and imaged using a multiphoton microscope. WT mice that received a dorsal column lesion after CL (F) have CTB-labeled axons crossing the lesion center; the arrow demarcates an axon growing past the lesion site (*). An adjacent section of the same WT mouse in F had several axons crossing the lesion center in a magnified region (H). However MT-deficient mice even with CL fail to regenerate even to the lesion site; the arrow demarcates the longest axons detected nearly at the lesion site (*) (G). Scale bar, 100 μm.
MT-I/II Promotes Axon Regeneration of Optic Nerve in Vivo
To assess whether MT-I/II can promote CNS axon regeneration in vivo, we delivered it to rat retinal ganglion cells (RGC) after optic nerve crush either directly to the neuronal cell bodies by intravitreal injections or by gel foam (GF) soaked with MT-I/II placed on top of the crushed axons. The optic nerve was crushed at 2 mm behind the eye, and immediately afterward a single intravitreal injection of MT-I/II was administered. We avoided LI, which in itself has a robust conditioning effect on regeneration of the RGC axons for one group of animals (Fig. 7, A–C and H) (28). Two weeks after the lesion, the animals were sacrificed, and the optic nerves were stained for GAP-43. We found that a single injection of MT-I/II promotes significant regeneration compared with saline injection. Fig. 7 shows the results from three animals, one that received saline (Fig. 7A), one that received MT-I/II injection (Fig. 7B), and one that had GF soaked with MT-I/II placed on top of the lesion site (Fig. 7C). Fibers up to 1 mm beyond the crush site are detected in the MT-I/II-injected animals but not in the saline-injected animals or with MT-I/II soaked in GF and applied to the injury site. There is a statistically significant increase in optic nerve axon regeneration in the MT-I/II-injected animals compared with saline-injected animals when we quantified the density of GAP-43 positive axons at 500- μm intervals (Fig. 7H). We found that the axonal density was significantly higher for MT-I/II-injected animals at 0–500 and 500–1000 distance from the lesion compared with saline-injected animals. Another group of animals with LI had received injections with saline (Fig. 7, D and F) or MT-I/II (Fig. 7, E and G). The group that received saline and LI had robust axonal regeneration, and a close-up taken from 1.2 mm past the crush site (Fig. 7F) shows the maximal extent of the growth. LI with MT-I/II had more pronounced axonal regeneration compared with the saline injection, and the close-up of that group had robust axonal regeneration almost 1.5 mm away from the crush (Fig. 7G) and significantly more than saline injection with LI (Fig. 7I). The finding without LI suggests that MT-I/II is sufficient to promote CNS axonal regeneration. Furthermore, MT-I/II can only promote axonal regeneration when delivered to the neuronal cell bodies, because applying GF soaked with MT-I/II to the lesioned optic nerve promoted no axonal regeneration. With LI, MT-I/II can enhance the regenerative effects compared with LI with saline injection. To confirm that a single injection of MT-I/II can promote axonal regeneration, we performed another optic nerve crush on a Fischer rat and injected intravitreally immediately afterward with MT-I/II. This time we labeled regenerating axons by intravitreally injecting with CTB 3 days prior to sacrificing the animals. After perfusion, we chemically cleared the nerve to make it transparent and to visualize regenerating axons in the whole optic nerve using a multiphoton microscope (29). In Fig. 7J, we see CTB-labeled regenerating fibers almost 1 mm from the crush site, and we also highlight an area with a white border where we reimaged at higher magnification (Fig. 7K). Fig. 7K shows regenerating axons with their usual tortured pattern growth, and we also point out with an arrow what appears to be a growth cone.
FIGURE 7.
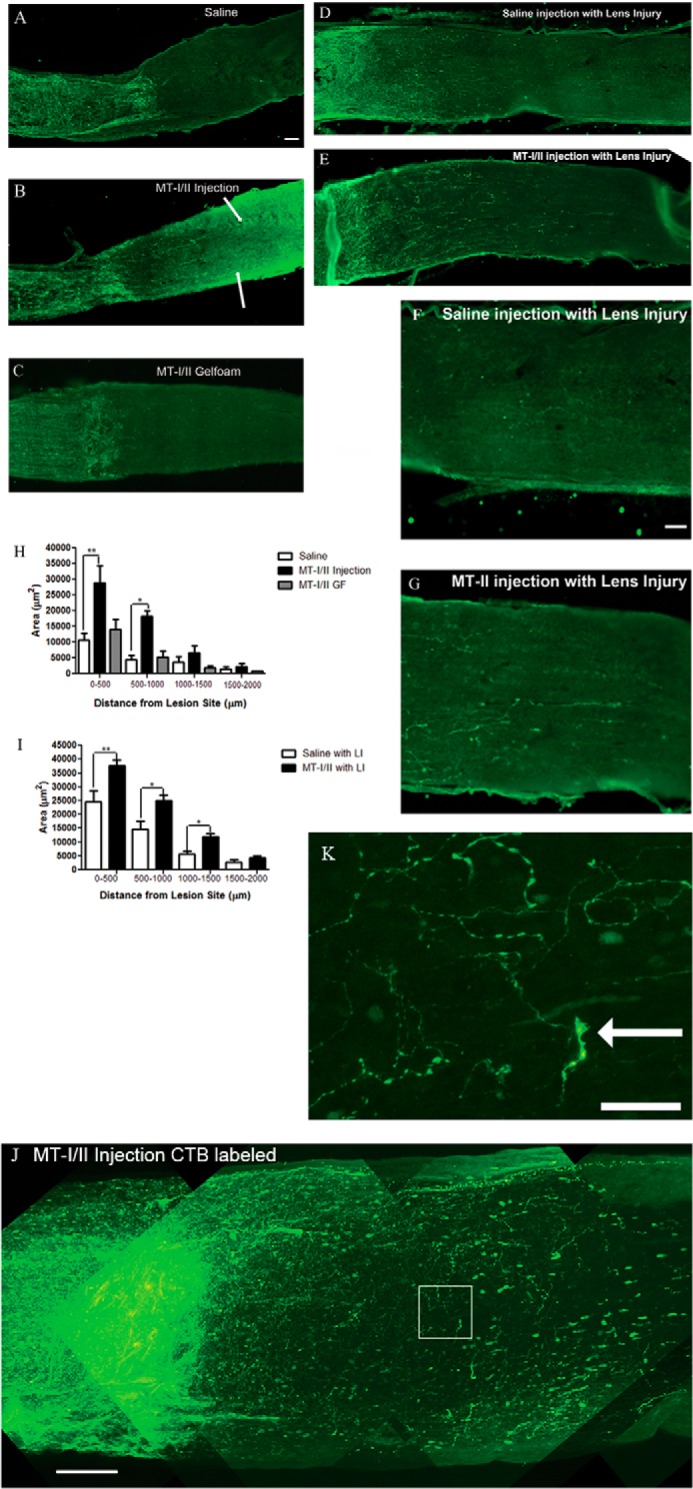
Intravitreal injection of MT-I/II-injected promotes optic nerve regeneration in vivo. Fischer rats with optic nerve injury were injected intravitreally with 5 μl of normal saline (A), MT-I/II (5 μg/μl) (B), or had MT-I/II-soaked GF (C) placed over the injured retinal axons immediately after crushing the optic nerve; animals A–C had no signs of lens injury. With LI these rats were injected intravitreally with 5 μl of normal saline (D), MT-I/II (5 μg/μl) (E), and a close-up view of the axons with either saline (F) or MT-I/II (G) injection ∼1.25 mm from the lesion center. Regenerating axons were evaluated 2 weeks later by staining for GAP-43. Quantification of axonal density in regions 0–500, 500–1000, 1000–1500, and 1500–2000 μm was distal to the lesion site. Graph depicts average axonal density (square micrometer) ± S.E. for animals with no LI (H) and for animals with LI (*, p < 0.05; **, p < 0.01) (I). Scale bar, 100 μm. Fischer rats received intravitreal injection with 5 μl of MT-I/II (5 μg/μl) immediately after crushing the optic nerve, and there was no signs of lens injury (J). To assess regenerating axons, we labeled with CTB coupled to Alexa-488 intravitreally injected 3 days prior to sacrifice. The whole optic nerve was chemically cleared, and we visualized CTB-labeled fibers using a multiphoton microscope scanning 250 μm of the optic nerve. Scale bar, 100 μm. The white box in J is an area we reimaged at higher magnification to show the extent of CTB-labeled regenerating fibers (K). The white arrow is pointing to what appears to be a growth cone. scale bar, 25 μm.
MT-I/II Overcomes MAG-mediated Inhibition of Neurite Outgrowth Independent of Zinc Chelation
MT-I/II can regulate levels of divalent cations, such as zinc, having been suggested to even chelate free zinc in the CNS. To determine whether MT-I/II's ability to promote neurite outgrowth in the presence of MAG could simply be due to functioning as a zinc chelator, we applied Ca-EDTA to our neurite outgrowth assay with the MAG-expressing CHO cells using primary cortical neurons (Fig. 8A). We used 1 mm Ca-EDTA to chelate extracellular zinc in primary cortical cultures, which is at a concentration known to abolish zinc-induced effects (34). The primary cortical neurons plated onto MAG-expressing CHO cells have 70% shorter neurites (Fig. 8C) compared with those on control CHO, which put out long neurites (Fig. 8B). The addition of MT-I/II (Fig. 8, F and G) or cAMP (Fig. 8E) overcomes this inhibitory effect of MAG; however, Ca-EDTA has no effect on overcoming MAG inhibition (Fig. 8D). Because the direct addition of the general zinc chelator Ca-EDTA is not sufficient to promote neurite outgrowth on MAG, this suggests to us that simply binding up all the free zinc is not efficient to overcome MAG inhibition. This finding with Ca-EDTA suggests that nonspecific zinc chelation by MT-I/II is unlikely to be the mechanism for overcoming myelin-mediated inhibitors. Rather an alternative mechanism for MT-I/II must be involved to overcome MAG inhibition.
FIGURE 8.
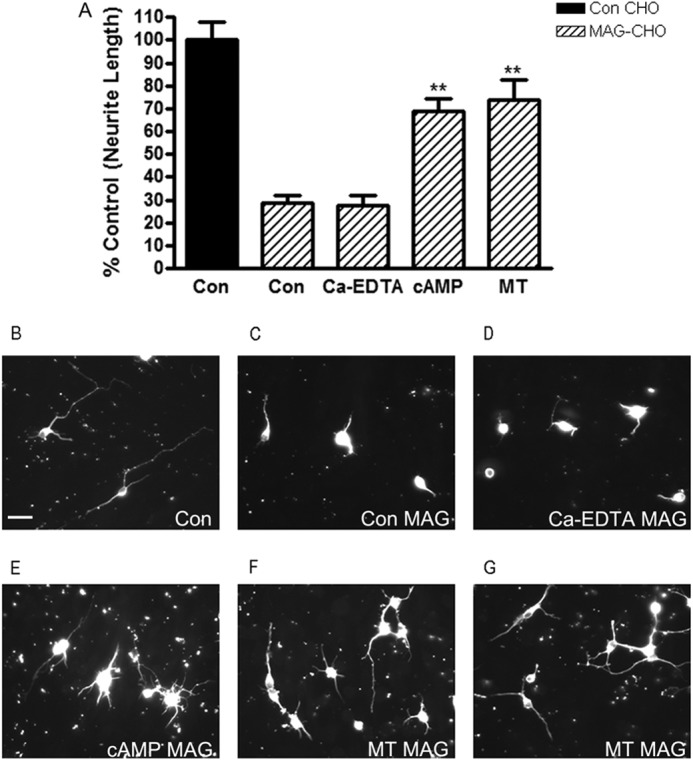
MT-I/II overcomes MAG-mediated inhibition of neurite outgrowth independent of zinc chelation. A, primary P1 cortical neurons are isolated plated on either control (Con) (black) or MAG-expressing (striped bars) CHO cells for 24 h, in the presence or absence of either PBS (control), 20 μg/ml MT-I/II, or 1 mm Ca-EDTA. Neurons plated onto MAG-expressing CHO cells have 70% shorter neurites compared with those on control CHO. The addition of MT-I/II strongly overcomes this inhibitory effect of MAG; however, Ca-EDTA has no effect on overcoming MAG inhibition suggesting that chelation of zinc alone is not sufficient to promote neurite outgrowth on MAG. Representative images of cortical neurons shown are plated on control (B) or MAG-expressing CHO (C) cells, Ca-EDTA-treated on MAG (D), cAMP-treated on MAG (E), and MT-I/II-treated on MAG (F and G). Statistics performed are by ANOVA.**, p < 0.01. Scale bar, 20 μm.
MT-I/II Inhibits α-Secretase (TACE) Activity and Is Down-regulated in WT DRG after Conditioning Lesion
To further investigate the mechanism behind MT-I/II to promote axonal regeneration in the presence of myelin-mediated inhibitors, we looked at the inhibitory cascade initiated by MAG binding to its receptor complexes. In the presence of MAG, the first enzyme that is activated is zinc-dependent membrane-bound α-secretase (35). Importantly, MT-I/II were implicated in the regulation of zinc-dependent proteins, such as zinc finger proteins that act in cellular signaling and transcription regulation (36, 37). To determine whether MT-I/II can regulate the proteolytic activity of α-secretase, we incubated the recombinant α-secretase candidate TACE, which can cleave α-secretase substrates, with MT-I/II (Fig. 9A). We found that MT-I/II can inhibit the proteolytic activity of TACE. This activity was specific to MT-I/II because if we used another protein, SLPI, which is similar in size and also promotes axonal regeneration in the presence of myelin-mediated inhibitors, but has no known zinc-chelating function, there was no change in activity of TACE (Fig. 9B). This finding implicates that MT-I/II can inhibit the activity of the α-secretase, TACE.
FIGURE 9.
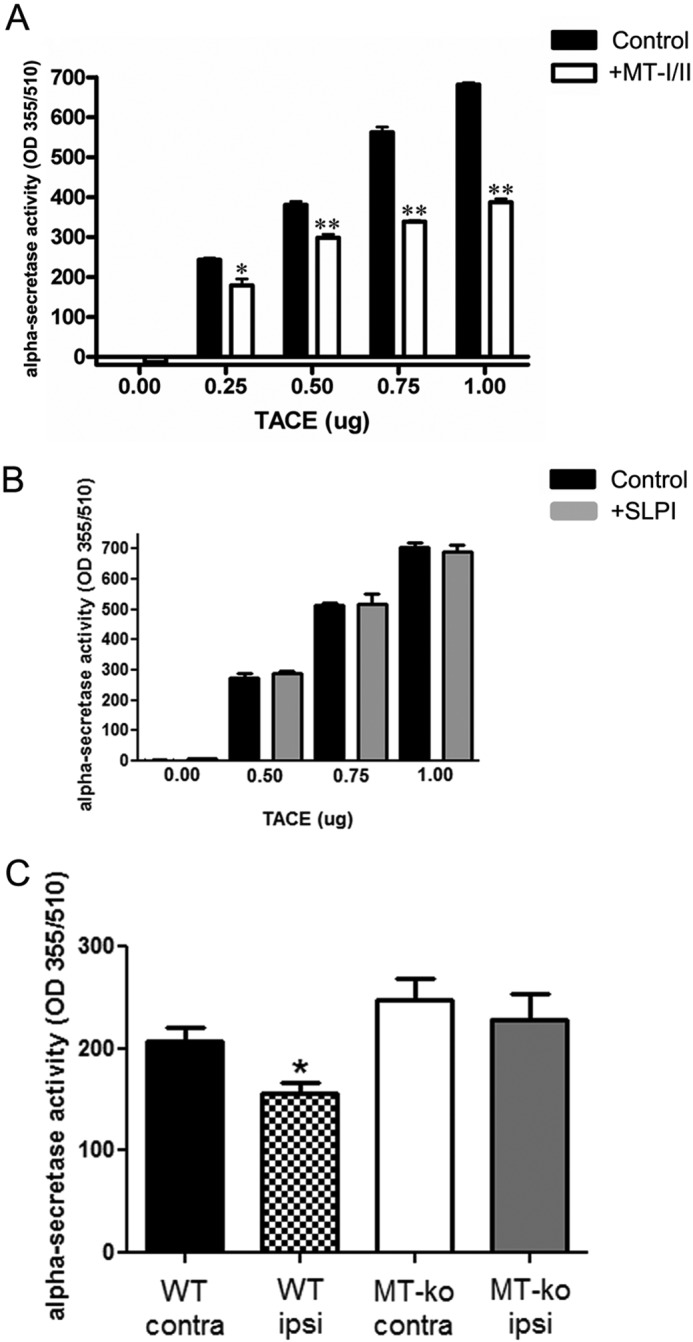
MT-I/II inhibits α-secretase (TACE) activity and is down-regulated in WT DRG after conditioning lesion. A, we used a kit to monitor the enzymatic activity of the α-secretase candidate TACE with or without MT-I/II; the graph is the average of three independent experiments. We determined that 60 min of incubation with 10 μg/ml MT-I/II at 37 °C inhibits the proteolytic activity of TACE by up to 43%. B, as a control, we also tested SLPI in the assay and found it had no effect on the enzymatic activity of TACE. C, age-matched WT and MT-I/II-deficient mice (MT-ko) had their L3–5 DRGs removed 3 h after conditioning lesion (ipsi), and for control we also collected the DRGs from the noninjured (contra) side, n = 5, for both genotypes; and the data are average ± S.E. We loaded 50 μg of total protein for each condition and monitored for α-secretase activity. We found that WT mice after conditioning lesion (checkered bar) had significantly less α-secretase activity compared with the contralateral side of DRGs (black bar) (t test, *, p < 0.05). However, MT-I/II-deficient mice had no significant change in α-secretase activity after conditioning lesion (white bar) compared with the contralateral side DRGs (gray bar).
To further elucidate whether indeed MT-I/II modulates α-secretase proteolytic activity, we collected DRGs from both the ipsilateral and contralateral sides of adult WT and MT-I/II-deficient mice after the conditioning lesion, and immediately after collection, we performed an α-secretase activity assay (Fig. 9C). We found that after the conditioning lesion, WT DRGs had significantly less α-secretase activity. Consistent with our in vitro finding, MT-I/II-deficient DRGs had no change in α-secretase activity after the conditioning lesion. This finding suggests that after the conditioning lesion, α-secretase activity is reduced in WT DRGs but is unaffected in MT-I/II-deficient DRGs.
MT-I/II Blocks the MAG-induced Phosphorylation of PKC and Activation of Rho
A key component of the downstream signaling pathway activated by myelin-associated inhibitory molecules is the phosphorylation of protein kinase C (PKC) and activation of Rho (9, 14). To assess whether MT-I/II could interfere with MAG-induced PKC activation, we treated cerebellar neurons with soluble MAG-Fc. 30 min of treatment with MAG-Fc was sufficient to induce activation of PKC; however, the addition of MT-I/II along with MAG-Fc blocked the activation of PKC (Fig. 10A). Treatment with MT-I/II alone was comparable with that seen in the unstimulated control. Further downstream of PKC activation by myelin inhibitors is the small GTPase Rho (14). All three of the major inhibitors found in myelin, MAG, oligodendrocyte-myelin glycoprotein, and Nogo, converge in the downstream activation of Rho (10–13). Using a Rho activation assay, we show that as before, Rho is activated in cortical neurons treated with MAG-Fc (Fig. 10B). However, MAG-Fc-induced activation of Rho is blocked in the presence of MT-I/II. Together, these results indicate that the ability of MT-I/II to overcome MAG inhibition is likely to be interfering with the MAG-induced activation of PKC and Rho.
FIGURE 10.

MT-I/II blocks the MAG-induced phosphorylation of PKC and activation of Rho. A, Western blots of P5 cerebellar neurons treated for 30 min with MAG-FC with or without 10 μg/ml MT-I/II. Membranes are probed for phospho-pan PKC and actin. Graphs depict average of four experiments with densitometric measurements expressed as fold change of control ± S.E. (*, p < 0.05; **, p < 0.01). Membranes are probed for pan-phospho-PKC and actin. B, Western blots of P1 cortical neurons are treated with 10 μg/ml MT-I/II for 60 min prior to exposing the neurons to 20 μg/ml MAG-Fc for 30 min and then used for a Rho-pulldown assay. Samples not processed for the pulldown are used to quantitate total Rho in the samples. Membranes are probed with RhoA. Graphs depict average of four experiments and densitometric measurements are expressed as fold change of control ± S.E. (*, p < 0.05; **, p < 0.01).
Discussion
We found that MT-I/II is up-regulated in DRG neurons after the conditioning lesion and that it can overcome MAG and myelin inhibition in three neuronal types. DRG neurons that had intrathecal delivery of MT-I/II in vivo are not inhibited by MAG when grown in culture. DRG neurons from WT mice with conditioning lesion overcome myelin-associated inhibitors, whereas the neurons from MT-I/II-deficient mice do not overcome MAG inhibition. In addition, MT-I/II-deficient mice have poor spinal axon regeneration after the conditioning lesion. Importantly, exogenous MT-I/II delivered by intravitreal injection promotes regeneration of RGC axons. Mechanistically, MT-I/II's ability to overcome MAG inhibition is transcription-dependent, as adult DRG neurons can no longer overcome MAG inhibition with the exogenous application of MT-I/II in the presence of the transcriptional inhibitor DRB. MT-I/II pro-regenerative effect is not due to its zinc-chelating function, as the addition of Ca-EDTA to cortical neurons does not overcome MAG inhibition. Rather than a global zinc chelation effect, MT-I/II seems to be more targeted in its response as it can inhibit the enzymatic activity of the zinc-dependent secretase TACE. Furthermore, we show that after a conditioning lesion, adult WT DRGs have significantly lower levels of α-secretase activity, but no apparent changes in α-secretase activity are detected in MT-I/II-deficient DRGS.
Previous studies have shown that MT-IIA promotes a significant increase in the rate of initial neurite elongation on a permissive substrate, but the effect on myelin-associated inhibitors was not addressed (20). We now add to these findings by demonstrating that MT-I/II overcomes MAG and myelin-mediated inhibition of neurite outgrowth in vitro for a variety of primary neurons, including hippocampal, cortical, and DRG (Figs. 2 and 3), and promotes axonal regeneration through an inhibitory environment in vivo.
Studies in the nervous system revealed that neurons, astrocytes, and microglia express MTs, and astrocytes secrete MT-I/II in injury models (21, 25, 38, 39). In focal cerebral ischemia models in mice, MT-I/II mRNA levels are strongly elevated (22). MT-I/II-deficient mice undergoing focal cerebral ischemia had significantly larger infarcts and neurological deficits compared with WT mice, suggesting a neuroprotective role for MT-I/II in CNS injury (22). One appealing aspect of MT-I/II is that it is both neuroprotective and pro-regenerative, and the neuroprotective effects could be attributed to its zinc-chelating ability (21). However, it is unlikely that the neuroprotective function of MT-I/II contributes to its ability to overcome inhibition by myelin-associated inhibitors. Using a TUNEL assay to measure cell death, we do not see a difference in survival among DRG neurons from either WT or MT-I/II-deficient mice following a conditioning lesion, and only 5% of neurons from each genotype undergo cell death (data not shown). Likewise, ArgI and polyamines are up-regulated in response to cAMP and a conditioning lesion. These agents have long been known to be neuroprotective, but like MT-I/II, this effect is unlikely to contribute to overcoming myelin-associated inhibitors and promoting axonal regeneration (30, 40, 41).
MT-I/II mRNA is elevated in the area within and just distal to the site of spinal cord transection as early as 6 h post-injury, and significantly elevated levels are sustained at 48 h post-lesion (42). This finding is consistent with other reports that MT-I/II is elevated after CNS injury. However, the elevation of MT-I/II levels at the injury site is not sufficient to promote axonal regeneration, as there is no regeneration without additional treatments. This could be the consequence of either insufficient elevation in the amounts of MT-I/II and/or MT-I/II failing to reach its site of action. We have shown that MT-I/II must reach the neuronal cell body to have its effect as demonstrated by the following findings. First, addition of MT-I/II overcomes MAG and myelin-associated inhibitors only when administered to freshly isolated neurons that have all their neurites severed by the isolation process. This also applies to the intrathecal delivery experiments where MT-I/II was delivered in vivo prior to isolating the neurons and performing the neurite outgrowth assay. More importantly, delivery of MT-I/II in vivo to the RGC cell bodies promotes axonal regeneration after optic nerve crush, but delivery of MT-I/II to the axons does not. This implicates that MT-I/II causes intrinsic changes in the neuronal cell bodies that allow them to overcome inhibition. This is supported by the observation that MT-I/II's ability to overcome MAG inhibition is transcription-dependent, as adding the transcriptional inhibitor DRB blocks MT-I/II's effect on neurite outgrowth on adult DRG neurons.
Metallothioneins' unique amino acid structure, composed of nearly one-third cysteines, can coordinate binding of up to seven zinc ions in two thiolate clusters, and it has a putative nuclear localization signal (43–46). It has been shown that metallothioneins are effective at sequestering metals, as MT-I/II-deficient mice are more prone to metal toxicity compared with wild-type mice (47). The ability of MT-I/II to overcome MAG inhibition does not appear to be dependent on global zinc chelation, as applying Ca-EDTA in our neurite outgrowth assay did not overcome MAG-mediated inhibition of cortical neurons. Metallothioneins are unique in having high thermodynamic stability when bound to zinc and also high kinetic lability, meaning that they can donate and/or accept zinc ions (36, 44). Interestingly, metallothioneins have also been shown to donate zinc ions to zinc-dependent proteins, including zinc finger proteins that act in cellular signaling and transcription regulation, suggesting a more targeted and specific interaction of MT-I/II and zinc ions (36, 37). It is therefore possible that the intrinsic changes induced by delivering MT-I/II in DRG neurons are related to the regulation of zinc-dependent proteins such as α-secretase. We show here that incubating TACE with MT-I/II inhibited the proteolytic activity of TACE, but by using another protein that is pro-regenerative but does not bind metals, SLPI does not alter TACE activity. This is further supported by our WT and MT-I/II-deficient mouse study for α-secretase activity 3 h after a conditioning lesion. WT mice ipsilateral DRGs have significantly lower levels of active α-secretase compared with the contralateral side, and we see no difference between the MT-I/II-deficient DRGs from the ipsilateral or contralateral sides. Furthermore, MT-I/II-deficient mice have poor spinal axon regeneration in the injured dorsal column even with a conditioning lesion, supporting that MT-I/II is one of the proteins required to promote axonal regeneration through the inhibitory environment of the CNS.
The signaling pathways for growth inhibitory molecules, such as myelin-associated inhibitors and chondroitin sulfate proteoglycans, converge on the activation of Rho GTPase for their inhibitory effect (48–50). Our data show that MT-I/II interferes with this pathway by not only blocking the MAG-induced activation of Rho, but also by blocking upstream of Rho, the activation of PKC. The inactivation of Rho to promote axonal regeneration in spinal cord injury has been extensively studied, and a recombinant Rho protein antagonist has now entered clinical trials for spinal cord injury (51).
Our work with MT-I/II is a proof of principle study, supporting the notion that it is necessary to reprogram the adult neurons to initiate growth. A single injection of MT-I/II promotes modest axonal regeneration and can enhance the robust regeneration observed with LI in the injured optic nerve. It is becoming evident that combinatorial approaches to overcoming the inhibitory environment in the injured CNS will be required to achieve substantial regenerative growth. Given the finding that MT-I/II can be delivered post-injury and in combination with LI promotes significant axonal regeneration warrants further study of MT-I/II to explore its therapeutic potential. Previously we showed that both SLPI and ArgI are also necessary and sufficient for axonal regeneration (32, 33). MT-I/II-deficient mice lack spinal axon regeneration after the conditioning lesion, so perhaps MT-I/II is necessary to initiate axonal regeneration, and other regenerative associated genes, such as slpi or arg I, are needed in combination to enhance the growth. It must be noted that when administered alone, MT-I/II, SLPI, and ArgI are not as robust as the conditioning lesion effect in their ability to promote axonal regeneration. Additional experiments are required to determine whether combinations of MT-I/II, SLPI, and/or ArgI, which have very different molecular mechanisms, could function synergistically. In addition, identifying optimal dosing regimens for all of them and elucidating potential redundancies in their downstream pathways are the important next steps to pursue.
Acknowledgments
Multiphoton microscopy was performed in the Microscopy CORE at the Icahn School of Medicine at Mount Sinai and was supported with funding from National Institutes of Health Shared Instrumentation Grant 1S10RR026639-01. We thank Saranna Belgrave and Dr. Nagarathnamma Chaudhry for their outstanding technical support and Drs. Rajiv R. Ratan (Burke-Cornell Medical Research Institute, White Plains, NY) and Ravi Iyengar (Mount Sinai School of Medicine, New York) for assistance with this manuscript. We also thank Drs. Larry Benowitz and Barbara Lorber (Harvard University, Cambridge, MA) for providing instruction in the optic nerve crush surgery and the GAP-43 antibody. We also thank Drs. Frank Bradke and Farida Hellal at Axonal Growth and Regeneration, German Center for Neurodegenerative Diseases (Bonn, Germany), for their instruction on performing chemical clearing.
This work was supported, in whole or in part, by National Institutes of Health Grants 3U54NS041073 from NINDS, RO1NS037060 from NINDS, RR03037 from NCRR, F32NS054511, and 1S10RR026639-01. The authors declare that they have no conflicts of interest with the contents of this article.
- MAG
- myelin-associated glycoprotein
- MT
- metallothionein
- DRG
- dorsal root ganglion
- Bt2cAMP
- dibutyryl cyclic AMP
- CTB
- cholera toxin B
- PFA
- paraformaldehyde
- DRB
- 5,6-dichlorobenzimidazole 1-β-d-ribofuranoside
- Arg
- arginase
- TACE
- tumor necrosis factor-α converting enzyme
- ANOVA
- analysis of variance
- LI
- lens injury
- PLL
- poly-l-lysine
- GF
- gel foam
- RGC
- retinal ganglion cell
- DAB
- 3,3′-diaminobenzidine
- CL
- conditioning (peripheral) lesion.
References
- 1. McKerracher L., David S., Jackson D. L., Kottis V., Dunn R. J., Braun P. E. (1994) Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron 13, 805–811 [DOI] [PubMed] [Google Scholar]
- 2. Mukhopadhyay G., Doherty P., Walsh F. S., Crocker P. R., Filbin M. T. (1994) A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron 13, 757–767 [DOI] [PubMed] [Google Scholar]
- 3. GrandPré T., Nakamura F., Vartanian T., Strittmatter S. M. (2000) Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature 403, 439–444 [DOI] [PubMed] [Google Scholar]
- 4. Prinjha R., Moore S. E., Vinson M., Blake S., Morrow R., Christie G., Michalovich D., Simmons D. L., Walsh F. S. (2000) Inhibitor of neurite outgrowth in humans. Nature 403, 383–384 [DOI] [PubMed] [Google Scholar]
- 5. Chen M. S., Huber A. B., van der Haar M. E., Frank M., Schnell L., Spillmann A. A., Christ F., Schwab M. E. (2000) Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 403, 434–439 [DOI] [PubMed] [Google Scholar]
- 6. Wang K. C., Koprivica V., Kim J. A., Sivasankaran R., Guo Y., Neve R. L., He Z. (2002) Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature 417, 941–944 [DOI] [PubMed] [Google Scholar]
- 7. Filbin M. T. (2003) Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat. Rev. Neurosci. 4, 703–713 [DOI] [PubMed] [Google Scholar]
- 8. Atwal J. K., Pinkston-Gosse J., Syken J., Stawicki S., Wu Y., Shatz C., Tessier-Lavigne M. (2008) PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science 322, 967–970 [DOI] [PubMed] [Google Scholar]
- 9. Lehmann M., Fournier A., Selles-Navarro I., Dergham P., Sebok A., Leclerc N., Tigyi G., McKerracher L. (1999) Inactivation of Rho signaling pathway promotes CNS axon regeneration. J. Neurosci. 19, 7537–7547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang K. C., Kim J. A., Sivasankaran R., Segal R., He Z. (2002) P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature 420, 74–78 [DOI] [PubMed] [Google Scholar]
- 11. Mi S., Lee X., Shao Z., Thill G., Ji B., Relton J., Levesque M., Allaire N., Perrin S., Sands B., Crowell T., Cate R. L., McCoy J. M., Pepinsky R. B. (2004) LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat. Neurosci. 7, 221–228 [DOI] [PubMed] [Google Scholar]
- 12. Shao Z., Browning J. L., Lee X., Scott M. L., Shulga-Morskaya S., Allaire N., Thill G., Levesque M., Sah D., McCoy J. M., Murray B., Jung V., Pepinsky R. B., Mi S. (2005) TAJ/TROY, an orphan TNF receptor family member, binds Nogo-66 receptor 1 and regulates axonal regeneration. Neuron 45, 353–359 [DOI] [PubMed] [Google Scholar]
- 13. Park J. B., Yiu G., Kaneko S., Wang J., Chang J., He X. L., Garcia K. C., He Z. (2005) A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron 45, 345–351 [DOI] [PubMed] [Google Scholar]
- 14. Sivasankaran R., Pei J., Wang K. C., Zhang Y. P., Shields C. B., Xu X. M., He Z. (2004) PKC mediates inhibitory effects of myelin and chondroitin sulfate proteoglycans on axonal regeneration. Nat. Neurosci. 7, 261–268 [DOI] [PubMed] [Google Scholar]
- 15. Qiu J., Cai D., Dai H., McAtee M., Hoffman P. N., Bregman B. S., Filbin M. T. (2002) Spinal axon regeneration induced by elevation of cyclic AMP. Neuron 34, 895–903 [DOI] [PubMed] [Google Scholar]
- 16. Richardson P. M., Issa V. M. (1984) Peripheral injury enhances central regeneration of primary sensory neurons. Nature 309, 791–793 [DOI] [PubMed] [Google Scholar]
- 17. Neumann S., Woolf C. J. (1999) Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron 23, 83–91 [DOI] [PubMed] [Google Scholar]
- 18. Neumann S., Bradke F., Tessier-Lavigne M., Basbaum A. I. (2002) Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron 34, 885–893 [DOI] [PubMed] [Google Scholar]
- 19. Gao Y., Deng K., Hou J., Bryson J. B., Barco A., Nikulina E., Spencer T., Mellado W., Kandel E. R., Filbin M. T. (2004) Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron 44, 609–621 [DOI] [PubMed] [Google Scholar]
- 20. Chung R. S., Vickers J. C., Chuah M. I., West A. K. (2003) Metallothionein-IIA promotes initial neurite elongation and postinjury reactive neurite growth and facilitates healing after focal cortical brain injury. J. Neurosci. 23, 3336–3342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. West A. K., Chuah M. I., Vickers J. C., Chung R. S. (2004) Protective role of metallothioneins in the injured mammalian brain. Rev. Neurosci. 15, 157–166 [DOI] [PubMed] [Google Scholar]
- 22. Trendelenburg G., Prass K., Priller J., Kapinya K., Polley A., Muselmann C., Ruscher K., Kannbley U., Schmitt A. O., Castell S., Wiegand F., Meisel A., Rosenthal A., Dirnagl U. (2002) Serial analysis of gene expression identifies metallothionein-II as major neuroprotective gene in mouse focal cerebral ischemia. J. Neurosci. 22, 5879–5888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carrasco J., Penkowa M., Hadberg H., Molinero A., Hidalgo J. (2000) Enhanced seizures and hippocampal neurodegeneration following kainic acid-induced seizures in metallothionein-I + II-deficient mice. Eur. J. Neurosci. 12, 2311–2322 [DOI] [PubMed] [Google Scholar]
- 24. Penkowa M., Hidalgo J. (2001) Metallothionein treatment reduces proinflammatory cytokines IL-6 and TNF-α and apoptotic cell death during experimental autoimmune encephalomyelitis (EAE). Exp. Neurol. 170, 1–14 [DOI] [PubMed] [Google Scholar]
- 25. Chung R. S., West A. K. (2004) A role for extracellular metallothioneins in CNS injury and repair. Neurosci. 123, 595–599 [DOI] [PubMed] [Google Scholar]
- 26. Siddiq M. M., Tsirka S. E. (2004) Modulation of zinc toxicity by tissue plasminogen activator. Mol. Cell. Neurosci. 25, 162–171 [DOI] [PubMed] [Google Scholar]
- 27. Cao Z., Gao Y., Bryson J. B., Hou J., Chaudhry N., Siddiq M., Martinez J., Spencer T., Carmel J., Hart R. B., Filbin M. T. (2006) The cytokine interleukin-6 is sufficient but not necessary to mimic the peripheral conditioning lesion effect on axonal growth. J. Neurosci. 26, 5565–5573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leon S., Yin Y., Nguyen J., Irwin N., Benowitz L. I. (2000) Lens injury stimulates axon regeneration in the mature rat optic nerve. J. Neurosci. 20, 4615–4626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ertürk A., Mauch C. P., Hellal F., Förstner F., Keck T., Becker K., Jährling N., Steffens H., Richter M., Hübener M., Kramer E., Kirchhoff F., Dodt H. U., Bradke F. (2012) Three-dimensional imaging of the unsectioned adult spinal cord to assess axon regeneration and glial responses after injury. Nat. Med. 18, 166–171 [DOI] [PubMed] [Google Scholar]
- 30. Cai D., Deng K., Mellado W., Lee J., Ratan R. R., Filbin M. T. (2002) Arginase I and polyamines act downstream from cyclic AMP in overcoming inhibition of axonal growth MAG and myelin in vitro. Neuron 35, 711–719 [DOI] [PubMed] [Google Scholar]
- 31. Cai D., Qiu J., Cao Z., McAtee M., Bregman B. S., Filbin M. T. (2001) Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J. Neurosci. 21, 4731–4739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hannila S. S., Siddiq M. M., Carmel J. B., Hou J., Chaudhry N., Bradley P. M., Hilaire M., Richman E. L., Hart R. P., Filbin M. T. (2013) Secretory leukocyte protease inhibitor reverses inhibition by CNS myelin, promotes regeneration in the optic nerve, and suppresses expression of the transforming growth factor-β signaling protein Smad2. J. Neurosci. 33, 5138–5151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Deng K., He H., Qiu J., Lorber B., Bryson J. B., Filbin M. T. (2009) Increased synthesis of spermidine as a result of upregulation of arginase I promotes axonal regeneration in culture and in vivo. J. Neurosci. 29, 9545–9552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Noh K. M., Kim Y. H., Koh J. Y. (1999) Mediation by membrane protein kinase C of zinc-induced oxidative neuronal injury in mouse cortical cultures. J. Neurochem. 72, 1609–1616 [DOI] [PubMed] [Google Scholar]
- 35. Domeniconi M., Zampieri N., Spencer T., Hilaire M., Mellado W., Chao M. V., Filbin M. T. (2005) MAG induces regulated intramembrane proteolysis of the p75 neurotrophin receptor to inhibit neurite outgrowth. Neuron 46, 849–855 [DOI] [PubMed] [Google Scholar]
- 36. Davis S. R., Cousins R. J. (2000) Metallothionein expression in animals: a physiological perspective on function. J. Nutr. 130, 1085–1088 [DOI] [PubMed] [Google Scholar]
- 37. Udom A. O., Brady F. O. (1980) Reactivation in vitro of zinc-requiring apo-enzymes by rat liver zinc-thionein. Biochem. J. 187, 329–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chung R. S., Penkowa M., Dittmann J., King C. E., Bartlett C., Asmussen J. W., Hidalgo J., Carrasco J., Leung Y. K., Walker A. K., Fung S. J., Dunlop S. A., Fitzgerald M., Beazley L. D., Chuah M. I., et al. (2008) Redefining the role of metallothionein within the injured brain: extracellular metallothioneins play an important role in the astrocyte-neuron response to injury. J. Biol. Chem. 283, 15349–15358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miyazaki I., Asanuma M., Kikkawa Y., Takeshima M., Murakami S., Miyoshi K., Sogawa N., Kita T. (2011) Astrocyte-derived metallothionein protects dopaminergic neurons from dopamine quinone toxicity. Glia 59, 435–451 [DOI] [PubMed] [Google Scholar]
- 40. Esch F., Lin K. I., Hills A., Zaman K., Baraban J. M., Chatterjee S., Rubin L., Ash D. E., Ratan R. R. (1998) Purification of a multipotent antideath activity from bovine liver and its identification as arginase: nitric oxide-independent inhibition of neuronal apoptosis. J. Neurosci. 18, 4083–4095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gilad G. M., Gilad V. H. (1988) Early polyamine treatment enhances survival of sympathetic neurons after postnatal axonal injury or immunosympathectomy. Brain Res. 466, 175–181 [DOI] [PubMed] [Google Scholar]
- 42. Carmel J. B., Galante A., Soteropoulos P., Tolias P., Recce M., Young W., Hart R. P. (2001) Gene expression profiling of acute spinal cord injury reveals inflammatory signals and neuron loss. Physiol. Genomics 7, 201–213 [DOI] [PubMed] [Google Scholar]
- 43. Vallee B. L., Coleman J. E., Auld D. S. (1991) Zinc fingers, zinc clusters, and zinc twists in DNA-binding protein domains. Proc. Natl. Acad. Sci. U.S.A. 88, 999–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zeng J., Vallee B. L., Kägi J. H. (1991) Zinc transfer from transcription factor IIIA fingers to thionein clusters. Proc. Natl. Acad. Sci. U.S.A. 88, 9984–9988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nishimura N., Nishimura H., Tohyama C. (1989) Localization of metallothionein in female reproductive organs of rat and guinea pig. J. Histochem. Cytochem. 37, 1601–1607 [DOI] [PubMed] [Google Scholar]
- 46. Suzuki K., Nakajima K., Otaki N., Kimura M. (1994) Metallothionein in developing human brain. Biol. Signals 3, 188–192 [DOI] [PubMed] [Google Scholar]
- 47. Kelly E. J., Quaife C. J., Froelick G. J., Palmiter R. D. (1996) Metallothionein I and II protect against zinc deficiency and zinc toxicity in mice. J. Nutr. 126, 1782–1790 [DOI] [PubMed] [Google Scholar]
- 48. Dergham P., Ellezam B., Essagian C., Avedissian H., Lubell W. D., McKerracher L. (2002) Rho signaling pathway targeted to promote spinal cord repair. J. Neurosci. 22, 6570–6577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Niederöst B., Oertle T., Fritsche J., McKinney R. A., Bandtlow C. E. (2002) Nogo-A and myelin-associated glycoprotein mediate neurite growth inhibition by antagonistic regulation of RhoA and Rac1. J. Neurosci. 22, 10368–10376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Monnier P. P., Sierra A., Schwab J. M., Henke-Fahle S., Mueller B. K. (2003) The Rho/ROCK pathway mediates neurite growth-inhibitory activity associated with the chondroitin sulfate proteoglycans of the CNS glial scar. Mol. Cell. Neurosci. 22, 319–330 [DOI] [PubMed] [Google Scholar]
- 51. Fehlings M. G., Theodore N., Harrop J., Maurais G., Kuntz C., Shaffrey C. I., Kwon B. K., Chapman J., Yee A., Tighe A., McKerracher L. (2011) A phase I/IIa clinical trial of a recombinant Rho protein antagonist in acute spinal cord injury. J. Neurotrauma 28, 787–796 [DOI] [PubMed] [Google Scholar]