

Abstract
Celiac disease (CD) is a T-cell mediated immune disease in which gliadin-derived peptides activate lamina propria effector CD4+ T cells. This activation leads to the release of cytokines, compatible with a Th1-like pattern, which play a crucial role in the pathogenesis of CD, controlling many aspects of the inflammatory immune response. Recent studies have shown that a novel subset of effector T cells, characterized by expression of high levels of IL-17A, termed Th17 cells, plays a pathogenic role in CD. While these effector T cell subsets produce proinflammatory cytokines, which cause substantial tissue injury in vivo in CD, recent studies have suggested the existence of additional CD4+ T cell subsets with suppressor functions. These subsets include type 1 regulatory T cells and CD25+CD4+ regulatory T cells, expressing the master transcription factor Foxp3, which have important implications for disease progression.
Keywords: Celiac disease, Th1 cells, Th17 cells, Treg cells
Core tip: Although it is evident that effector gliadin-specific Th1 cells play an important role in celiac disease (CD) pathogenesis, recent studies have implicated Th17 effector cells in the disease process. Contrasting evidence has been reported on the ability of gliadin-specific cells to produce IL-17A. In addition, regulatory T cells, formerly known as suppressor T cells, have been identified in intestinal biopsy specimens of patients with active CD. Nevertheless, despite the presence of an endogenous counter-regulatory mechanism in the intestinal mucosa of celiac patients, the inflammatory response is not suppressed. In this review, we provide an overview of the current knowledge of the contribution of both Th1 and Th17 effector T cells and Treg cells in controlling mucosal inflammation in CD.
INTRODUCTION
Celiac disease (CD) is an immune-mediated disease triggered by the ingestion of wheat gliadin and related prolamins from other toxic cereals, such as barley and rye, and is responsible for enteropathy in genetically susceptible individuals. Much of the genetic susceptibility maps to the HLA region on chromosome 6, as approximately 95% of CD patients carry an almost identical HLA DQ2 heterodimer (DQA1*0501 DQB1*0201). In most individuals who are negative for this haplotype, this disease is associated with the class II alleles DQA1*0301 and DQB1*0302, which encode a DQ8 molecule linked to DR4 haplotypes[1]. Consistently, gliadin-specific CD4+T cells isolated from intestinal biopsies of CD patients respond to gliadin peptides presented via HLA-DQ2 or HLA-DQ8. Such gliadin-reactive T cells recognize a variety of different epitopes, which generally are much better recognized by the T cells after deamidation through the enzyme transglutaminase 2[1].
The immune response against prolamins of toxic cereals is mediated through cytokines produced via both innate and adaptive immune branches[2]. In the early phase of CD, epithelial cells are likely destroyed via toxic gliadin peptides, such as 19-mer, that might activate the innate immune system, thereby up-regulating interleukin (IL)-15 secretion[3]. Therefore, immunoadaptive peptides, such as the 33-mer, can enter the lamina propria, where the HLA class II molecules DQ2+ or DQ8+ present these peptides to T cells, which activate gluten-reactive T helper (Th)1 cells and produce high levels of proinflammatory cytokines[4].
Although T-cell receptor (TCR)-mediated T-cell activation by gliadin peptides has been well documented for CD4+ cells restricted by the HLA class II molecules DQ2 and DQ8, we have identified a peptide recognized in the context of HLA class I molecules by CD8+ T lymphocytes isolated from CD mucosa[5]. This peptide induces interferon (IFN)-γ production and the lysis of target cells through specific CD8+ T cells[6].
Recently, consistent with our data[7], other studies have reported that IL-17 is highly produced in the inflamed gut of patients with CD[8-10], confirming the involvement of a novel subset of effector T cells, termed Th17 cells, in CD pathogenesis. Th17 cells, which are highly enriched in the intestine, have been implicated in the pathogenesis of various immune-mediated disorders[11].
Concomitantly with the pro-inflammatory response, high amounts of the anti-inflammatory cytokines IL-10 and transforming growth factor-β (TGF-β) are also produced in the untreated intestinal mucosa[12-14]. This apparent paradoxical milieu of both pro-inflammatory and suppressive cytokines strongly suggests that regulatory mechanisms might operate to counterbalance the gliadin-triggered, abnormal immune activation in untreated CD[15]. Importantly, we have observed that celiac intestinal mucosa harbors two subsets of regulatory T cells (Tregs), known as type 1 regulatory T cells (Tr1) and Foxp3+ Tregs, which, through the release of both IL-10 and TGF-β, inhibit the pathogenic response to in vitro gliadin challenge[16,17].
Herein, we provide an overview of the current knowledge about the immune-mediated effects of cytokines produced by effector Th1 and Th17 cells and suppressor Treg cells in CD.
TH1 CELLS
Although the innate immune response is a prerequisite for the excessive activation of adaptive immunity, the latter is the more proximate driver of the tissue damage that manifests in CD patients. Upon activation, gliadin-specific CD4+ T cells polarize along the T helper (Th) 1-type pathway, substantiated by an ability to produce large amounts of IFN-γ, the signature cytokine of Th1 responses[18]. Indeed, mRNA for IFN-γ is more increased in untreated disease than the message for IL-2, IL-18, and TNF-α. Furthermore, the IFN-γ mRNA levels in the biopsies of treated patients have been demonstrated to reach that of untreated patients by in vitro stimulation with gliadin[18]. Therefore, IFN-γ might be much more involved in the generation of gliadin-driven mucosal damage in CD, as indicated by the efficacy of anti-IFN-γ antibodies in preventing villous atrophy[19].
The biological effects of IFN-γ primarily rely on the activity of the transcription factor signal transducer and activator of transcription (STAT) 1 and the intracellular levels of suppressor of cytokine signaling (SOCS)-1, a negative regulator that controls the amplitude and duration of STAT-1 activation[20,21]. Once activated through IFN-γ, STAT-1 dimers migrate to the nucleus and bind to the γ-activated sequence (GAS) element contained within the promoters of IFN-γ-inducible immune-inflammatory genes (e.g., MHC class II antigens, CD86, IRF-1, INOS, and ICAM-1)[20]. SOCS-1 is also induced through the IFN-γ/STAT-1 pathway and therefore participates in a negative feedback loop[21]. We have observed that STAT-1 phosphorylation is enhanced in CD mucosa, whereas no SOCS-1 protein was detected[22]. These data suggest that exaggerated IFN-γ and defective SOCS-1 protein expression result in persistent STAT1 activation in CD, thereby contributing to the expansion and maintenance of the local inflammatory response.
Although there is evidence that effector T cells in the human small intestine produce enteropathy[23], the final effector molecules involved in mucosal damage in CD remain poorly understood. Matrix metalloproteinases (MMPs) are a family of endopeptidases that play a key role in tissue remodeling under both physiological and pathological conditions. The levels of MMP expression are increased in CD[24,25], and importantly, studies have shown that IFN-γ induces the secretion of certain MMPs from fibroblasts with consequent degradation of extracellular matrix components[25]. Therefore, IFN-γ might be responsible for the production of particular MMPs, thereby contributing to the characteristic mucosal lesions of CD, i.e., villous atrophy and crypt hyperplasia.
Concerning intestinal damage mechanisms, recent studies have demonstrated the marked expression of the stress molecule MICA in enterocytes in patients with active disease and the involvement of the MICA-NKG2D interaction in enterocyte lysis through CD8+ intraepithelial lymphocytes (IELs)[26,27]. Interestingly, the activation of these killer IELs, although gliadin-triggered, is TCR independent and most likely mediated through IL-15, an inflammatory cytokine that is highly upregulated in patient-derived mucosa, produced from both enterocytes and dendritic cells[28]. Although TCR-mediated T-cell activation through gliadin peptides has only been demonstrated for CD4+ cells restricted by the HLA class II molecules DQ2 and DQ8, our recent evidence also supports the involvement of HLA class I -restricted CD8+ T cells[5,6]. We previously reported the TCR-dependent activation of intestinal CD8+ T cells through a gliadin-derived peptide (pA2) in HLA-A2+ CD patients[5]. This peptide induced IFN-γ production and the lysis of target cells through specific CD8+ T cells. Furthermore, we showed that the intestinal mucosa of HLA-A2+ CD patients harbors CD8+ T cells, activated through pA2 gliadin peptide following the in vitro challenge of intestinal mucosa. These cells recognize gliadin peptide when presented by epithelial cells, produce IFN-γ, and contribute to the lesion of the intestinal epithelial layer by inducing enterocyte apoptosis[6]. Therefore, these data show that a gliadin peptide activates mucosa infiltrating CD8+ T cells in the context of HLA class I restriction, suggesting a role for these cells in CD epithelial cell death.
In summary, these observations demonstrate that IFN-γ triggers various effector mechanisms, such as the upregulation of HLA expression facilitating T cell priming and expansion, the secretion of tissue-damaging MMPs from fibroblasts, the heightened cytotoxicity of IELs against enterocytes with increased enterocyte apoptosis and villous flattening.
The mediators of the polarization of naïve T-cells into Th1 cells in CD are T-bet[29], IFN-α[30] and IL-21[31], as shown by the high expression of these molecules in the biopsies of CD patients compared with controls. T-bet is a member of the T-box family of transcription factors that not only direct Th1 lineage commitment[32] but are also essential for IFN-γ production in CD4+ T cells[33]. In 2002, Monteleone and colleagues revealed that T-bet was upregulated in untreated CD mucosa[34]. Subsequently, the same authors observed that IFN-γ induced T-bet through the STAT-1 signaling pathway in vitro[29], suggesting that in CD mucosa, optimal IFN-γ/STAT-1 signaling is necessary to expand and stabilize the committed Th1 cell phenotype through the enhanced expression of T-bet (Figure 1).
Figure 1.
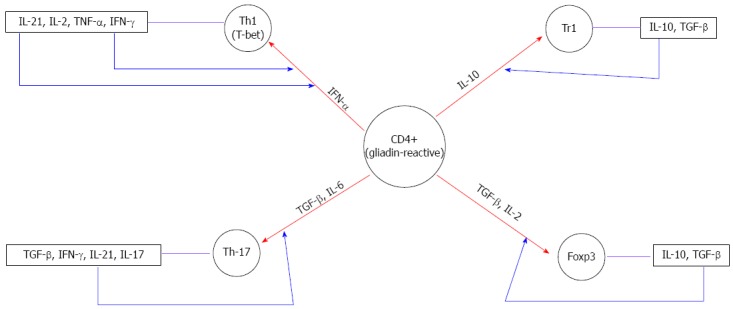
Influence of the distinct cytokine milieu in the differentiation of gliadin reactive CD4+T cells. The red arrows show the differentiation of gliadin-reactive cells in the presence of particular cytokines. The blue arrows represent the autocrine amplification loop. IL: Interleukin; TNF: Tumor necrosis factor; TGF: Tumor growth factor; IFN: Interferon.
Some studies have identified two other cytokines, IFN-α and IL-21, as possible candidates to drive and/or amplify the Th1 response in CD. IFN-α, a Type I IFN, exerts potent antiviral and immuneregulating activity, promoting the differentiation and maintenance of Th1 cells[35]. In a previous study, consistent with others reports[34,36], we showed the enhanced production of IFN-α and IL-18 in CD mucosa[30]. Both IFN-α and IL-18, produced from dendritic cells, can enhance Th1 polarization and promote IFN-γ synthesis in human beings[37]. Notably, blocking IFN-α, but not IL-18, inhibits IFN-γ transcripts induced through gliadin in cultured celiac biopsy specimens[30]. These findings suggest a role for IFN-α in inducing and maintaining the local Th1 inflammatory response in celiac disease (Figure 1). Nevertheless, the molecular mechanisms underlying the effects of gluten on the upregulation of IFN-α remain elusive.
IL-21 is a member of the IL-2 family of cytokines that is involved in both the differentiation of Th17 cells and the maintenance of ongoing Th1 responses[38]. Recently, it has been shown that IL-21 expression is increased in the intestinal mucosa of patients with active CD[31]. Importantly, the stimulation of treated CD biopsies with gliadin enhanced IL-21 expression in CD4+ T cells, and the neutralization of IL-21 activity decreased the expression of IFN-γ and T-bet. Because IFN-γ enhances T-bet, these findings suggest that IL-21 enhances IFN-γ production by lamina propria CD4+ T cells through an autocrine amplification loop, thus facilitating the amplification and stabilization of the committed Th1 cell phenotype in CD (Figure 1).
Together, these studies indicate that IFN-α, produced from innate cells, and IL-21, produced from CD4+ T cells, might act in concert to promote the proinflammatory TH1 response in CD.
TH17 CELLS
The heterogeneity of CD4 cells was expanded by the recent description of a novel subpopulation characterized by the production of a distinct set of proinflammatory cytokines, including IL-17A, IL-17F, IL-21 and IL-22. These cells are generally referred to as Th17[39]. Th17 cells are involved in several autoimmune or inflammatory diseases[40]. In 2008, Castellanos-Rubio[8] implicated the Th17 immune response in CD pathogenesis, evidenced by the increased expression of several Th17-related cytokines in patients with active CD. Subsequently, additional studies have confirmed that IL-17A is highly produced in the inflamed gut of patients with CD[7,9,10,41-43]. In particular, Monteleone and colleagues showed that CD4+ and CD4+ CD8+ T cells are major sources of this cytokine[9]. Moreover, these authors showed that IL-17A-producing T cells also coexpress IFN-γ, and blocking IL-21 activity using a neutralizing IL-21 Ab reduced IL-17A expression in cultures of active CD, suggesting that IL-21 is important for the development of Th17 cells, through an autocrine amplification loop (Figure 1).
Contrasting evidence of the ability of gliadin-specific cells to produce IL-17A opens this issue for further investigation. In 2010, Bood and colleagues[41] reported that IL-17, in contrast with IL-21 and IFN-γ, is not produced in gliadin-specific CD4+ T cells, suggesting that the activation of Th17 is generated through bystander effects. In contrast, gluten-specific IL-17A-producing cells have been recently observed in the duodenum of CD patients[42], thus supporting an active role for Th17 cells in the pathogenesis of CD. Furthermore, it was shown that gliadin-specific Th17 cells also produce IFN-γ and IL-21 proinflammatory cytokines, mucosa-protective IL-22, and regulatory TGF-β. This finding suggests that Th17 cells in CD, secreting both proinflammatory and anti-inflammatory cytokines, show functional plasticity. Several studies have reported that TGF-β is a critical signaling cytokine in Th17 differentiation[44,45]. However, TGF-β signaling pathways also play a significant role in the development of Treg. Th17 and Treg are antagonistically related. At high concentrations, TGF-β alone can divert lineage differentiation towards Treg development through the induction of Foxp3[46,47] (Figure 1). At low concentrations and in the presence of IL6, TGF-β induces Th17 differentiation and IL21 production in the CD mucosa[8,10,18,30,48-50] (Figure 1). Therefore, these data suggest that the importance of the intestine as the site of Th17 and Treg cells development might reflect the prevailing cytokine environment. However, in CD, TGF-β activity is impaired due to IL-15, thus arguing against the possibility that this cytokine might play a role in Th17 differentiation.
In summary, although it is clear that Th17 cells have emerged as a novel subset of a key immune cell population, a role for these cells in CD pathogenesis remains ambiguous, and further studies should be conducted to clarify the potential of Th17 cells for therapeutic intervention.
REGULATORY T CELLS
CD results from a break of tolerance in which the regulation of the mucosal immune response to dietary gliadin might be altered. Several Tregs subsets are involved in immune tolerance[51]. These subsets include natural Treg cells expressing the forkhead box P3 (Foxp3) transcription factor, which are selected in the thymus, and antigen-induced Foxp3+ cells, generated in the periphery[52]. Tr1 cells, that downregulate naive and memory T-cell responses upon the local secretion of IL-10 and TGF-β[53] and TGF-β-producing Treg cells (Th3)[54] are other important subsets, induced in the periphery, that possess regulatory properties. TGF-β and IL-10 play an essential role in the differentiation of Foxp3+ and Tr1 cells, respectively[46,55] (Figure 1).
In the untreated intestinal mucosa, concomitantly with the pro-inflammatory response, high amounts of the anti-inflammatory cytokines IL-10 and TGF-β are also produced[12-14]. This apparent paradoxical milieu of both pro-inflammatory and suppressive cytokines strongly suggests that regulatory mechanisms might operate to counterbalance the gluten-triggered, abnormal immune activation in untreated mucosa[15].
Among cytokines with regulatory properties, IL10 is a molecule with a strong immunomodulatory activity. IL10 is a potent immunoregulatory cytokine that suppresses the T cell-mediated immune response through either the inhibition of costimulatory molecule expression on antigen-presenting cells[56] or the direct induction of the long-term unresponsiveness of specific T cells[57].
Several groups, including our group, have reported higher IL10 mRNA and protein levels in the intestinal mucosa of untreated celiac patients than in controls; however, these patients also show significantly higher levels of proinflammatory cytokines, such as IFN-γ[58]. Moreover, the addition of exogenous IL10 to the organ culture of biopsies from untreated celiac patients does not control the inflammatory process[14]. Nonetheless, the addition of IL10 in the organ culture system of treated celiac mucosa challenged with gliadin downregulates the specific mucosal immune response to gliadin in terms of the reduced densities of CD25+ cells, reduced expression of CD80/CD86 costimulatory molecules and mRNA for inflammatory cytokines[14]. Furthermore, we have shown that the production of IFN-γ upon gliadin stimulation is absent at 3 weeks after the isolation of gliadin-specific T-cell lines (iTCLs) from biopsies cultured with gliadin in presence of IL10, suggesting that IL-10 treatment induces an anergic state of mucosa-derived, gliadin-reactive T cells[14]. Interestingly, when the functions of IL-10 and TGF-β (the two main Tr1 cytokines) are blocked using specific neutralizing antibodies, an increased immune activation to gliadin stimulation is observed in the vast majority of iTCLs generated, suggesting the existence of endogenous anti-inflammatory mechanisms in celiac disease mucosa to control local gliadin-induced inflammation. Subsequently, the cell cloning of gliadin-specific iTCLs revealed that the celiac intestinal mucosa harbors gliadin-reactive Tr1 cells that show a low proliferative rate to gliadin stimuli, but suppress pathogenic T cells through the release of both IL-10 and TGF-β[16]. Collectively, these ex vivo and in vitro results suggest that gliadin-specific Tr1 cells differentiate in vivo most likely as a consequence of the marked IL-10 production in inflamed celiac disease mucosa (Figure 1).
Although Tr1 cells have some properties similar to those of Treg cells, these cells do not express Foxp3[59]; this phenomenon suggests that Tr1 cells are functionally distinct and might represent another level of regulation of the inflammatory response. Foxp3 has been identified as a Treg marker, and subsequently several studies have demonstrated that the development of Foxp3+ Tregs in vivo occurs through a TGF-β-dependent mechanism[60] (Figure 1). Similarly, IL-2 plays an essential role in the differentiation of Foxp3+ Tregs[61] (Figure 1).
The exact mechanism of suppression through Foxp3+ Tregs remains uncertain. In vitro studies have shown that the suppressive function is cell contact dependent and cytokine independent[62,63]. However, additional studies involving animal models have suggested a role for IL-10 in the suppression through Foxp3+ Treg[64,65]. More recently, Huber and colleagues[66] demonstrated that Foxp3+ Tregs control Th17 and Th17/Th1 cells in an IL-10-dependent manner in vivo.
Several studies have found that the number of Foxp3+ T cells are significantly increased in the small intestinal mucosa with active CD compared with both treated CD and non-CD controls[67,68]. In general, these data suggest that Foxp3 expression is associated with the Th1-driven mucosal immune response to gliadin. Indeed, the expansion of this subset, proportional to the intensity of local inflammation, could play a role in the negative feedback loop of T-cell activation. Recently, in in vitro gliadin challenge systems, we provided evidence that Foxp3+ Treg cell can be expanded locally during gliadin-specific stimulation in the CD intestinal mucosa as a likely attempt to curtail the mucosal immune response[17]. Moreover, we showed that CD4+ CD25+ Foxp3+ T cells isolated from intestinal samples of active CD patients exert regulatory effects in vitro in terms of the inhibition of proliferation and IFN-γ secretion of CD4+ CD25- responder T (Tresp) cells[17].
These data suggest that intestinal Treg cells of untreated CD patients are not functionally deficient and could control the ongoing immune response to gliadin and consequent inflammation. Despite the increased frequency and suppressive activity in vitro, Treg cells do not control the development of inflammation in the small intestinal mucosa with active CD, suggesting a defect in the activation of regulatory mechanisms. The Cerf-bensussan group showed that IL-15 not only plays a pleiotropic role at the interface between innate and adaptive immunity in CD, but also interferes with two important immunoregulatory mechanisms. First, IL-15 was involved in the local downregulation of TGF-β signaling[69], required to maintain Tregs and sustain the suppressor functions and Foxp3 expression in these cells[70]. Second, IL-15 renders effector T cells resistant to suppression through Tregs[71,72]. In addition, we showed that IL-15 impairs the functions of Treg cells, making Tresp cells refractory to the regulatory effects of CD4+ CD25+ T cells, in terms of the proliferation and production of IFN-γ[17]. The sensitivity of these cells to IL-15 action likely reflects the enhanced expression of the IL-15 receptor[17], which is consistent with other studies[73] in CD patients. In support of this hypothesis, we recently showed that in “potential” CD (patients with positive CD serology, low local inflammation and increased number of Foxp3-expressing cells[67]), the suppressive effects of intestinal CD4+ CD25+ Foxp3+ T cells were not impaired through IL-15 and that the reduced sensitivity to IL-15 in potential CD patients likely reflects the reduced expression of IL-15 receptor[74]. Therefore, the low-grade inflammation in potential CD patients could reflect active regulatory mechanisms, preventing progression toward mucosal damage.
Based on these results and the finding that IL-15 is overexpressed in the intestinal mucosa of patients with active CD, suggesting that in target tissues, the function of Tregs might be substantially limited through these cytokines, and therapies aimed at neutralizing these cytokines might not only decrease bystander T-cell activation but also reconstitute the suppressor function of regulatory T cells. Furthermore, it would be of great importance to investigate strategies to boost the numbers and/or function of gliadin-specific Tr1 cells, giving rise to new therapeutic avenues for CD. Thus, in Crohn’s disease, the administration of antigen-specific Tregs was well tolerated with dose-related efficacy[75], suggesting Treg-based therapy as a therapeutic option to restore or induce tolerance in T-cell-mediated diseases.
CONCLUSION
In conclusion, some key aspects of the immunopathogenesis of CD are well understood. We know that peptide HLA-DQ complexes induce adaptive Th1 responses that increase the production of cytokines, particularly IFN-γ, a key cytokine in the downstream initiation of mucosal damage. However, there is accumulating evidence of a relevant role for the novel Th17 immune response in CD, thus highlighting the complexity of the mucosal cytokine network and suggesting that anti-cytokine approaches that target a single pro-inflammatory cytokine will have major limitations in terms of offering an effective therapy for CD.
Furthermore, Tregs have also been found in intestinal biopsy specimens of patients with celiac disease, but the regulatory properties of these cells are influenced through the signals received from the tissue environment. Indeed, the pro-inflammatory microenvironment identified in intestinal biopsies from CD subjects might promote the differentiation and development of Th1 and Th17 cells, thus restricting Treg activity. Therefore, therapeutic approaches to re-establish regulatory and effector cell homeostasis by boosting the number of Treg cells or Treg cell functions might be effective for the treatment of CD.
Footnotes
Conflict-of-interest: I have no competing interests to declare.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: January 29, 2015
First decision: March 10, 2015
Article in press: May 4, 2015
P- Reviewer: Shimizu Y, Wakim-Fleming J S- Editor: Qi Y L- Editor: A E- Editor: Liu XM
References
- 1.Sollid LM, Jabri B. Celiac disease and transglutaminase 2: a model for posttranslational modification of antigens and HLA association in the pathogenesis of autoimmune disorders. Curr Opin Immunol. 2011;23:732–738. doi: 10.1016/j.coi.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sollid LM, Jabri B. Triggers and drivers of autoimmunity: lessons from coeliac disease. Nat Rev Immunol. 2013;13:294–302. doi: 10.1038/nri3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maiuri L, Ciacci C, Ricciardelli I, Vacca L, Raia V, Auricchio S, Picard J, Osman M, Quaratino S, Londei M. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet. 2003;362:30–37. doi: 10.1016/S0140-6736(03)13803-2. [DOI] [PubMed] [Google Scholar]
- 4.Koning F, Schuppan D, Cerf-Bensussan N, Sollid LM. Pathomechanisms in celiac disease. Best Pract Res Clin Gastroenterol. 2005;19:373–387. doi: 10.1016/j.bpg.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 5.Gianfrani C, Troncone R, Mugione P, Cosentini E, De Pascale M, Faruolo C, Senger S, Terrazzano G, Southwood S, Auricchio S, et al. Celiac disease association with CD8+ T cell responses: identification of a novel gliadin-derived HLA-A2-restricted epitope. J Immunol. 2003;170:2719–2726. doi: 10.4049/jimmunol.170.5.2719. [DOI] [PubMed] [Google Scholar]
- 6.Mazzarella G, Stefanile R, Camarca A, Giliberti P, Cosentini E, Marano C, Iaquinto G, Giardullo N, Auricchio S, Sette A, et al. Gliadin activates HLA class I-restricted CD8+ T cells in celiac disease intestinal mucosa and induces the enterocyte apoptosis. Gastroenterology. 2008;134:1017–1027. doi: 10.1053/j.gastro.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sapone A, Lammers KM, Mazzarella G, Mikhailenko I, Cartenì M, Casolaro V, Fasano A. Differential mucosal IL-17 expression in two gliadin-induced disorders: gluten sensitivity and the autoimmune enteropathy celiac disease. Int Arch Allergy Immunol. 2010;152:75–80. doi: 10.1159/000260087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castellanos-Rubio A, Santin I, Irastorza I, Castaño L, Carlos Vitoria J, Ramon Bilbao J. TH17 (and TH1) signatures of intestinal biopsies of CD patients in response to gliadin. Autoimmunity. 2009;42:69–73. doi: 10.1080/08916930802350789. [DOI] [PubMed] [Google Scholar]
- 9.Monteleone I, Sarra M, Del Vecchio Blanco G, Paoluzi OA, Franzè E, Fina D, Fabrizi A, MacDonald TT, Pallone F, Monteleone G. Characterization of IL-17A-producing cells in celiac disease mucosa. J Immunol. 2010;184:2211–2218. doi: 10.4049/jimmunol.0901919. [DOI] [PubMed] [Google Scholar]
- 10.Lahdenperä AI, Hölttä V, Ruohtula T, Salo HM, Orivuori L, Westerholm-Ormio M, Savilahti E, Fälth-Magnusson K, Högberg L, Ludvigsson J, et al. Up-regulation of small intestinal interleukin-17 immunity in untreated coeliac disease but not in potential coeliac disease or in type 1 diabetes. Clin Exp Immunol. 2012;167:226–234. doi: 10.1111/j.1365-2249.2011.04510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stockinger B, Veldhoen M, Martin B. Th17 T cells: linking innate and adaptive immunity. Semin Immunol. 2007;19:353–361. doi: 10.1016/j.smim.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 12.Lahat N, Shapiro S, Karban A, Gerstein R, Kinarty A, Lerner A. Cytokine profile in coeliac disease. Scand J Immunol. 1999;49:441–446. doi: 10.1046/j.1365-3083.1999.00523.x. [DOI] [PubMed] [Google Scholar]
- 13.Hansson T, Ulfgren AK, Lindroos E, DannAEus A, Dahlbom I, Klareskog L. Transforming growth factor-beta (TGF-beta) and tissue transglutaminase expression in the small intestine in children with coeliac disease. Scand J Immunol. 2002;56:530–537. doi: 10.1046/j.1365-3083.2002.01157.x. [DOI] [PubMed] [Google Scholar]
- 14.Salvati VM, Mazzarella G, Gianfrani C, Levings MK, Stefanile R, De Giulio B, Iaquinto G, Giardullo N, Auricchio S, Roncarolo MG, et al. Recombinant human interleukin 10 suppresses gliadin dependent T cell activation in ex vivo cultured coeliac intestinal mucosa. Gut. 2005;54:46–53. doi: 10.1136/gut.2003.023150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forsberg G, Hernell O, Hammarström S, Hammarström ML. Concomitant increase of IL-10 and pro-inflammatory cytokines in intraepithelial lymphocyte subsets in celiac disease. Int Immunol. 2007;19:993–1001. doi: 10.1093/intimm/dxm077. [DOI] [PubMed] [Google Scholar]
- 16.Gianfrani C, Levings MK, Sartirana C, Mazzarella G, Barba G, Zanzi D, Camarca A, Iaquinto G, Giardullo N, Auricchio S, et al. Gliadin-specific type 1 regulatory T cells from the intestinal mucosa of treated celiac patients inhibit pathogenic T cells. J Immunol. 2006;177:4178–4186. doi: 10.4049/jimmunol.177.6.4178. [DOI] [PubMed] [Google Scholar]
- 17.Zanzi D, Stefanile R, Santagata S, Iaffaldano L, Iaquinto G, Giardullo N, Lania G, Vigliano I, Vera AR, Ferrara K, et al. IL-15 interferes with suppressive activity of intestinal regulatory T cells expanded in Celiac disease. Am J Gastroenterol. 2011;106:1308–1317. doi: 10.1038/ajg.2011.80. [DOI] [PubMed] [Google Scholar]
- 18.Nilsen EM, Jahnsen FL, Lundin KE, Johansen FE, Fausa O, Sollid LM, Jahnsen J, Scott H, Brandtzaeg P. Gluten induces an intestinal cytokine response strongly dominated by interferon gamma in patients with celiac disease. Gastroenterology. 1998;115:551–563. doi: 10.1016/s0016-5085(98)70134-9. [DOI] [PubMed] [Google Scholar]
- 19.Przemioslo RT, Lundin KE, Sollid LM, Nelufer J, Ciclitira PJ. Histological changes in small bowel mucosa induced by gliadin sensitive T lymphocytes can be blocked by anti-interferon gamma antibody. Gut. 1995;36:874–879. doi: 10.1136/gut.36.6.874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horvath CM, Darnell JE. The state of the STATs: recent developments in the study of signal transduction to the nucleus. Curr Opin Cell Biol. 1997;9:233–239. doi: 10.1016/s0955-0674(97)80067-1. [DOI] [PubMed] [Google Scholar]
- 21.Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, Corbin JE, Cornish AL, Darwiche R, Owczarek CM, et al. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- 22.Mazzarella G, MacDonald TT, Salvati VM, Mulligan P, Pasquale L, Stefanile R, Lionetti P, Auricchio S, Pallone F, Troncone R, et al. Constitutive activation of the signal transducer and activator of transcription pathway in celiac disease lesions. Am J Pathol. 2003;162:1845–1855. doi: 10.1016/S0002-9440(10)64319-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacDonald TT, Spencer J. Evidence that activated mucosal T cells play a role in the pathogenesis of enteropathy in human small intestine. J Exp Med. 1988;167:1341–1349. doi: 10.1084/jem.167.4.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daum S, Bauer U, Foss HD, Schuppan D, Stein H, Riecken EO, Ullrich R. Increased expression of mRNA for matrix metalloproteinases-1 and -3 and tissue inhibitor of metalloproteinases-1 in intestinal biopsy specimens from patients with coeliac disease. Gut. 1999;44:17–25. doi: 10.1136/gut.44.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ciccocioppo R, Di Sabatino A, Bauer M, Della Riccia DN, Bizzini F, Biagi F, Cifone MG, Corazza GR, Schuppan D. Matrix metalloproteinase pattern in celiac duodenal mucosa. Lab Invest. 2005;85:397–407. doi: 10.1038/labinvest.3700225. [DOI] [PubMed] [Google Scholar]
- 26.Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, Raulet DH, Lanier LL, Groh V, Spies T, et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity. 2004;21:357–366. doi: 10.1016/j.immuni.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 27.Hüe S, Mention JJ, Monteiro RC, Zhang S, Cellier C, Schmitz J, Verkarre V, Fodil N, Bahram S, Cerf-Bensussan N, et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity. 2004;21:367–377. doi: 10.1016/j.immuni.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 28.Mention JJ, Ben Ahmed M, Bègue B, Barbe U, Verkarre V, Asnafi V, Colombel JF, Cugnenc PH, Ruemmele FM, McIntyre E, et al. Interleukin 15: a key to disrupted intraepithelial lymphocyte homeostasis and lymphomagenesis in celiac disease. Gastroenterology. 2003;125:730–745. doi: 10.1016/s0016-5085(03)01047-3. [DOI] [PubMed] [Google Scholar]
- 29.Monteleone I, Monteleone G, Del Vecchio Blanco G, Vavassori P, Cucchiara S, MacDonald TT, Pallone F. Regulation of the T helper cell type 1 transcription factor T-bet in coeliac disease mucosa. Gut. 2004;53:1090–1095. doi: 10.1136/gut.2003.030551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Sabatino A, Pickard KM, Gordon JN, Salvati V, Mazzarella G, Beattie RM, Vossenkaemper A, Rovedatti L, Leakey NA, Croft NM, et al. Evidence for the role of interferon-alfa production by dendritic cells in the Th1 response in celiac disease. Gastroenterology. 2007;133:1175–1187. doi: 10.1053/j.gastro.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 31.Fina D, Sarra M, Caruso R, Del Vecchio Blanco G, Pallone F, MacDonald TT, Monteleone G. Interleukin 21 contributes to the mucosal T helper cell type 1 response in coeliac disease. Gut. 2008;57:887–892. doi: 10.1136/gut.2007.129882. [DOI] [PubMed] [Google Scholar]
- 32.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 33.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 2002;295:338–342. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 34.Salvati VM, MacDonald TT, Bajaj-Elliott M, Borrelli M, Staiano A, Auricchio S, Troncone R, Monteleone G. Interleukin 18 and associated markers of T helper cell type 1 activity in coeliac disease. Gut. 2002;50:186–190. doi: 10.1136/gut.50.2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Havenar-Daughton C, Kolumam GA, Murali-Krishna K. Cutting Edge: The direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J Immunol. 2006;176:3315–3319. doi: 10.4049/jimmunol.176.6.3315. [DOI] [PubMed] [Google Scholar]
- 36.Monteleone G, Pender SL, Wathen NC, MacDonald TT. Interferon-alpha drives T cell-mediated immunopathology in the intestine. Eur J Immunol. 2001;31:2247–2255. doi: 10.1002/1521-4141(200108)31:8<2247::aid-immu2247>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 37.O’Garra A, Arai N. The molecular basis of T helper 1 and T helper 2 cell differentiation. Trends Cell Biol. 2000;10:542–550. doi: 10.1016/s0962-8924(00)01856-0. [DOI] [PubMed] [Google Scholar]
- 38.Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 39.Korn T, Oukka M, Kuchroo V, Bettelli E. Th17 cells: effector T cells with inflammatory properties. Semin Immunol. 2007;19:362–371. doi: 10.1016/j.smim.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 41.Bodd M, Ráki M, Tollefsen S, Fallang LE, Bergseng E, Lundin KE, Sollid LM. HLA-DQ2-restricted gluten-reactive T cells produce IL-21 but not IL-17 or IL-22. Mucosal Immunol. 2010;3:594–601. doi: 10.1038/mi.2010.36. [DOI] [PubMed] [Google Scholar]
- 42.Fernández S, Molina IJ, Romero P, González R, Peña J, Sánchez F, Reynoso FR, Pérez-Navero JL, Estevez O, Ortega C, et al. Characterization of gliadin-specific Th17 cells from the mucosa of celiac disease patients. Am J Gastroenterol. 2011;106:528–538. doi: 10.1038/ajg.2010.465. [DOI] [PubMed] [Google Scholar]
- 43.Lahdenperä AI, Fälth-Magnusson K, Högberg L, Ludvigsson J, Vaarala O. Expression pattern of T-helper 17 cell signaling pathway and mucosal inflammation in celiac disease. Scand J Gastroenterol. 2014;49:145–156. doi: 10.3109/00365521.2013.863966. [DOI] [PubMed] [Google Scholar]
- 44.Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupé P, Barillot E, Soumelis V. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650–657. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 46.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Przemioslo RT, Kontakou M, Nobili V, Ciclitira PJ. Raised pro-inflammatory cytokines interleukin 6 and tumour necrosis factor alpha in coeliac disease mucosa detected by immunohistochemistry. Gut. 1994;35:1398–1403. doi: 10.1136/gut.35.10.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kontakou M, Przemioslo RT, Sturgess RP, Limb AG, Ciclitira PJ. Expression of tumour necrosis factor-alpha, interleukin-6, and interleukin-2 mRNA in the jejunum of patients with coeliac disease. Scand J Gastroenterol. 1995;30:456–463. doi: 10.3109/00365529509093307. [DOI] [PubMed] [Google Scholar]
- 50.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 51.Lan RY, Mackay IR, Gershwin ME. Regulatory T cells in the prevention of mucosal inflammatory diseases: patrolling the border. J Autoimmun. 2007;29:272–280. doi: 10.1016/j.jaut.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ito T, Hanabuchi S, Wang YH, Park WR, Arima K, Bover L, Qin FX, Gilliet M, Liu YJ. Two functional subsets of FOXP3+ regulatory T cells in human thymus and periphery. Immunity. 2008;28:870–880. doi: 10.1016/j.immuni.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roncarolo MG, Bacchetta R, Bordignon C, Narula S, Levings MK. Type 1 T regulatory cells. Immunol Rev. 2001;182:68–79. doi: 10.1034/j.1600-065x.2001.1820105.x. [DOI] [PubMed] [Google Scholar]
- 54.Miller A, Lider O, Roberts AB, Sporn MB, Weiner HL. Suppressor T cells generated by oral tolerization to myelin basic protein suppress both in vitro and in vivo immune responses by the release of transforming growth factor beta after antigen-specific triggering. Proc Natl Acad Sci USA. 1992;89:421–425. doi: 10.1073/pnas.89.1.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gregori S, Tomasoni D, Pacciani V, Scirpoli M, Battaglia M, Magnani CF, Hauben E, Roncarolo MG. Differentiation of type 1 T regulatory cells (Tr1) by tolerogenic DC-10 requires the IL-10-dependent ILT4/HLA-G pathway. Blood. 2010;116:935–944. doi: 10.1182/blood-2009-07-234872. [DOI] [PubMed] [Google Scholar]
- 56.de Waal Malefyt R, Haanen J, Spits H, Roncarolo MG, te Velde A, Figdor C, Johnson K, Kastelein R, Yssel H, de Vries JE. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J Exp Med. 1991;174:915–924. doi: 10.1084/jem.174.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Groux H, Bigler M, de Vries JE, Roncarolo MG. Interleukin-10 induces a long-term antigen-specific anergic state in human CD4+ T cells. J Exp Med. 1996;184:19–29. doi: 10.1084/jem.184.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Salvati VM, Troncone R, Mazzarella G. High levels of IFN-g mRNA in untreated celaics and in treated mucosa in vitro cultured with gliadin. Ital J Gastroenterol Hepatol. 1999;31:559. [Google Scholar]
- 59.Vieira PL, Christensen JR, Minaee S, O’Neill EJ, Barrat FJ, Boonstra A, Barthlott T, Stockinger B, Wraith DC, O’Garra A. IL-10-secreting regulatory T cells do not express Foxp3 but have comparable regulatory function to naturally occurring CD4+CD25+ regulatory T cells. J Immunol. 2004;172:5986–5993. doi: 10.4049/jimmunol.172.10.5986. [DOI] [PubMed] [Google Scholar]
- 60.Liang S, Alard P, Zhao Y, Parnell S, Clark SL, Kosiewicz MM. Conversion of CD4+ CD25- cells into CD4+ CD25+ regulatory T cells in vivo requires B7 costimulation, but not the thymus. J Exp Med. 2005;201:127–137. doi: 10.1084/jem.20041201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Davidson TS, DiPaolo RJ, Andersson J, Shevach EM. Cutting Edge: IL-2 is essential for TGF-beta-mediated induction of Foxp3+ T regulatory cells. J Immunol. 2007;178:4022–4026. doi: 10.4049/jimmunol.178.7.4022. [DOI] [PubMed] [Google Scholar]
- 62.Earle KE, Tang Q, Zhou X, Liu W, Zhu S, Bonyhadi ML, Bluestone JA. In vitro expanded human CD4+CD25+ regulatory T cells suppress effector T cell proliferation. Clin Immunol. 2005;115:3–9. doi: 10.1016/j.clim.2005.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, Shimizu J, Takahashi T, Nomura T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 64.Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med. 1999;190:995–1004. doi: 10.1084/jem.190.7.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Annacker O, Pimenta-Araujo R, Burlen-Defranoux O, Barbosa TC, Cumano A, Bandeira A. CD25+ CD4+ T cells regulate the expansion of peripheral CD4 T cells through the production of IL-10. J Immunol. 2001;166:3008–3018. doi: 10.4049/jimmunol.166.5.3008. [DOI] [PubMed] [Google Scholar]
- 66.Huber S, Gagliani N, Esplugues E, O’Connor W, Huber FJ, Chaudhry A, Kamanaka M, Kobayashi Y, Booth CJ, Rudensky AY, et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3 and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity. 2011;34:554–565. doi: 10.1016/j.immuni.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tiittanen M, Westerholm-Ormio M, Verkasalo M, Savilahti E, Vaarala O. Infiltration of forkhead box P3-expressing cells in small intestinal mucosa in coeliac disease but not in type 1 diabetes. Clin Exp Immunol. 2008;152:498–507. doi: 10.1111/j.1365-2249.2008.03662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vorobjova T, Uibo O, Heilman K, Rägo T, Honkanen J, Vaarala O, Tillmann V, Ojakivi I, Uibo R. Increased FOXP3 expression in small-bowel mucosa of children with coeliac disease and type I diabetes mellitus. Scand J Gastroenterol. 2009;44:422–430. doi: 10.1080/00365520802624177. [DOI] [PubMed] [Google Scholar]
- 69.Benahmed M, Meresse B, Arnulf B, Barbe U, Mention JJ, Verkarre V, Allez M, Cellier C, Hermine O, Cerf-Bensussan N. Inhibition of TGF-beta signaling by IL-15: a new role for IL-15 in the loss of immune homeostasis in celiac disease. Gastroenterology. 2007;132:994–1008. doi: 10.1053/j.gastro.2006.12.025. [DOI] [PubMed] [Google Scholar]
- 70.Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:1061–1067. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ben Ahmed M, Belhadj Hmida N, Moes N, Buyse S, Abdeladhim M, Louzir H, Cerf-Bensussan N. IL-15 renders conventional lymphocytes resistant to suppressive functions of regulatory T cells through activation of the phosphatidylinositol 3-kinase pathway. J Immunol. 2009;182:6763–6770. doi: 10.4049/jimmunol.0801792. [DOI] [PubMed] [Google Scholar]
- 72.Hmida NB, Ben Ahmed M, Moussa A, Rejeb MB, Said Y, Kourda N, Meresse B, Abdeladhim M, Louzir H, Cerf-Bensussan N. Impaired control of effector T cells by regulatory T cells: a clue to loss of oral tolerance and autoimmunity in celiac disease? Am J Gastroenterol. 2012;107:604–611. doi: 10.1038/ajg.2011.397. [DOI] [PubMed] [Google Scholar]
- 73.Bernardo D, Garrote JA, Allegretti Y, León A, Gómez E, Bermejo-Martin JF, Calvo C, Riestra S, Fernández-Salazar L, Blanco-Quirós A, et al. Higher constitutive IL15R alpha expression and lower IL-15 response threshold in coeliac disease patients. Clin Exp Immunol. 2008;154:64–73. doi: 10.1111/j.1365-2249.2008.03743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Borrelli M, Salvati VM, Maglio M, Zanzi D, Ferrara K, Santagata S, Ponticelli D, Aitoro R, Mazzarella G, Lania G, et al. Immunoregulatory pathways are active in the small intestinal mucosa of patients with potential celiac disease. Am J Gastroenterol. 2013;108:1775–1784. doi: 10.1038/ajg.2013.303. [DOI] [PubMed] [Google Scholar]
- 75.Desreumaux P, Foussat A, Allez M, Beaugerie L, Hébuterne X, Bouhnik Y, Nachury M, Brun V, Bastian H, Belmonte N, et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease. Gastroenterology. 2012;143:1207–1217.e1-e2. doi: 10.1053/j.gastro.2012.07.116. [DOI] [PubMed] [Google Scholar]