

Background: Cyclic di-AMP inactivates the potassium transport activity of KtrA.
Results: Cyclic di-AMP binding to KrtA induced conformational changes.
Conclusion: Cyclic di-AMP selectively binds to the KtrA RCK_C domain and signals the inactivation of potassium transport.
Significance: The molecular basis for the role of cyclic di-AMP in potassium channel activity was investigated.
Keywords: bacterial signal transduction, crystal structure, cyclic diadenosine monophosphate (c-di-AMP), isothermal titration calorimetry (ITC), potassium transport, Staphylococcus aureus (S. aureus)
Abstract
Although it was only recently identified as a second messenger, c-di-AMP was found to have fundamental importance in numerous bacterial functions such as ion transport. The potassium transporter protein, KtrA, was identified as a c-di-AMP receptor. However, the co-crystallization of c-di-AMP with the protein has not been studied. Here, we determined the crystal structure of the KtrA RCK_C domain in complex with c-di-AMP. The c-di-AMP nucleotide, which adopts a U-shaped conformation, is bound at the dimer interface of RCK_C close to helices α3 and α4. c-di-AMP interacts with KtrA RCK_C mainly by forming hydrogen bonds and hydrophobic interactions. c-di-AMP binding induces the contraction of the dimer, bringing the two monomers of KtrA RCK_C into close proximity. The KtrA RCK_C was able to interact with only c-di-AMP, but not with c-di-GMP, 3′,3-cGAMP, ATP, and ADP. The structure of the KtrA RCK_C domain and c-di-AMP complex would expand our understanding about the mechanism of inactivation in Ktr transporters governed by c-di-AMP.
Introduction
Although research on the bacterial second messenger c-di-GMP is being actively carried out, little is known about bis-(3′,5′)-cyclic dimeric adenosine monophosphate (c-di-AMP),4 which is an equally essential, ubiquitous bacterial second messenger. c-di-AMP was first discovered during a structural study on Thermotoga maritima DNA integrity scanning protein (DisA), which is a bacterial checkpoint protein that checks for DNA damage during sporulation (1). c-di-AMP is synthesized from two molecules of ATP by diadenylyl cyclases (DACs) and degraded to the 5′-phosphoadenylyl-(3′-5′)-adenosine (pApA) by phosphodiesterases (PDEs). The DACs commonly contain a DAC domain that has no amino acid or structural similarity to the GGDEF domain of c-di-GMP synthesizing enzymes. The DAC domain also seems unrelated to the recently identified dinucleotide cyclase DncV, which produces 3′-5′-linked cyclic GMP-AMP (cGAMP(3′-5′)) in Vibrio cholera (2). So far, the DAC domain is the only known c-di-AMP synthesizing enzyme (3). In Bacillus subtilis, the PDE enzyme YybT, which degrades c-di-AMP, was discovered (4). Interestingly, this protein contains a highly modified GGDEF domain that can be generally found in c-di-GMP synthesizing enzyme. However, this GGDEF domain does not function as a c-di-GMP synthesizing motif, but rather controls the activity of the PDE domain.
Similar to the c-di-GMP signaling system, it is expected that the concentration of c-di-AMP is controlled by DACs and PDEs upon internal and external cues. When a high concentration of c-di-AMP is reached, it binds to receptor or target proteins. This interaction leads to a functional change of downstream effector proteins and enables control of specific cellular pathways. Although it was only recently identified as a second messenger, c-di-AMP was found to have central importance in numerous bacterial functions including stress responses, antibiotic resistance, cellular morphology, bacterial growth, and virulence (5–12). Also, c-di-AMP secretion by Listeria monocytogenes, Mycobacterium tuberculosis, and Chlamydia trachomatis during infection induces an IFN-β-mediated host immune response (13–15). Despite its significance, many details of the c-di-AMP signaling network still remain to be elucidated.
Since the first c-di-AMP receptor protein, the transcription factor DarR was identified in Mycobacterium smegmatis (7), four additional Staphylococcus aureus proteins including the potassium transporter KtrA were identified as c-di-AMP receptors using an affinity pulldown assay (16). KtrA is a member of the RCK (Regulator of conductance of K+) domain family of proteins, which are known to regulate the gating of ion channels (17, 18). Using the differential radial capillary action of ligand assay (DRaCALA), the interaction between KtrA and c-di-AMP was confirmed and the RCK_C domain was identified as the binding motif for c-di-AMP (16). Corrigan et al. (16) investigated the involvement of KtrA and c-di-AMP in the growth of S. aureus under low potassium conditions. It was shown that the ktrA mutant had growth defects under osmotic stress and that c-di-AMP binding to KtrA disabled its potassium transport activity.
The structure of the KtrAB potassium transporter was previously determined by x-ray crystallography (19). The KtrAB complex consists of the homodimeric KtrB membrane protein and a cytoplasmic octameric KtrA ring. The KtrA octameric ring has a 4-fold symmetry, with the RCK_N lobe creating the ring and the RCK_C lobe located at the periphery. An ATP molecule is bound to the ligand-binding site in the RCK_N lobe. Interestingly, the structure of KtrA differs depending on the bound nucleotide. The KtrA·ATP complex structure adopts a 4-fold symmetric square conformation, whereas the KtrA·ADP complex adopts a 2-fold symmetric diamond conformation. The superposition of the two structures revealed that the conformational change from the ADP-bound ring to ATP-bound ring requires an asymmetric contraction of the ring. It was suggested that this conformational change would facilitate opening of the cytoplasmic pore and ion transport activity.
Although the structure of KtrAB provides the molecular basis for potassium transport activity, the molecular details of the c-di-AMP-bound state are unknown. In this study, the structure of the KtrA RCK_C domain in complex with c-di-AMP was determined by x-ray crystallography and the binding affinity was determined by isothermal titration calorimetry (ITC). The holo-KtrA RCK_C domain structure reveals that c-di-AMP binds to the dimeric interface of KtrA RCK_C domain. c-di-AMP interacts with KtrA RCK_C mainly by forming hydrogen bonds and through a hydrophobic patch. Based on the binding specificity and induced conformational changes upon binding, it is expected that c-di-AMP binding inactivates the potassium transport activity of KtrA.
Experimental Procedures
Reagent and Protein Sample Preparation
c-di-AMP was purchased from BIOLOG (Bremen, Germany). The concentration of c-di-AMP was determined by UV spectroscopy using the estimate of the extinction coefficient at 259 nm (27,000 m−1 cm−1).
The coding sequences of S. aureus KtrA, KtrA RCK_N domain, and KtrA RCK_C domain were amplified by PCR using the pGEM-B1 KtrA plasmid as the template and cloned into the 2M-T vector (Addgene) using the LIC cloning method. The mutants were generated by the QuikChange XL Site-directed Mutagenesis Kit (Stratagene) following the manufacturer's protocol. The proteins were expressed in the BL21(DE3) GOLD cells and purified using nickel-nitrilotriacetic acid affinity chromatography (Qiagen). The MBP (maltose-binding protein) tag was cleaved by tobacco etch virus digestion during dialysis against 20 mm Tris-HCl, pH 7.5, 100 mm NaCl, 5 mm β-mercaptoethanol. The proteins were purified further by a Q-Sepharose column (GE Healthcare) with a linear gradient of 0–1 m NaCl in the same buffer. The fractions containing protein were loaded onto the nickel-nitrilotriacetic acid column again to remove the maltose-binding protein tag. The protein fractions were pooled and purified using a Superdex 75 (GE Healthcare) gel-filtration column equilibrated with buffer containing 20 mm Tris-HCl, pH 7.5, 100 mm NaCl, 10 mm MgCl2, and 5 mm β-mercaptoethanol.
ITC Experiments
The binding affinity of c-di-AMP binding was measured with a VP-ITC microcalorimeter (GE Healthcare) in buffer containing 20 mm Tris-HCl, pH 7.5, 100 mm NaCl, 10 mm MgCl2, and 5 mm β-mercaptoethanol at 298 K. Protein and c-di-AMP were loaded into the reaction cell and the syringe, respectively, at concentrations of 10 and 100 μm, respectively. Thirty injections were done per experiment: 1 μl for the first and 10 μl for the additional 29 injections. The stirring velocity of the syringe was fixed at 350 rpm. Data analysis was done using the Origin software.
Protein Crystallization
The initial screening of holo-KtrA yielded two crystallization conditions but the crystal was not reproduced during the improvement steps. Thus, we used the RCK_C domain to determine the holo-form crystal structure. The KtrA RCK_C protein was concentrated to ∼5 mg ml−1 in 20 mm Tris-HCl, pH 7.5, 100 mm NaCl, 10 mm MgCl2, and 5 mm β-mercaptoethanol using Amicon Ultra-10 filtration unit (Millipore). A 3-fold molar excess of c-di-AMP was added to the protein sample and used for the crystallization of the holo-KtrA RCK_C domain. The final crystal was grown in 100 mm BisTris, pH 6.0, 24.5% (w/v) PEG6000, and 250 mm lithium acetate. The crystal was flash frozen in liquid nitrogen using 25% glycerol as a cryo-protectant.
Data Collection and Structure Determination
X-ray diffraction data for the S. aureus holo-KtrA RCK_C domain dimer were collected on beamline 7A at Pohang Accelerator Laboratory in Korea, and the highest resolution was 2.7 Å. The data were indexed and integrated using the HKL-2000 (HKL Research) processing software. The space group of the holo-KtrA RCK_C domain dimer crystal was P43212. The simple molecular replacement (MR) phasing using B. subtilis KtrA RCK_C domain (PDB code 4J91) (19) was not successful. Therefore, we modified the MR probe based on alignment data from HHPred (20) and the DALI server (21). Briefly, we selected several candidates that might be used as MR probes from these servers, and aligned these candidate structures to the B. subtilis KtrA RCK_C domain structure. From the alignment result, we found that the N-terminal region of the RCK_C domain showed large structural variations. Also, we thought the loop between β3 and β4 might cause a problem in the MR calculation. Therefore, we removed the N-terminal segment of the B. subtilis KtrA RCK_C domain prior to the β1 strand, which showed large structural flexibility, and the loop between β3 and β4. The modified B. subtilis KtrA RCK_C domain structure was used as the MR probe, and the program PHASER (22) was used to perform the MR calculation. LigandFit wizard (23) and eLBOW wizard (24) were then used to fit c-di-AMP and generate restraints, respectively. To refine the structure, phenix.refine wizard (25) from the PHENIX suite (26) and COOT (27) were used (final Rcryst of 22.85% and Rfree of 27.40%), respectively. The crystallographic data and statistics are summarized in Table 1.
TABLE 1.
Summary of the crystallographic data for S. aureus holo-KtrA RCK_C domain dimer
Values for the outer shell are given in parentheses.
| Beamline | 7A, Pohang accelerator laboratory |
|---|---|
| Processing program | HKL2000 |
| Space group | P43212 |
| a, b, c (Å) | 84.976, 84.976, 85.575 |
| α, β, γ (°) | 90, 90, 90 |
| Mosaicity (°) | 0.9 |
| Resolution range (Å) | 28.35 – 2.7 (2.8 − 2.7) |
| Unique reflections | 8969 (873) |
| Completeness (%) | 99.8 (100) |
| Redundancy | 11.3 (9.7) |
| 〈I/σ(I)〉 | 20.4 (3.5) |
| Rsym | 0.145 (0.641) |
| Rfree test set size (%) | 10 |
| Refinement statistics | |
| Final Rcryst (%) | 22.85 (25.37) |
| Final Rfree (%) | 27.40 (33.30) |
| No. of non-H atoms | |
| Protein | 1330 |
| Ligand | 44 |
| Water | 20 |
| Total | 1394 |
| Root mean square deviations | |
| Bonds (Å) | 0.006 |
| Angles (°) | 0.99 |
| Average B factors (Å2) | |
| Protein | 42.40 |
| Ligand | 26.10 |
| Water | 33.50 |
| Ramachandran plot | |
| Most favored (%) | 94.1 |
| Allowed (%) | 5.9 |
Results
Overall Structure of S. aureus Holo-KtrA RCK_C Domain
The holo-KtrA RCK_C domain is composed of a c-di-AMP molecule and a RCK_C homodimer, which is consistent with the results from gel filtration chromatography (the expected elution time of the 19.5-kDa dimer is 86 min). Each RCK_C monomer contains 4 α-helices and 4 β-sheets, and the two monomers in the dimer interact extensively with each other. The c-di-AMP nucleotide is bound at the dimer interface in proximity to helices α3 and α4 (Fig. 1B). The c-di-AMP ligand adopts a U-shaped conformation (Fig. 1A). This U-shaped conformation is previously observed in the binding mode of c-di-GMP and c-di-AMP in some proteins (28–34).
FIGURE 1.

Structure of the KtrA RCK_C domain in complex with c-di-AMP. A, |2Fo − Fc| omit map of the bound c-di-AMP contoured at 1.5 σ. B, the c-di-AMP adopts a U-shaped conformation and is bound to the dimeric interface of the KtrA RCK_C domain. The monomers A and B are colored in green and cyan, respectively.
Superimposition of the S. aureus holo-KtrA RCK_C monomer and other RCK_C monomers from different bacterial species revealed that there were no significant structural differences between them except in the N-terminal region (Fig. 2). The N-terminal region of the S. aureus RCK_C domain is α-helical, whereas that of the B. subtilis RCK_C domain is a β-sheet. However, the previously solved calcium-gated potassium channel MthK (PDB code 3KXD) (35) has an α-helix structure at the N terminus of its RCK_C domain, as in the S. aureus RCK_C domain. This implies that the linkage between the RCK_N and RCK_C domains may adopt various structures, and the S. aureus RCK_C domain shows structural similarities to the MthK RCK_C domain in this segment.
FIGURE 2.
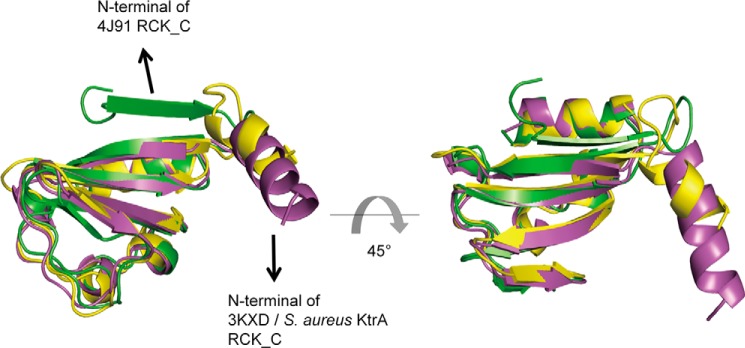
The superimposition of S. aureus KtrA RCK_C domain with other RCK_C domains. S. aureus KtrA RCK_C domain, B. subtilis KtrA RCK_C domain (PDB code 4J91), and MthK RCK_C domain (PDB code 3XKD) are shown in lavender, green, and yellow, respectively. The N termini of S. aureus KtrA RCK_C domain and MthK RCK_C domain adopt an α-helix structure, whereas the N terminus of B. subtilis KtrA RCK_C domain has a β-sheet structure.
Analysis of the c-di-AMP Binding Site and Dimeric Interface of Holo-KtrA RCK_C Domain
The c-di-AMP ligand binds to the KtrA RCK_C domain by hydrogen bonding and hydrophobic interactions (Fig. 3). The side chain of Asp-167 forms a hydrogen bond with the hydroxyl of the sugar ring of c-di-AMP. Another hydrogen bond is formed by the interaction between the side chain of Arg-169 and the oxygen atom of the phosphate group of c-di-AMP. Asp-167 and Arg-169 of each monomer interact with c-di-AMP from opposite sides. One adenine of c-di-AMP interacts with the oxygen atom of Asn-175 of monomer B. In addition, there are several hydrophobic interactions between c-di-AMP and the RCK_C domain. When the residues of KtrA RCK_C were colored by their hydrophobicity (Fig. 4), the c-di-AMP binding pocket was lined with hydrophobic residues Ile-163, Ile-164, Ile-176, and Ile-177, which form a hydrophobic patch.
FIGURE 3.
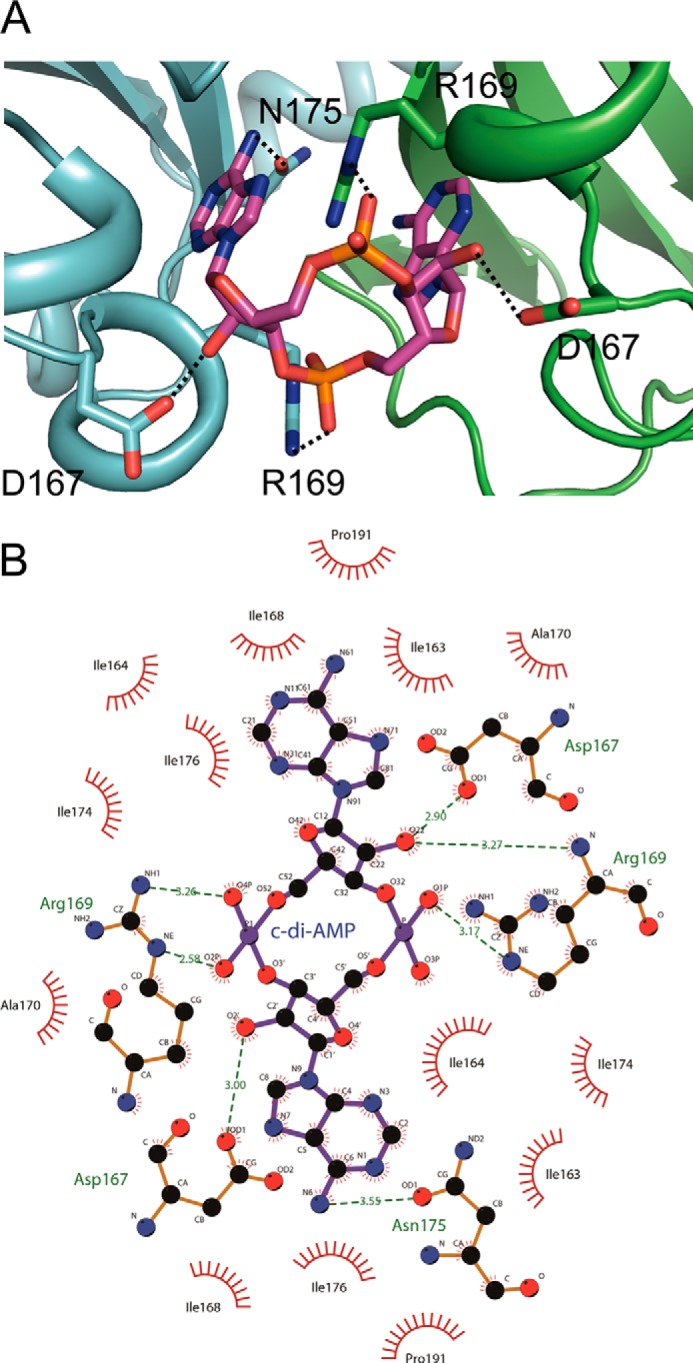
Recognition of c-di-AMP by KtrA RCK_C domain. A, the binding site residues that form direct hydrogen bonds are shown. The hydrogen bonds are indicated by dashed lines. B, the hydrogen bond formation and hydrophobic interaction between the KtrA RCK_C domain and c-di-AMP are illustrated schematically using LIGPLOT+ (47). Hydrogen bonds are shown as green dotted lines, whereas the spoked arcs indicate the residues that make non-bonded contacts with c-di-AMP.
FIGURE 4.
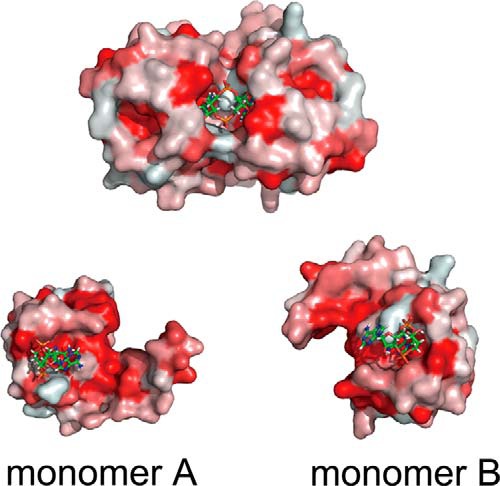
The hydrophobicity of KtrA RCK_C. The hydrophobicity of KtrA RCK_C is color-coded. The most hydrophobic residues are colored in red, whereas the least hydrophobic residues are in white. The c-di-AMP binding pocket is lined with isoleucine residues forming the hydrophobic patch.
The contact area of the S. aureus holo-KtrA RCK_C domain spans 1131.3 Å2, with a calculated solvation free energy gain upon formation of the dimer of −16.5 kcal/mol as determined by PISA (36). The α1 helix, β1 and β2 strands, and the loop between β3 and β4 are predominantly involved in the intermolecular interaction. The residues involved in the dimeric interface mostly interact with each other by forming hydrogen bonds and salt bridges. It is notable that there are some residues that are involved in forming multiple hydrogen bonds. For example, Asn-175 of monomer B makes hydrogen bonds with Ile-176, Ile-177, Asn-175, and Ser-188 of monomer A. Because Asn-175 forms a hydrogen bond at both the c-di-AMP binding site and the dimeric interface, this residue is expected to play an important role in c-di-AMP binding site formation. Ser-188 of monomer B also contributes to dimer formation by making hydrogen bonds with Asn-175 and Ile-204 of monomer A. Also, there is a salt bridge between Lys-180 of monomer A and Asp-139 of monomer B.
Structural Change upon c-di-AMP Binding
To investigate the conformational change induced by c-di-AMP binding, we solved the crystal structure of the apo-KtrA RCK_C dimer and compared the structure with holo-KtrA RCK_C and other RCK domain-containing proteins. However, unlike S. aureus holo-KtrA RCK_C dimer and other K+ transporter proteins, the S. aureus apo-KtrA RCK_C dimer structure shows largely different dimer interface. Furthermore, the N-terminal helix direction of apo-KtrA RCK_C is significantly different from other RCK domain-containing proteins (data not shown). Thus, instead of apo-KtrA RCK_C, we used the B. subtilis KtrA RCK_C domain structure (PDB code 4J91) to indirectly predict the conformational change that occurred by binding of c-di-AMP.
Upon c-di-AMP binding, there was dramatic change in the dimeric interface of the S. aureus RCK_C domain. The distance between key residues that make important contacts with c-di-AMP is shortened when c-di-AMP binds to the RCK_C domain (Fig. 5, Table 2). Compared with the apo form, the distance between Asp-167 of the holo-KtrA RCK_C domain is shortened about 14.5 Å. The distance between the Cα of Arg-169 in the two monomers is shortened by 10.3 Å. Also, Ile-163/Ile-164 and Ile-176/Ile-177, which make up the hydrophobic patch, are also moved closer to each other upon binding of c-di-AMP. Based on these data, we conclude that RCK_C monomers are brought into close proximity with each other upon binding of c-di-AMP.
FIGURE 5.

The Cα-Cα distance change measured upon c-di-AMP binding. The Cα-Cα distances of binding site residues (A) Asp-167, (B) Arg-169, and (C) Ile-164 are shown. The residue numbers in parentheses indicate the corresponding residue in B. subtilis KtrA. Upon c-di-AMP binding, the Cα-Cα distance between the residues dramatically decreased, indicating the tightening of the two KtrA RCK_C domain monomers.
TABLE 2.
Cα-Cα distance change of the key residues for binding of c-di-AMP
| Residuea | Distance (Cα-Cα) |
|
|---|---|---|
| Apo-KtrA RCK_C domain (PDB code 4J91) | Holo-KtrA RCK_C domain | |
| Ile-163 (Ile-167) | 28.1 | 18.2 |
| Ile-164 (Ile-168) | 30.9 | 14.6 |
| Asp-167 (Asn-171) | 29.9 | 15.4 |
| Arg-169 (Arg-173) | 23.1 | 12.8 |
| Ile-176 (Ile-180) | 15.3 | 9.9 |
| Ile-177 (Leu-181) | 9.9 | 6.9 |
a The corresponding residues of B. subtilis apo-KtrA RCK_C domain are indicated in parentheses.
Determination of c-di-AMP Binding Constants by Isothermal Titration Calorimetry
ITC experiments were carried out to determine c-di-AMP binding affinity and stoichiometry. The Kd between KtrA and c-di-AMP was 664.3 ± 164.3 nm (data not shown). The RCK_C domain showed higher binding affinity with a Kd value of 43.1 ± 16.0 nm (Fig. 6A, Table 3). To identify the residues that are crucial for c-di-AMP binding, the residues that form hydrogen bonds with c-di-AMP were mutated to Ala or to the opposite charge and subjected to ITC experiments (Fig. 6). The mutations of Arg-169 and Asn-175, which directly interact with c-di-AMP, resulted in the complete loss of c-di-AMP binding. However, the mutation of Asp-167, which also forms a direct hydrogen bond with c-di-AMP, did not affect binding and showed a comparable Kd value to that of the wild-type protein. The mutations of Ile-163/Ile-164 and Ile-176/Ile-177, which are distributed in the hydrophobic patch, showed no apparent binding of c-di-AMP. It is interesting that Arg-169 and the Ile residues are well conserved in other species (Fig. 7). This implies the importance of these residues in c-di-AMP binding across species. Thus, we concluded that the hydrogen bonds formed by Arg-169 and Asn-175 and the hydrophobic patch formed by the isoleucine residues are significant for c-di-AMP binding.
FIGURE 6.

c-di-AMP binding of KtrA proteins and its variants are measured by ITC. A, KtrA RCK_C domain wild-type, (B) D167A, (C) D167K, (D) N175A, (E) R169A, (F) R169E, (G) I163A/I164A, and (H) I176A/I177A. Original titration data and integrated heat measurements are shown in the upper and lower plots, respectively.
TABLE 3.
Kd values for c-di-AMP binding to KtrA proteins and its variants
| Protein | Kd |
|---|---|
| nm | |
| KtrA | 664.3 ± 164.3 |
| RCK_N domain | NBa |
| RCK_C domain | 43.1 ± 16.0 |
| D167A | 83.1 ± 12.2 |
| D167K | 32.2 ± 7.8 |
| R169A | NB |
| R169E | NB |
| N175A | NB |
| I163A/I164A | NB |
| I176A/I177A | NB |
a NB, no binding.
FIGURE 7.
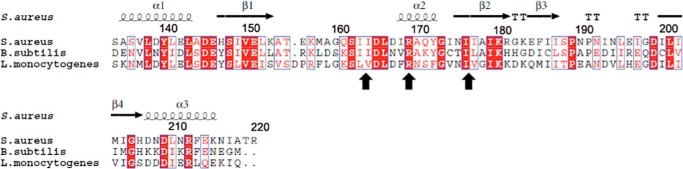
Conservation of residues in the c-di-AMP binding site. The alignment was generated by ESPript and adjusted manually (48). The conserved residues among S. aureus, L. monocytogenes, and B. subtilis RCK_C were marked with black arrows. Arg-169, which is involved in the hydrogen bonding, and the hydrophobic patches formed by isoleucine residues are conserved among other species.
Analysis of Nucleotide Binding and Specificity
We performed ITC experiments to test the potential binding of other dicyclic nucleotides and mononucleotides: c-di-GMP, 3′3′-cGAMP, ADP, and ATP (Fig. 8). c-di-GMP is another important cyclic dinucleotide second messenger that controls a broad range of cellular functions in bacteria. Some c-di-GMP receptors also interact with c-di-AMP. For example, c-di-AMP can inhibit the interaction between c-di-GMP and STING, suggesting that STING is also a receptor for c-di-AMP (37). In addition, c-di-GMP and c-di-AMP were both demonstrated to bind the DEAD box domain of DDX41 (38). We therefore tested the binding of c-di-GMP to KtrA, but the result showed that c-di-GMP did not interact with KtrA RCK_C. Next, we tested the binding of 3′,3′-cGAMP, which was reported to bind to another c-di-AMP receptor protein, DarA (39). Unlike DarA, we could not detect any binding between 3′,3′-cGAMP and the KtrA RCK_C domain. We then tested binding of the mononucleotides ADP and ATP. The binding of ADP/ATP to the RCK_N domain is known to regulate potassium transport activity by inducing a conformational change in KtrA (19), but neither ADP nor ATP bound to KtrA RCK_C domain. Thus, it can be concluded that c-di-AMP is the only nucleotide that specifically interacts with the KtrA RCK_C domain.
FIGURE 8.

Binding of other nucleotides determined by ITC. A, c-di-GMP; B, 3′,3′-cGAMP; C, ATP; and D, ADP. The results show that none of the nucleotides tested bound to the KtrA RCK_C domain and that only c-di-AMP specifically interacts with KtrA RCK_C domain.
Discussion
KtrA ion transporters are important in potassium transport systems in many bacteria. They play a crucial role in resistance to osmotic stress and high salinity by mediating the early uptake of potassium ions (40–42). The Ktr transporters consist of two components, which are a membrane protein (KtrB or KtrD) and a cytosolic regulatory protein (KtrA or KtrC). Previous studies showed that KtrA forms an octameric ring and that binding of ATP and ADP induce a conformational change in KtrA that in turn changes the conformation of KtrB (19). Such a conformational change controls the opening of the intramembrane gate, and is considered to be the basis for the regulation of potassium transport. The structural basis for the interaction of the KtrA protein with c-di-AMP, however, was previously unknown.
The structure of the S. aureus KtrA RCK_C domain in complex with c-di-AMP was determined by molecular replacement using B. subtilis KtrA as the probe. The superimposition of the two structures illustrates that the N-terminal region of S. aureus KtrA RCK_C domain differs from that of B. subtilis KtrA (Fig. 2). Rather, the N-terminal structure of S. aureus is similar to the calcium-gated potassium channel MthK RCK domain (35). This indicates that the N-terminal structure of RCK_C and the relative orientation between the domains can vary depending on the species. The holo-KtrA RCK_C domain forms a dimer and the c-di-AMP molecule, which forms a U-shaped conformation, binds at the dimeric interface (Fig. 1). This c-di-AMP conformation and binding mode is reminiscent of the c-di-GMP binding of the PilZ domain (28) and the human STING protein (29–33). The U-shaped conformation of c-di-AMP is found in the structure of the c-di-AMP binding PII-like signal transduction protein DarA. c-di-AMP is bound by DarA in a pocket located in the space between two subunits (39). It is also observed in the crystal structure of the L. monocytogenes pyruvate carboxylase·c-di-AMP complex (34). The U-shaped c-di-AMP is bound at the dimer interface of two carboxyltransferase domains in the pyruvate carboxylase structure. The KtrA and pyruvate carboxylase structures also have in common that the c-di-AMP is aligned with the 2-fold symmetry axis of the dimer.
ITC experiments were carried out to confirm the interaction between KtrA proteins and c-di-AMP. The Kd between KtrA and c-di-AMP was 664.3 ± 164.3 nm, and the RCK_C domain showed higher binding affinity with a Kd value of 43.1 ± 16.0 nm. A previous study showed that the Kd values of c-di-AMP binding for KtrA and the RCK_C domain were 64.4 ± 3.4 and 369.0 ± 44 nm, respectively (16). This discrepancy might be the result of using different types of experiments to measure the binding constant. The DraCALA and equilibrium dialysis were used to measure the Kd in the previous study. These methods take the fraction bound into account, whereas ITC measures Kd by fitting the integrated heat measurements per mole of injectant produced by direct titration. In addition, Gundlach et al. (39) suggested that a detailed analysis of the thermodynamic parameters was not straightforwardly possible because the c-di-AMP could be in a multistate oligomeric equilibrium with the monomer, dimer, tetramer, and octamer species existing at the concentration used in the experiments. However, both the previous study and this study consistently showed that the RCK_N domain does not interact with c-di-AMP. No change in integrated heat measurement was detected upon the titration of c-di-AMP into RCK_N domain (data not shown). Also, we again confirmed that the interactions between c-di-AMP and RCK_C domain or full-length KtrA are in the nanomolar range.
The key residues for c-di-AMP binding in the KtrA RCK_C domain are Arg-169 and Asn-175, which form direct hydrogen bonds with c-di-AMP (Fig. 3, 6). The sandwiching arginine in the c-di-AMP binding pocket is also seen in the c-di-GMP binding PilZ domain protein (28) and another c-di-AMP-binding protein, DarA (39). The carbonyl group of arginine forms a polar interaction with one adenine of c-di-AMP in the DarA·c-di-AMP complex. The recently reported PstA·c-di-AMP complex structure also shows that arginine residue forms a π-cation interaction with the adenine ring (43). Apart from common features of c-di-AMP binding shared with other proteins, KtrA also has a unique c-di-AMP binding mode. The isoleucine residues that form the hydrophobic patch inside the c-di-AMP binding site play a crucial role in c-di-AMP binding. Mutation of these isoleucine residues resulted in the complete loss of c-di-AMP binding, indicating these residues are indispensable for c-di-AMP binding. Because the c-di-AMP binding modes identified in this and previous studies are rather different from one another, further investigation is needed to specify the binding motif of c-di-AMP.
Although the crystal structure of the KtrA RCK_C domain and c-di-AMP in complex shows some structural similarities to the c-di-GMP-binding proteins, the KtrA RCK_C domain was unable to bind c-di-GMP in ITC experiments (Fig. 8). The mixed dinucleotide 3′,3′-cGAMP and the mononucleotides ADP and ATP were also unable to interact with the KtrA RCK_C domain. This makes c-di-AMP the only effector that can specifically interact with KtrA RCK_C among the nucleotides tested.
The B. subtilis KtrA structure showed that ADP and ATP interact with the RCK_N domain of KtrA to activate the intramembrane gate, regulating its potassium uptake (19). Other RCK domains control the activity of K+ channels and transporters, including Ca2+-gated K+ channels MthK and Maxi-K+ (BK). In these channels, the RCK domains transduce Ca2+ binding to gate transmembrane K+ flux in response to signaling events. In MthK, the RCK domains exist in a range of conformations with Ca2+ binding sites empty. Once Ca2+ ions bind to the C1 sites in the N-lobe, a partially activated conformation is stabilized. The increase in calcium concentration leads to Ca2+ binding to the C3 sites in the C-lobe, resulting in complete activation of the RCK domain and maximal stabilization of channel opening (44). In maxi-K+ channel, four RCK dimers form a gating ring structure that can expand or constrict depending on the cytosolic concentration of calcium. The membrane potential and cytosolic [Ca2+] synergistically act to induce conformational changes that results in opening of the channel (45).
Although there have been numerous studies about the activation mechanism of the RCK domain associated with K+ transporters and channels, the inactivation mechanism of the RCK domain proteins was not extensively investigated. It is notable that ATP and ADP only bind to the RCK_N domain, and not to the RCK_C domain of KtrA. Meanwhile, c-di-AMP interacts with the RCK_C domain specifically and does not interact with the RCK_N domain. This structural arrangement provides a separation of function, because c-di-AMP binding to KtrA was shown to disable potassium transport (16). Previously, Bai et al. (46) identified CabP as a c-di-AMP-binding protein and demonstrated that c-di-AMP reduced potassium uptake via interference of the interaction between CabP and KtrB ortholog, SPD0076, in S. pneumoniae. It is expected that c-di-AMP binds to the RCK_C domain of KtrA to inactivate the potassium transport in a similar manner to CabP. c-di-AMP binding to the KtrA RCK_C domain might interfere with the interaction between KtrA and KtrB, which in turn results in significantly reduced potassium uptake. As our attempts to determine the structure of holo-KtrA (including both RCK_N and RCK_C domains) have been unsuccessful so far, any discussion about the structural changes in the RCK_C domain upon c-di-AMP binding and the details of potassium transport inactivation will be fundamentally incomplete. Therefore, it would be worthwhile to determine the entire KtrAB structure complexed with c-di-AMP to provide the molecular basis for inactivation of the Ktr potassium channel by c-di-AMP.
In summary, we have determined the structure of the potassium transport protein KtrA RCK_C domain in complex with c-di-AMP to identify the molecular basis for the role of c-di-AMP in potassium channel activity. The crystal structure shows that the KtrA RCK_C domain is a homodimer and that c-di-AMP is bound at the dimeric interface of the KtrA RCK_C domain. The key binding residues are Arg-169 and Asn-175, which form direct hydrogen bonds with c-di-AMP. The hydrophobic patch formed by 4 isoleucine residues is also crucial for c-di-AMP binding. Upon c-di-AMP binding, the intermonomeric distance between these residues decreased, indicating the overall structure becomes more compact. KtrA RCK_C domain was capable of binding c-di-AMP only, but not c-di-GMP, 3′,3′-cGAMP, ATP, or ADP, as demonstrated by ITC experiments. Thus, it is expected that c-di-AMP selectively binds to the KtrA RCK_C domain and signals the inactivation of potassium transport.
Acknowledgments
We thank the staff of the beam lines 7A at the Pohang Accelerator Laboratory and 23-ID-D at the Advanced Photon Source. We also thank Dr. Melissa Stauffer for editing the manuscript.
This work was supported by the National Research Foundation of Korea Grant 2011-0020322 and High Risk High Return project by KAIST (to B-S. C.) and the Intelligent Synthetic Biology Center of Global Frontier Project funded by the Ministry of Science, ICT and Future Planning Grant 2011-0031955 (to J. L.). The authors declare that they have no conflicts of interest with the contents of this article.
- c-di-AMP
- bis-(3′-5′)-cyclic dimeric adenosine monophosphate
- DAC
- diadenylyl cyclase
- pApA
- 5′-phosphoadenylyl-(3′-5′)-adenosine
- RCK
- regulator of conductance of K+
- ITC
- isothermal titration calorimetry
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- MR
- molecular replacement
- ITC
- isothermal titration calorimetry
- PDE
- phosphodiesterase.
References
- 1. Witte G., Hartung S., Büttner K., Hopfner K. P. (2008) Structural biochemistry of a bacterial checkpoint protein reveals diadenylate cyclase activity regulated by DNA recombination intermediates. Mol. Cell 30, 167–178 [DOI] [PubMed] [Google Scholar]
- 2. Davies B. W., Bogard R. W., Young T. S., Mekalanos J. J. (2012) Coordinated regulation of accessory genetic elements produces cyclic di-nucleotides for V. cholerae virulence. Cell 149, 358–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Corrigan R. M., Gründling A. (2013) Cyclic di-AMP: another second messenger enters the fray. Nat. Rev. Microbiol. 11, 513–524 [DOI] [PubMed] [Google Scholar]
- 4. Rao F., See R. Y., Zhang D., Toh D. C., Ji Q., Liang Z. X. (2010) YybT is a signaling protein that contains a cyclic dinucleotide phosphodiesterase domain and a GGDEF domain with ATPase activity. J. Biol. Chem. 285, 473–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oppenheimer-Shaanan Y., Wexselblatt E., Katzhendler J., Yavin E., Ben-Yehuda S. (2011) c-di-AMP reports DNA integrity during sporulation in Bacillus subtilis. EMBO Rep. 12, 594–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Corrigan R. M., Abbott J. C., Burhenne H., Kaever V., Gründling A. (2011) c-di-AMP is a new second messenger in Staphylococcus aureus with a role in controlling cell size and envelope stress. PLoS Pathog. 7, e1002217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang L., Li W., He Z. G. (2013) DarR, a TetR-like transcriptional factor, is a cyclic di-AMP-responsive repressor in Mycobacterium smegmatis. J. Biol. Chem. 288, 3085–3096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mehne F. M., Gunka K., Eilers H., Herzberg C., Kaever V., Stülke J. (2013) Cyclic di-AMP homeostasis in bacillus subtilis: both lack and high level accumulation of the nucleotide are detrimental for cell growth. J. Biol. Chem. 288, 2004–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Campos S. S., Ibarra-Rodriguez J. R., Barajas-Ornelas R. C., Ramírez-Guadiana F. H., Obregón-Herrera A., Setlow P., Pedraza-Reyes M. (2014) Interaction of apurinic/apyrimidinic endonucleases Nfo and ExoA with the DNA integrity scanning protein DisA in the processing of oxidative DNA damage during Bacillus subtilis spore outgrowth. J. Bacteriol. 196, 568–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Luo Y., Helmann J. D. (2012) Analysis of the role of Bacillus subtilis σ(M) in β-lactam resistance reveals an essential role for c-di-AMP in peptidoglycan homeostasis. Mol. Microbiol. 83, 623–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pozzi C., Waters E. M., Rudkin J. K., Schaeffer C. R., Lohan A. J., Tong P., Loftus B. J., Pier G. B., Fey P. D., Massey R. C., O'Gara J. P. (2012) Methicillin resistance alters the biofilm phenotype and attenuates virulence in Staphylococcus aureus device-associated infections. PLoS Pathog. 8, e1002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith W. M., Pham T. H., Lei L., Dou J., Soomro A. H., Beatson S. A., Dykes G. A., Turner M. S. (2012) Heat resistance and salt hypersensitivity in Lactococcus lactis due to spontaneous mutation of llmg_1816 (gdpP) induced by high-temperature growth. Appl. Environ. Microbiol. 78, 7753–7759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Woodward J. J., Iavarone A. T., Portnoy D. A. (2010) c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328, 1703–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Barker J. R., Koestler B. J., Carpenter V. K., Burdette D. L., Waters C. M., Vance R. E., Valdivia R. H. (2013) STING-dependent recognition of cyclic di-AMP mediates type I interferon responses during Chlamydia trachomatis infection. mBio 4, e00018–00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang J., Bai Y., Zhang Y., Gabrielle V. D., Jin L., Bai G. (2014) Deletion of the cyclic di-AMP phosphodiesterase gene (cnpB) in Mycobacterium tuberculosis leads to reduced virulence in a mouse model of infection. Mol. Microbiol. 93, 65–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Corrigan R. M., Campeotto I., Jeganathan T., Roelofs K. G., Lee V. T., Gründling A. (2013) Systematic identification of conserved bacterial c-di-AMP receptor proteins. Proc. Natl. Acad. Sci. U.S.A. 110, 9084–9089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Albright R. A., Ibar J. L., Kim C. U., Gruner S. M., Morais-Cabral J. H. (2006) The RCK domain of the KtrAB K+ transporter: multiple conformations of an octameric ring. Cell 126, 1147–1159 [DOI] [PubMed] [Google Scholar]
- 18. Hänelt I., Tholema N., Kröning N., Vor der Brüggen M., Wunnicke D., Bakker E. P. (2011) KtrB, a member of the superfamily of K+ transporters. Eur. J. Cell Biol. 90, 696–704 [DOI] [PubMed] [Google Scholar]
- 19. Vieira-Pires R. S., Szollosi A., Morais-Cabral J. H. (2013) The structure of the KtrAB potassium transporter. Nature 496, 323–328 [DOI] [PubMed] [Google Scholar]
- 20. Söding J., Biegert A., Lupas A. N. (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Terwilliger T. C., Klei H., Adams P. D., Moriarty N. W., Cohn J. D. (2006) Automated ligand fitting by core-fragment fitting and extension into density. Acta Crystallogr. D Biol. Crystallogr. 62, 915–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moriarty N. W., Grosse-Kunstleve R. W., Adams P. D. (2009) Electronic ligand builder and optimization workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. D Biol. Crystallogr. 65, 1074–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 28. Benach J., Swaminathan S. S., Tamayo R., Handelman S. K., Folta-Stogniew E., Ramos J. E., Forouhar F., Neely H., Seetharaman J., Camilli A., Hunt J. F. (2007) The structural basis of cyclic diguanylate signal transduction by PilZ domains. EMBO J. 26, 5153–5166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang Y. H., Liu X. Y., Du X. X., Jiang Z. F., Su X. D. (2012) The structural basis for the sensing and binding of cyclic di-GMP by STING. Nat. Struct. Mol. Biol. 19, 728–730 [DOI] [PubMed] [Google Scholar]
- 30. Ouyang S., Song X., Wang Y., Ru H., Shaw N., Jiang Y., Niu F., Zhu Y., Qiu W., Parvatiyar K., Li Y., Zhang R., Cheng G., Liu Z. J. (2012) Structural analysis of the STING adaptor protein reveals a hydrophobic dimer interface and mode of cyclic di-GMP binding. Immunity 36, 1073–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shang G., Zhu D., Li N., Zhang J., Zhu C., Lu D., Liu C., Yu Q., Zhao Y., Xu S., Gu L. (2012) Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nat. Struct. Mol. Biol. 19, 725–727 [DOI] [PubMed] [Google Scholar]
- 32. Shu C., Yi G., Watts T., Kao C. C., Li P. (2012) Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat. Struct. Mol. Biol. 19, 722–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yin Q., Tian Y., Kabaleeswaran V., Jiang X., Tu D., Eck M. J., Chen Z. J., Wu H. (2012) Cyclic di-GMP sensing via the innate immune signaling protein STING. Mol. Cell 46, 735–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sureka K., Choi P. H., Precit M., Delince M., Pensinger D. A., Huynh T. N., Jurado A. R., Goo Y. A., Sadilek M., Iavarone A. T., Sauer J. D., Tong L., Woodward J. J. (2014) The cyclic dinucleotide c-di-AMP is an allosteric regulator of metabolic enzyme function. Cell 158, 1389–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dvir H., Valera E., Choe S. (2010) Structure of the MthK RCK in complex with cadmium. J. Struct. Biol. 171, 231–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 37. Burdette D. L., Monroe K. M., Sotelo-Troha K., Iwig J. S., Eckert B., Hyodo M., Hayakawa Y., Vance R. E. (2011) STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Parvatiyar K., Zhang Z., Teles R. M., Ouyang S., Jiang Y., Iyer S. S., Zaver S. A., Schenk M., Zeng S., Zhong W., Liu Z. J., Modlin R. L., Liu Y. J., Cheng G. (2012) The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat. Immunol. 13, 1155–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gundlach J., Dickmanns A., Schröder-Tittmann K., Neumann P., Kaesler J., Kampf J., Herzberg C., Hammer E., Schwede F., Kaever V., Tittmann K., Stülke J., Ficner R. (2015) Identification, characterization, and structure analysis of the cyclic di-AMP-binding PII-like signal transduction protein DarA. J. Biol. Chem. 290, 3069–3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Berry S., Esper B., Karandashova I., Teuber M., Elanskaya I., Rögner M., Hagemann M. (2003) Potassium uptake in the unicellular cyanobacterium Synechocystis sp. strain PCC 6803 mainly depends on a Ktr-like system encoded by slr1509 (ntpJ). FEBS Lett. 548, 53–58 [DOI] [PubMed] [Google Scholar]
- 41. Holtmann G., Bakker E. P., Uozumi N., Bremer E. (2003) KtrAB and KtrCD: two K+ uptake systems in Bacillus subtilis and their role in adaptation to hypertonicity. J. Bacteriol. 185, 1289–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Matsuda N., Kobayashi H., Katoh H., Ogawa T., Futatsugi L., Nakamura T., Bakker E. P., Uozumi N. (2004) Na+-dependent K+ uptake Ktr system from the cyanobacterium Synechocystis sp. PCC 6803 and its role in the early phases of cell adaptation to hyperosmotic shock. J. Biol. Chem. 279, 54952–54962 [DOI] [PubMed] [Google Scholar]
- 43. Campeotto I., Zhang Y., Mladenov M. G., Freemont P. S., Gründling A. (2015) Complex structure and biochemical characterization of the Staphylococcus aureus cyclic diadenylate monophosphate (c-di-AMP)-binding protein PstA, the founding member of a new signal transduction protein family. J. Biol. Chem. 290, 2888–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Smith F. J., Pau V. P., Cingolani G., Rothberg B. S. (2013) Structural basis of allosteric interactions among Ca2+-binding sites in a K+ channel RCK domain. Nat. Commun. 4, 2621. [DOI] [PubMed] [Google Scholar]
- 45. Horrigan F. T., Heinemann S. H., Hoshi T. (2005) Heme regulates allosteric activation of the Slo1 BK channel. J. Gen. Physiol. 126, 7–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bai Y., Yang J., Zarrella T. M., Zhang Y., Metzger D. W., Bai G. (2014) Cyclic di-AMP impairs potassium uptake mediated by a cyclic di-AMP binding protein in Streptococcus pneumoniae. J. Bacteriol. 196, 614–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Laskowski R. A., Swindells M. B. (2011) LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Infor. Model. 51, 2778–2786 [DOI] [PubMed] [Google Scholar]
- 48. Gouet P., Courcelle E., Stuart D. I., Metoz F. (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308 [DOI] [PubMed] [Google Scholar]