

Abstract
AIM: To examine whether lysophosphatidic acid (LPA) induces phosphorylation of c-Met and epidermal growth factor receptor (EGFR), both of which have been proposed as prognostic markers of colorectal cancer, and whether LPA induces cyclooxygenase-2 (COX-2) expression in human colon cancer cells.
METHODS: Using a human colon cancer cell line, LoVo cells, we performed immunoprecipitation analysis, followed by Western blot analysis. We also examined whether LPA induced COX-2 expression, by Western blot analysis.
RESULTS: Immunoprecipitation analysis revealed that 10 µmol/L LPA induced tyrosine phosphorylation of c-Met and EGFR in LoVo cells within a few minutes. We found that c-Met tyrosine phosphorylation induced by LPA was not attenuated by pertussis toxin or a matrix metalloproteinase inhibitor, in marked contrast to the results for EGFR. In addition, 0.2-40 µmol/L LPA induced COX-2 expression in a dose-dependent manner.
CONCLUSION: Our results suggest that LPA acts upstream of various receptor tyrosine kinases (RTKs) and COX-2, and thus may act as a potent stimulator of colorectal cancer.
Keywords: Lysophosphatidic acid, c-Met, EGFR, Transactivation, Cyclooxygenase-2, Colon cancer
INTRODUCTION
Although the incidence of colorectal cancer was substantially lower in Asia than that in the USA in the mid-twentieth century, the incidence in Japan and China has been rapidly increasing[1,2]. Thus, colorectal cancer is now a major cause of cancer death worldwide. Surgery is the main modality of treatment for colorectal cancer, other modalities like chemotherapy being adjuvant in nature. Whereas fluorouracil has been reported to be the only effective systemic therapeutic agent for colorectal cancer until recently, considerable improvement has been achieved in the last decade with the development of newer chemotherapeutic agents, such as irinotecan and oxaliplatin. In addition to these conventional cytotoxic agents, growing interest and significant advances have been made in the use of targeted therapy for colorectal cancer during the last few years. Bevacizumab, a mAb against vascular endothelial growth factor (VEGF), and cetuximab, a chimeric mAb directed against epidermal growth factor receptor (EGFR), were shown to improve the survival of patients with metastatic colorectal cancer in large randomized clinical trials[3-5]. Now that molecular target therapy for colorectal cancer has started in clinical settings, new and more effective molecular targets are eagerly being investigated.
From this standpoint, we have focused on lysophosphatidic acid (LPA, 1- or 2-acyl-sn-glycero- 3-phosphate). LPA, the simplest glycerophospholipid, has been shown to interact with at least three G protein-coupled receptors (GPCRs), such as LPA1/Edg-2, LPA2/Edg-4, and LPA3/Edg-7[6,7]. Through these receptors, LPA mediates a broad range of cellular responses, including smooth muscle cell contraction, platelet aggregation, neurite retraction/cell rounding, regulation of cell proliferation, protection from apoptosis, modulation of chemotaxis and transcellular migration[8-10]. Some of these cellular responses implicate LPA as a mediator of tumor progression[11]. We have previously reported several experimental studies using cell lines and surgically resected specimens. Using various human colon cancer cells, we revealed that LPA stimulated migration, adhesion, proliferation, and secretion of angiogenic factors in vitro, although the responses induced showed some differences among cells, which seemed to be based on their expression pattern of LPA receptors[12]. We also revealed aberrant expression of LPA receptors in human colorectal cancer tissues; i.e., overexpression of LPA2 and underexpression of LPA1 in colorectal cancer compared with normal mucosa[13]. Thus, LPA and its receptors play crucial roles in the development and progression of colorectal cancer.
As for colorectal cancer, it is well known that the use of nonsteroidal anti-inflammatory drugs (NSAIDs) has been linked to a 40-50% reduction in relative risk for cancer development[14]. The molecular basis for this striking chemopreventive effect has been attributed to inhibition of cyclooxygenases (COX)[14]. Consistent with this, approximately 80% of colorectal cancers show increased levels of COX-2[14,15]. COX-2 is responsible for the synthesis of prostaglandin E 2 (PGE2), a bioactive lipid molecule with diverse biological activities, including growth- promoting actions on the gastrointestinal mucosa. Although the precise mechanism of these trophic actions of PGs had remained unclear for a long time, it was recently demonstrated that PGE2 could phosphorylate EGFR and trigger the extracellular signal- regulated kinase 2 (ERK2)- mitogenic signaling pathway, which reveals a previously unknown mechanism by which PGE2 mediates a trophic action resulting in the growth of colonic polyps and cancers[16].
Thus, tyrosine phosphorylation of various receptor tyrosine kinases (RTKs) in response to activation of many GPCRs, which was designated as “transactivation”, has been shown to have important physiological consequences and has received considerable attention in recent years. In addition to EGFR, PGE2 was also shown to transactivate c-Met, the receptor for hepatocyte growth factor (HGF) resulting in increased invasiveness of colon cancer cells[16,17]. In colorectal cancer, c-Met expression level has been shown to be a predictor of tumor invasion and lymph node metastasis, suggesting that c-Met signals contribute to colorectal cancer progression[18,19].
LPA has been reported to transactivate EGFR in several cell types, and, in some epithelial cells, LPA has also been reported to induce COX-2 gene expression[13,20-24]. However, to the authors’ knowledge, LPA signaling pathways in colon cancer cells have not been fully understood as yet. In the present study, using colon cancer LoVo cells which are known to express abundant c-Met at both mRNA and protein levels[25] , we investigated whether LPA induces phosphorylation of c-Met as well as EGFR, and whether LPA induces COX-2 expression.
MATERIALS AND METHODS
Materials
A selective inhibitor of COX-2, NS-398, was purchased from Cayman Chemicals (Ann Arbor, MI, USA), and 1-oleoyl-LPA was purchased from Sigma Chemical Co. (St. Louis, MO). Recombinant human HGF was purchased from R&D Systems, Inc. (Minneapolis, MN, USA) and recombinant human EGF was purchased from PeproTechEC Ltd. (London, UK). Rabbit polyclonal anti-human c-Met antibody (C-28), rabbit polyclonal anti-human EGFR antibody, and mouse monoclonal anti- human phosphotyrosine antibody (PY20) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Mouse monoclonal anti-human COX-2 antibody was purchased from Exalpha Biologicals, Inc. (Boston, MA, USA). Pertussis toxin (PTX) and the broad-spectrum matrix metalloproteinase (MMP) inhibitor GM6001 were purchased from Calbiochem (La Jolla, CA, USA).
Cell culture
The human colon cancer cell line, LoVo cells, were obtained from American Type Culture Collection (Manassas, VA, USA) and maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 100 mL/L fetal calf serum (Sigma, St. Louis, MO, USA), 100 U/mL penicillin, and 100 µg/mL streptomycin (Gibco BRL Co., Grand Island, NY, USA).
Immunoprecipitation and Western blot analysis of RTK transactivation
Immunoprecipitation and Western blot analysis were performed as described previously[13,26,27]. In brief, LoVo cells were starved in serum-free medium for 24 h and then LPA was added to the culture. After incubation of starved LoVo cells with 10 µmol/L LPA for various times, cellular protein lysates were obtained and then proteins were incubated with antibodies against two RTKs, c-Met and EGFR. Immunoprecipitates were collected with protein A-agarose. Immunoprecipitated proteins were electrophoresed on sodium dodecyl sulfate (SDS)- 75 g/L polyacrylamide gel for 35 min at 200 V. Then, the protein was transferred onto an Immobilon transfer membrane (Millipore Co., Bedford, MA, USA) for sequential incubation with 50 g/L reconstituted non-fat milk powder to block nonspecific sites, dilutions of rabbit polyclonal anti-phosphotyrosine antibody, and then horseradish peroxidase-labeled donkey anti-rabbit IgG, prior to the development with a standard ECL kit (Amersham, Inc., Buckinghamshire, UK). Some cells were pretreated with 100 ng/mL PTX for 24 h before stimulation, and other cells were pretreated with 5-50 µmol/L GM6001 for 30 min before stimulation. All membranes were stripped and immunoblotted with antibodies against RTKs as a control.
Western blot analysis of cyclooxygenase-2
LoVo cells were starved in serum- free medium overnight and then various concentrations of LPA were added to the culture. After incubation of starved LoVo cells with LPA for 24 h, cellular protein lysates were obtained. Then, cell proteins (15 µg per lane) were electrophoresed on SDS-150 g/L polyacrylamide gel for 45 min at 200 V. Then, the protein was transferred onto an Immobilon transfer membrane (Millipore Co.) for sequential incubation with 50 g/L reconstituted non-fat milk powder to block nonspecific sites, dilutions of mouse monoclonal anti-human COX-2 antibody, and then horseradish peroxidase- labeled donkey anti-rabbit IgG, prior to the development with a standard ECL kit (Amersham, Inc.). The same membrane was stripped and immunoblotted with antibodies against β-actin as a control.
Proliferation assay
LoVo cells (5×103 cells in 100 µL solution per well), which were pretreated with DMSO or 100 µmol/L NS- 398, were seeded in a 96 -well plate in DMEM containing 1 g/L fatty acid-free BSA with various concentrations of LPA, which was added every 24 h. After incubation at 37°C in 50 mL/L CO2 for 120 h, the number of living cells was measured using an MTS assay (Promega Co., Madison, WI, USA) according to the manufacturer’s instructions. Briefly, MTS solution was added to each well and the cells were further incubated for 3 h. The number of living cells was determined by measuring optical density at 490 nm.
RESULTS
LPA-induced tyrosine phosphorylation of c-Met in colon cancer cells
In order to investigate whether tyrosine phosphorylation of c-Met was induced by LPA, LoVo cells were incubated with 10 µmol/L LPA for 2-60 min. As shown in Figure 1, LPA induced a significant level of tyrosine phosphorylation of c -Met in LoVo cells. Time course experiments revealed that phosphorylation was maximal after 2 min of stimulation and declined thereafter, suggesting that LPA-induced rapid and transient tyrosine phosphorylation of c- Met. After stripping, c-Met in LoVo cells was detected by anti-human c-Met antibody at 190 ku. The amount of c-Met immunoprecipitation was not different with or without LPA (Figure 1). In reverse experiments, phosphotyrosine immunoprecipitation and anti- c-Met immunoblotting revealed the same results (Figure 1). Tyrosine phosphorylation of c-Met induced by 10 µmol/L LPA was as strong as that induced by 10 ng/mL HGF (Figure 2).
Figure 1.
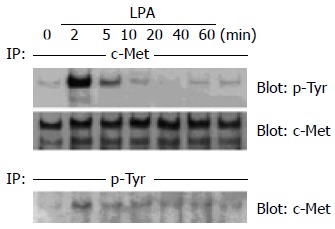
Rapid tyrosine phosphorylation of c-Met in response to LPA in human colon cancer cells. (A) Human colon cancer LoVo cells were serum -starved for 24 h and then incubated with 10 µmol/L LPA for 2-60 min. After cell lysis, c -Met was immunoprecipitated (IP) using polyclonal anti-c -Met antibody, and immunoprecipitates were immunoblotted with monoclonal anti-phosphotyrosine antibody. Then the membrane was stripped and immunoblotted with anti-c-Met as a control. (B) Human colon cancer LoVo cells were serum-starved for 24 h and then incubated with 10 µmol/L LPA for 2-60 min. These cell lysates were IP with anti-phosphotyrosine, and the immunoprecipitates were immunoblotted with anti-c-Met.
Figure 2.

Effect of PTX, a Gi inhibitor, on LPA-stimulated c-Met transactivation in colon cancer cells. Human colon cancer LoVo cells were serum-starved for 24 h with or without 100 ng/mL PTX and then stimulated with 10 µmol/L LPA or 10 ng/mL HGF for 2 min. Cell lysates were IP with anti-c- Met. Immunoprecipitates were immunoblotted with anti-phosphotyrosine. Then the membrane was stripped and immunoblotted with anti-c -Met as a control. The same experiments were performed using anti-EGFR as a control for the inhibitory effect of PTX.
Tyrosine phosphorylation of c-Met induced by LPA was not inhibited by pertussis toxin
As shown in Figure 2, pretreatment with PTX partly decreased the LPA- induced phosphorylation of EGFR in LoVo cells. This is compatible with previous reports that LPA-induced EGFR transactivation in Cos-7 cells, and head and neck squamous cell carcinoma cells (HNSCC) was PTX sensitive, suggesting the involvement of Gi protein in this signaling pathway[28,29]. To address the question of whether a Gi -dependent mechanism is also involved in LPA-induced c-Met transactivation in colon cancer cells, we examined the effect of PTX in our system. In contrast to EGFR transactivation, PTX did not significantly inhibit tyrosine phosphorylation of c-Met in response to LPA in any experimental condition (24-48 h pretreatment with 100-200 ng/mL PTX) (Figure 2). Thus, LPA-induced c-Met transactivation in colon cancer cells seems to be Gi-independent.
Tyrosine phosphorylation of c-Met induced by LPA was not dependent on MMP activation
Previous reports have shown that GPCR-induced EGFR transactivation requires MMP activation and cleavage of the membrane-anchored growth factor precursor pro-HB-EGF in COS-7 cells, HEK-293 cells and HNSCC[20,30]. As shown in Figure 3, pretreatment with an MMP inhibitor, GM6001, decreased the phosphorylation of EGFR by LPA in LoVo cells in a dose-dependent manner, which is compatible with previous reports. In contrast to EGFR transactivation, GM6001 did not significantly inhibit LPA-induced tyrosine phosphorylation of c-Met at any concentration (Figure 3). Thus, LPA-induced c-Met transactivation in colon cancer cells does not require MMP activation.
Figure 3.
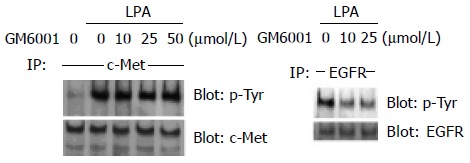
Effect of GM6001, an MMP inhibitor, on LPA-stimulated c-Met transactivation in colon cancer cells. Human colon cancer LoVo cells were serum-starved for 24 h and pre-incubated with the indicated concentrations of GM6001 for 30 min. After incubation with 10 µmol/L LPA for 2 min, cell lysates were obtained and IP with anti-c-Met. Immunoprecipitates were immunoblotted with anti-phosphotyrosine. Then the membrane was stripped and immunoblotted with anti-c -Met as a control. The same experiments were performed using anti-EGFR as a control for the inhibitory effect of GM6001.
LPA induced COX-2 expression in colon cancer cells
LPA has been shown to induce COX-2 expression in some epithelial cells[13,20,21]. However, as for cancer cells, to our knowledge, there is no report of LPA-induced COX-2 expression. Hence, we examined whether LPA induces COX-2 expression in LoVo cells. When LoVo cells were stimulated with various concentrations of LPA for 24 h, the 72-ku COX-2 protein was increased in a concentration-dependent manner (Figure 4). This effect was maximal at an LPA concentration of 40 µmol/L.
Figure 4.

Effect of LPA on COX-2 expression in colon cancer cells. Human colon cancer LoVo cells were serum-starved for 24 h, and then treated with 0.02-40 µmol/L LPA for 24 h. After cell lysis, the expression level of COX-2 protein was examined by Western blotting using anti-COX-2 antibody. Then the membrane was stripped and immunoblotted with anti-β-actin as a control.
COX-2 inhibitor reduced LPA-induced proliferation in colon cancer cells
LPA has been shown to be a potent mitogen for various cell types. In order to investigate the role of LPA-induced COX-2 expression, we performed a proliferation assay using NS398, a selective COX-2 inhibitor, with the measurement by MTS assay. LPA itself induced a dose-dependent increase in proliferation, showing significant growth stimulation at 0.2 µmol/L and a stronger effect at 20 µmol/L (5.2±0.6 -fold increase compared with control) (Figure 5). NS398 alone did not show a significant effect in control conditions without LPA; however, NS398 markedly inhibited LPA-induced proliferation at 20 µmol/L LPA (51±6% inhibition) (Figure 5). Thus, NS398 significantly inhibited LPA-induced LoVo cell proliferation.
Figure 5.
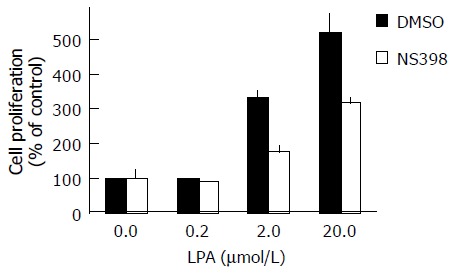
Inhibition of LPA-induced proliferation of LoVo cells by NS398, a COX-2 inhibitor. Human colon cancer LoVo cells, after being pretreated with DMSO or NS398, were seeded with the indicated concentrations of LPA. After incubation for 120 h, the cell proliferation was measured using MTS assay. Data are expressed as a percentage of control, where control indicates cell proliferation in the absence of LPA without NS398 pretreatment (100%).
DISCUSSION
In this study, using LoVo cells, we revealed LPA induced transactivation of c-Met as well as EGFR. Since both c-Met and EGFR serve as prognostic markers in human colorectal cancer, LPA may be a potent stimulator of colorectal cancer progression that acts upstream of these RTK signaling pathways (Figure 5)[18,31]. We also revealed that LPA induced COX-2 expression. Interestingly, PGE2, which is synthesized by COX-2, is known to transactivate both c-Met and EGFR[16,17]. Taking these results together, LPA may act upstream of these RTK signaling pathways in a dual manner; one is a direct effect and the other an indirect effect accomplished via COX-2 upregulation (Figure 6).
Figure 6.
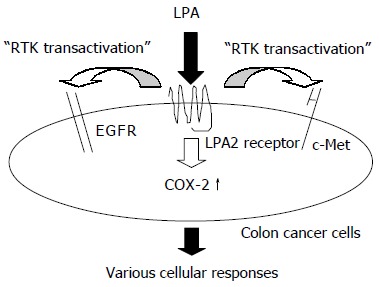
Mechanism of the effects of LPA on human colon cancer cells.
EGFR is well-known to not only transduce EGF signals, but also integrate diverse stimuli, such as LPA, bombesin, thrombin, and S1P[28,30,32-34]. This transactivation of EGFR represents a paradigm for cross-talk between GPCRs and RTK signaling pathways. From this knowledge, a novel picture of EGFR as a central cellular network element has been established in recent years, which broadens both its physiological as well as pathological significance[35-37]. In addition to EGFR transactivation, Pai et al[17] recently reported c-Met transactivation which was induced by PGE2 in colon cancer cells. They also showed that, whereas HGF induces autophosphorylation of tyrosine residues in c-Met and can enhance invasion into the extracellular matrix, ligand-independent activation of the c-Met signaling pathway could cause similar changes and increase colon cancer cell invasion[17]. Thus, c-Met signals, which were activated by various stimuli, also seem to play an important role in colorectal cancer progression. In this study, we revealed that tyrosine phosphorylation of c-Met induced by LPA was rapid and transient, which is exactly the same pattern as that of EGFR. Thus, LPA utilizes EGFR and c-Met as a downstream signaling partner in colon cancer cells.
Previous studies indicated that LPA-stimulated transactivation of EGFR in COS-7 cells and HNSCC was mediated partly via Gi protein and required MMP activation[28-30]. In our study of colon cancer cells, we also confirmed that LPA-induced transactivation of EGFR was mediated partly via Gi protein and required MMP activation. In contrast, LPA- induced transactivation of c-Met did not show significant inhibition by either PTX or GM6001. These data indicated that c-Met transactivation by LPA does not require Gi proteins and MMP activation, in marked contrast to EGFR transactivation induced by LPA. Since Gq is known to be coupled with LPA receptors and plays a role in EGFR transactivation, this Gq or other G proteins could be involved in the transactivation of c-Met. The contribution of MMP activation in EGFR transactivation has been shown to induce proteolytic cleavage of transmembrane precursors, such as pro-HB- EGF, which subsequently activates the EGFR family[20,30]. Our results, however, suggested that HGF or other c-Met ligands do not exist as a membrane-bound form, such as HB-EGF.
Accumulating evidence indicates that COX-2 plays key roles in intestinal tumorigenesis. In addition, COX-2 overexpression in colon cancer cells was shown to promote cell motility and stimulate the production of angiogenic factors[38]. Thus, the expression level of COX-2 may determine, at least in part, the aggressiveness of cancer. In this study, we demonstrated LPA induced COX-2 expression in a dose-dependent manner. We also revealed that a COX-2 inhibitor significantly reduced LPA -induced proliferation of LoVo cells. These results suggest that LPA uses COX-2 as a downstream signaling partner, which results in making cancer cells aggressive.
We have previously demonstrated that LoVo cells exclusively expressed LPA2 among the three LPA receptors, by both Northern blot analysis and real-time RT-PCR method[12,13]. Therefore, LPA-induced transactivation of c-Met and EGFR in LoVo cells, as well as LPA-induced COX- 2 expression, might be presumably mediated by the LPA2 receptor. We have previously demonstrated that, in colorectal cancer tissue, the LPA2 receptor is markedly overexpressed as compared with normal mucosa[12,13]. Moreover, overexpression of the LPA2 receptor was also shown in ovarian cancer, thyroid cancer, and breast cancer as compared to normal tissue[39-42]. Since, in general, the targets of newly developed drugs are mostly GPCRs, the LPA2 receptor, a member of GPCR, could be a novel molecular target for cancer therapy.
In summary, this study demonstrates that LPA, at a physiological concentration, induces transactivation of c-Met as well as EGFR, and induces COX -2 expression. Colorectal cancers are often associated with local bleeding and thus, at the tumor site, platelets are activated and secrete lysophospholipids such as lysophosphatidylcholine (LPC), lysophosphatidylethanolamine (LPE), and lysophosph-atidylserine (LPS). These lysophospholipids are subsequently converted to LPA by lysophospholipase D[43]. Therefore, a significant amount of extracellular LPA is thought to be present in colorectal cancer tissue, and is able to induce various important biological responses in cancer tissues. Our findings indicate that the development of an antagonist for LPA or the appropriate spectrum of LPA receptors may be a new and effective therapy for colon cancer.
Footnotes
Science Editor Guo SY and Kumar M Language Editor Elsevier HK
Supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan and a grant from the Ministry of Health, Labour and Welfare of Japan
References
- 1.Lu JB, Sun XB, Dai DX, Zhu SK, Chang QL, Liu SZ, Duan WJ. Epidemiology of gastroenterologic cancer in Henan Province, China. World J Gastroenterol. 2003;9:2400–2403. doi: 10.3748/wjg.v9.i11.2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yiu HY, Whittemore AS, Shibata A. Increasing colorectal cancer incidence rates in Japan. Int J Cancer. 2004;109:777–781. doi: 10.1002/ijc.20030. [DOI] [PubMed] [Google Scholar]
- 3.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 4.Saltz LB, Meropol NJ, Loehrer PJ, Needle MN, Kopit J, Mayer RJ. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22:1201–1208. doi: 10.1200/JCO.2004.10.182. [DOI] [PubMed] [Google Scholar]
- 5.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 6.An S, Bleu T, Hallmark OG, Goetzl EJ. Characterization of a novel subtype of human G protein-coupled receptor for lysophosphatidic acid. J Biol Chem. 1998;273:7906–7910. doi: 10.1074/jbc.273.14.7906. [DOI] [PubMed] [Google Scholar]
- 7.Bandoh K, Aoki J, Hosono H, Kobayashi S, Kobayashi T, Murakami-Murofushi K, Tsujimoto M, Arai H, Inoue K. Molecular cloning and characterization of a novel human G-protein-coupled receptor, EDG7, for lysophosphatidic acid. J Biol Chem. 1999;274:27776–27785. doi: 10.1074/jbc.274.39.27776. [DOI] [PubMed] [Google Scholar]
- 8.Moolenaar WH. Lysophosphatidic acid, a multifunctional phospholipid messenger. J Biol Chem. 1995;270:12949–12952. doi: 10.1074/jbc.270.22.12949. [DOI] [PubMed] [Google Scholar]
- 9.Moolenaar WH. Bioactive lysophospholipids and their G protein-coupled receptors. Exp Cell Res. 1999;253:230–238. doi: 10.1006/excr.1999.4702. [DOI] [PubMed] [Google Scholar]
- 10.Goetzl EJ, An S. Diversity of cellular receptors and functions for the lysophospholipid growth factors lysophosphatidic acid and sphingosine 1-phosphate. FASEB J. 1998;12:1589–1598. [PubMed] [Google Scholar]
- 11.Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3:582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 12.Shida D, Kitayama J, Yamaguchi H, Okaji Y, Tsuno NH, Watanabe T, Takuwa Y, Nagawa H. Lysophosphatidic acid (LPA) enhances the metastatic potential of human colon carcinoma DLD1 cells through LPA1. Cancer Res. 2003;63:1706–1711. [PubMed] [Google Scholar]
- 13.Shida D, Watanabe T, Aoki J, Hama K, Kitayama J, Sonoda H, Kishi Y, Yamaguchi H, Sasaki S, Sako A, et al. Aberrant expression of lysophosphatidic acid (LPA) receptors in human colorectal cancer. Lab Invest. 2004;84:1352–1362. doi: 10.1038/labinvest.3700146. [DOI] [PubMed] [Google Scholar]
- 14.DuBois RN, Giardiello FM, Smalley WE. Nonsteroidal anti-inflammatory drugs, eicosanoids, and colorectal cancer prevention. Gastroenterol Clin North Am. 1996;25:773–791. doi: 10.1016/s0889-8553(05)70274-0. [DOI] [PubMed] [Google Scholar]
- 15.Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S, DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994;107:1183–1188. doi: 10.1016/0016-5085(94)90246-1. [DOI] [PubMed] [Google Scholar]
- 16.Pai R, Soreghan B, Szabo IL, Pavelka M, Baatar D, Tarnawski AS. Prostaglandin E2 transactivates EGF receptor: a novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat Med. 2002;8:289–293. doi: 10.1038/nm0302-289. [DOI] [PubMed] [Google Scholar]
- 17.Pai R, Nakamura T, Moon WS, Tarnawski AS. Prostaglandins promote colon cancer cell invasion; signaling by cross-talk between two distinct growth factor receptors. FASEB J. 2003;17:1640–1647. doi: 10.1096/fj.02-1011com. [DOI] [PubMed] [Google Scholar]
- 18.Takeuchi H, Bilchik A, Saha S, Turner R, Wiese D, Tanaka M, Kuo C, Wang HJ, Hoon DS. c-MET expression level in primary colon cancer: a predictor of tumor invasion and lymph node metastases. Clin Cancer Res. 2003;9:1480–1488. [PubMed] [Google Scholar]
- 19.Di Renzo MF, Olivero M, Giacomini A, Porte H, Chastre E, Mirossay L, Nordlinger B, Bretti S, Bottardi S, Giordano S. Overexpression and amplification of the met/HGF receptor gene during the progression of colorectal cancer. Clin Cancer Res. 1995;1:147–154. [PubMed] [Google Scholar]
- 20.Gschwind A, Prenzel N, Ullrich A. Lysophosphatidic acid-induced squamous cell carcinoma cell proliferation and motility involves epidermal growth factor receptor signal transactivation. Cancer Res. 2002;62:6329–6336. [PubMed] [Google Scholar]
- 21.Kue PF, Taub JS, Harrington LB, Polakiewicz RD, Ullrich A, Daaka Y. Lysophosphatidic acid-regulated mitogenic ERK signaling in androgen-insensitive prostate cancer PC-3 cells. Int J Cancer. 2002;102:572–579. doi: 10.1002/ijc.10734. [DOI] [PubMed] [Google Scholar]
- 22.Ershov AV, Bazan NG. Induction of cyclooxygenase-2 gene expression in retinal pigment epithelium cells by photoreceptor rod outer segment phagocytosis and growth factors. J Neurosci Res. 1999;58:254–261. [PubMed] [Google Scholar]
- 23.Goppelt-Struebe M, Fickel S, Reiser CO. The platelet-derived-growth-factor receptor, not the epidermal-growth-factor receptor, is used by lysophosphatidic acid to activate p42/44 mitogen-activated protein kinase and to induce prostaglandin G/H synthase-2 in mesangial cells. Biochem J. 2000;345 Pt2:217–224. [PMC free article] [PubMed] [Google Scholar]
- 24.Hahn A, Barth H, Kress M, Mertens PR, Goppelt-Struebe M. Role of Rac and Cdc42 in lysophosphatidic acid-mediated cyclo-oxygenase-2 gene expression. Biochem J. 2002;362:33–40. doi: 10.1042/0264-6021:3620033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nabeshima K, Shimao Y, Inoue T, Itoh H, Kataoka H, Koono M. Hepatocyte growth factor/scatter factor induces not only scattering but also cohort migration of human colorectal-adenocarcinoma cells. Int J Cancer. 1998;78:750–759. doi: 10.1002/(sici)1097-0215(19981209)78:6<750::aid-ijc13>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 26.Shida D, Kitayama J, Yamaguchi H, Hama K, Aoki J, Arai H, Yamashita H, Mori K, Sako A, Konishi T, et al. Dual mode regulation of migration by lysophosphatidic acid in human gastric cancer cells. Exp Cell Res. 2004;301:168–178. doi: 10.1016/j.yexcr.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 27.Shida D, Kitayama J, Yamaguchi H, Yamashita H, Mori K, Watanabe T, Yatomi Y, Nagawa H. Sphingosine 1-phosphate transactivates c-Met as well as epidermal growth factor receptor (EGFR) in human gastric cancer cells. FEBS Lett. 2004;577:333–338. doi: 10.1016/j.febslet.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 28.Daub H, Wallasch C, Lankenau A, Herrlich A, Ullrich A. Signal characteristics of G protein-transactivated EGF receptor. EMBO J. 1997;16:7032–7044. doi: 10.1093/emboj/16.23.7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gschwind A, Hart S, Fischer OM, Ullrich A. TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. EMBO J. 2003;22:2411–2421. doi: 10.1093/emboj/cdg231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- 31.Resnick MB, Routhier J, Konkin T, Sabo E, Pricolo VE. Epidermal growth factor receptor, c-MET, beta-catenin, and p53 expression as prognostic indicators in stage II colon cancer: a tissue microarray study. Clin Cancer Res. 2004;10:3069–3075. doi: 10.1158/1078-0432.ccr-03-0462. [DOI] [PubMed] [Google Scholar]
- 32.Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 33.Endo A, Nagashima K, Kurose H, Mochizuki S, Matsuda M, Mochizuki N. Sphingosine 1-phosphate induces membrane ruffling and increases motility of human umbilical vein endothelial cells via vascular endothelial growth factor receptor and CrkII. J Biol Chem. 2002;277:23747–23754. doi: 10.1074/jbc.M111794200. [DOI] [PubMed] [Google Scholar]
- 34.Kim JH, Kim JH, Song WK, Kim JH, Chun JS. Sphingosine 1-phosphate activates Erk-1/-2 by transactivating epidermal growth factor receptor in rat-2 cells. IUBMB Life. 2000;50:119–124. doi: 10.1080/713803698. [DOI] [PubMed] [Google Scholar]
- 35.Zwick E, Hackel PO, Prenzel N, Ullrich A. The EGF receptor as central transducer of heterologous signalling systems. Trends Pharmacol Sci. 1999;20:408–412. doi: 10.1016/s0165-6147(99)01373-5. [DOI] [PubMed] [Google Scholar]
- 36.Gschwind A, Zwick E, Prenzel N, Leserer M, Ullrich A. Cell communication networks: epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene. 2001;20:1594–1600. doi: 10.1038/sj.onc.1204192. [DOI] [PubMed] [Google Scholar]
- 37.Fischer OM, Hart S, Gschwind A, Ullrich A. EGFR signal transactivation in cancer cells. Biochem Soc Trans. 2003;31:1203–1208. doi: 10.1042/bst0311203. [DOI] [PubMed] [Google Scholar]
- 38.Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998;93:705–716. doi: 10.1016/s0092-8674(00)81433-6. [DOI] [PubMed] [Google Scholar]
- 39.Fujita T, Miyamoto S, Onoyama I, Sonoda K, Mekada E, Nakano H. Expression of lysophosphatidic acid receptors and vascular endothelial growth factor mediating lysophosphatidic acid in the development of human ovarian cancer. Cancer Lett. 2003;192:161–169. doi: 10.1016/s0304-3835(02)00713-9. [DOI] [PubMed] [Google Scholar]
- 40.Fang X, Schummer M, Mao M, Yu S, Tabassam FH, Swaby R, Hasegawa Y, Tanyi JL, LaPushin R, Eder A, et al. Lysophosphatidic acid is a bioactive mediator in ovarian cancer. Biochim Biophys Acta. 2002;1582:257–264. doi: 10.1016/s1388-1981(02)00179-8. [DOI] [PubMed] [Google Scholar]
- 41.Schulte KM, Beyer A, Köhrer K, Oberhäuser S, Röher HD. Lysophosphatidic acid, a novel lipid growth factor for human thyroid cells: over-expression of the high-affinity receptor edg4 in differentiated thyroid cancer. Int J Cancer. 2001;92:249–256. doi: 10.1002/1097-0215(200102)9999:9999<::aid-ijc1166>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 42.Kitayama J, Shida D, Sako A, Ishikawa M, Hama K, Aoki J, Arai H, Nagawa H. Over-expression of lysophosphatidic acid receptor-2 in human invasive ductal carcinoma. Breast Cancer Res. 2004;6:R640–R646. doi: 10.1186/bcr935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aoki J, Taira A, Takanezawa Y, Kishi Y, Hama K, Kishimoto T, Mizuno K, Saku K, Taguchi R, Arai H. Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J Biol Chem. 2002;277:48737–48744. doi: 10.1074/jbc.M206812200. [DOI] [PubMed] [Google Scholar]