

Abstract
Aims
Accumulating evidence suggest that sarcomere signalling complexes play a pivotal role in cardiomyocyte hypertrophy by communicating stress signals to the nucleus to induce gene expression. Ankyrin repeat domain 1 (ANKRD1) is a transcriptional regulatory protein that also associates with sarcomeric titin; however, the exact role of ANKRD1 in the heart remains to be elucidated. We therefore aimed to examine the role of ANKRD1 in cardiomyocyte hypertrophic signalling.
Methods and results
In neonatal rat ventricular myocytes, we found that ANKRD1 is part of a sarcomeric signalling complex that includes ERK1/2 and cardiac transcription factor GATA4. Treatment with hypertrophic agonist phenylephrine (PE) resulted in phosphorylation of ERK1/2 and GATA4 followed by nuclear translocation of the ANKRD1/ERK/GATA4 complex. Knockdown of Ankrd1 attenuated PE-induced phosphorylation of ERK1/2 and GATA4, inhibited nuclear translocation of the ANKRD1 complex, and prevented cardiomyocyte growth. Mice lacking Ankrd1 are viable with normal cardiac function. Chronic PE infusion in wild-type mice induced significant cardiac hypertrophy with reactivation of the cardiac fetal gene program which was completely abrogated in Ankrd1 null mice. In contrast, ANKRD1 does not play a role in haemodynamic overload as Ankrd1 null mice subjected to transverse aortic constriction developed cardiac hypertrophy comparable to wild-type mice.
Conclusion
Our study reveals a novel role for ANKRD1 as a selective regulator of PE-induced signalling whereby ANKRD1 recruits and localizes GATA4 and ERK1/2 in a sarcomeric macro-molecular complex to enhance GATA4 phosphorylation with subsequent nuclear translocation of the ANKRD1 complex to induce hypertrophic gene expression.
Keywords: CARP, GATA4, Titin, Sarcomere, Hypertrophy
1. Introduction
Pathological hypertrophic cardiac growth occurs in response to various stress stimuli such as hypertension, ischaemia, and neurohormonal activation and can culminate in heart failure if left untreated.1 These stress stimuli can trigger multiple intracellular signalling pathways that target pro-growth genes to induce cardiomyocyte growth.2 Accumulating evidence suggests that sarcomeric signalling complexes play a pivotal role in inducing genes that regulate the cardiomyocyte hypertrophic response.
Ankyrin repeat domain 1 (ANKRD1, also known as cardiac ankyrin repeat protein, cardiac adriamycin responsive protein, CARP) is a member of a family of conserved muscle ankyrin repeat proteins (MARPs) that include ANKRD2 and diabetes ankyrin repeat protein (DARP, ANKRD23).3–6 ANKRD1 is highly expressed in cardiomyocytes, interacts with the N2A region in the titin spring domain, and localizes to the nucleus.5,6 This dual sub-cellular localization suggests that ANKRD1 could be a component of the titin spring complex that is capable of sensing and relaying biomechanical stress signals to regulate gene expression. A previous study has shown that ANKRD1 interacts with myopalladin and that this complex could be important in maintaining sarcomere integrity.7 ANKRD1 is up-regulated in animal models of cardiac hypertrophy induced by pressure overload and adrenergic stimulation, and in patients with dilated cardiomyopathy.8–10 Recently, several missense mutations in ANKRD1 were identified in patients with dilated and hypertrophic cardiomyopathy, and in vitro studies of some of these mutations suggest disruption of ANKRD1 localization and cardiac stretch-based signalling.11–13
GATA4, a member of the GATA family of zinc finger transcription factors, is crucial for normal cardiac development,14 and in the adult heart GATA4 is centrally involved in regulating cardiac hypertrophy.15 GATA4 has been shown to be phosphorylated by ERK leading to enhanced GATA4-DNA binding and activation of the hypertrophic gene program.16 We have previously shown that both ANKRD1 and GATA4 maintain sarcomere integrity by regulating myofilament gene transcription, and that loss of either ANKRD1 or GATA4 in vitro directly contributes to myofibrillar disarray.17
In the present study, we set out to further dissect the ANKRD1/GATA4 signalling pathway and uncovered a novel role for ANKRD1 as a selective regulator of the α-adrenergic pathway whereby ANKRD1 recruits and localizes ERK1/2 and GATA4 in a sarcomeric complex to enhance GATA4 phosphorylation, followed by translocation of the ANKRD1 complex to the nucleus to induce gene activation and cardiac hypertrophy.
2. Methods
A detailed discussion of the Methods with lists of antibodies, constructs, and primers is provided in the Supplementary material online. All experiments involving animals were approved by the Animal Care and Use Committee at Vanderbilt University Medical Center, and carried out in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
2.1. Generation of Ankrd1−/− (KO) mice
Global deletion of Ankrd1 was achieved in offspring generated by crossing of a Sox2-Cre mouse and an Ankrd1fl/fl mouse, which had been generated by recombineering of loxP sites 600 bp upstream of transcription initiation and within intron 2 of a BAC clone derived from a 192B6 library.18
2.2. Cardiac hypertrophy induction in vivo
Male Ankrd1−/ − mice (12–14 weeks, ∼20–25 g) and age- and body-weight-matched male wild-type mice were used for the study.
For chronic infusion of phenylephrine (PE), Alzet miniosmotic pumps (1002, Durect Corp) containing PE (70 mg/kg/day) or PBS vehicle were surgically implanted dorsally and subcutaneously in each mice under isoflurane anaesthesia (1.5–2%). Following 2 weeks of PE infusion, mice were anaesthetized with isoflurane (>5%) and hearts harvested for assessment of hypertrophy.
For the TAC procedure, mice were anaesthetized with sodium pentobarbital (50 mg/kg, i.p.), intubated and placed on a ventilator. The aortic branch was exposed via left thoracotomy and a 27G blunt needle was placed parallel to the transverse aorta. A 7–0 ligature was tied against the needle and aorta, and the needle removed to yield an aortic constriction of ∼0.4 mm in diameter. In sham-operated mice, the entire procedure was identical except for constriction of the aorta. One day following TAC or sham, mice were subjected to pulsed-wave Doppler echocardiography (Vevo2100, VisualSonics), to measure transverse aortic flow velocities distal to the site of constriction. The pressure gradient was estimated using the modified Bernouilli equation (Δp = 4 × velocity2). Following 4 weeks of TAC, mice were anaesthetized with isoflurane (>5%) and hearts harvested for assessment of hypertrophy.
2.3. Echocardiography
Echocardiography was performed in conscious mice using the Vevo2100 System with an 18–38 MHz linear array probe. Standard short-axis images were obtained in the B- and M-mode and image measurements were performed off-line.
2.4. Isolation of cardiac myocytes and cell culture
Neonatal Sprague–Dawley pups (1–2 days old) were sacrificed by swift decapitation and hearts harvested for enzymatic isolation of neonatal rat ventricular myocytes (NRVM) as previously described.17,19 This method of euthanasia is in accordance with the guidelines of the NIH and Vanderbilt University Medical Center.
2.5. Electrophoretic mobility shift assay
NRVM were harvested and enriched nuclear protein fractions (NE-PER, Thermo Scientific) were incubated with a biotinylated probe from the α-MHC promoter.20 The gel-shift assay was performed according to manufacturer's protocol (LightShift Chemiluminscent EMSA kit, Thermo Scientific).
2.6. Gel electrophoresis, immunoblotting, and IP
NRVM were lysed, resolved on 10% Tris–HCl polyacrylamide gels (Bio-Rad) and transferred to PVDF (Amersham Biosciences) membranes for immunoblotting as previously described.17,19 For immunoprecipitation (IP), NRVM were harvested in IP lysis buffer and nuclear and cytoplasmic extracts were prepared according to manufacturer's protocol (Thermo Scientific).
2.7. Immunofluorescence imaging and proximity ligation assay
Cardiomyocytes were fixed with 4% paraformaldehyde/PBS, permeabilized with 0.2% Triton-X/PBS, and incubated with primary antibodies as previously described.17,19 To detect and localize specific protein complexes within the cells, proximity ligation assay (PLA) was performed according to manufacturer's protocol (Duolink, Sigma).
2.8. Quantitative RT–PCR
Total RNA was isolated from left-ventricular tissue via TRIzol Reagent and DNase treated per manufacturer's instructions (Life Technologies). RNA was converted to cDNA and RT–PCR was performed using SYBR Green and specified fetal gene and 18S control primers in a CFX96 Real-Time PCR detection system (Bio-Rad).
2.9. Statistical analysis
Data are reported as mean ± SEM. Where appropriate, results were either analysed by Student's t-test or ANOVA with a Bonferroni's multiple comparison post-hoc test. P < 0.05 was considered statistically significant.
3. Results
3.1. ANKRD1 interacts with GATA4
ANKRD1 is a known cofactor for several transcription factors including YB-1, p53, and nucleolin.6,21,22 We have previously shown that both ANKRD1 and GATA4 maintain cardiomyocyte sarcomere integrity by regulating myofilament gene transcription.17 This led us to examine if ANKRD1 directly interacts with GATA4. Whole neonatal rat ventricular myocyte (NRVM) extracts were subjected to immunoprecipitation (IP) with anti-GATA4 antibody followed by western blot for ANKRD1. As shown in Supplementary material online, Figure S1A, ANKRD1 was detected in the immunoprecipitate indicating that GATA4 and ANKRD1 interact in NRVM. A faster migrating band was also detected, which could be a calpain-mediated cleaved ANKRD1 product as previously reported.23 The interaction was corroborated by reverse IP with anti-ANKRD1 followed by western blot for GATA4. IP using nuclear and cytoplasmic NRVM extracts confirmed an interaction between ANKRD1 and GATA4 in both fractions. This was further supported by PLA showing an interaction between ANKRD1 and GATA4 in both nuclear and cytosolic compartments (see Supplementary material online, Figure S1B).
3.2. ANKRD1 enhances GATA4 activity and DNA binding affinity in cardiomyocytes
To determine whether ANKRD1 is a transcriptional co-activator/repressor of GATA4, we transfected a GATA4-dependent luciferase reporter in NRVMs that were previously transfected with Ankrd1 siRNA. Phenylephrine (PE, 100 µM), a hypertrophic agonist known to stimulate GATA4 activity in cardiomyocytes, induced an expected increase in GATA4-luciferase activity (Figure 1A). Anrkd1 siRNA significantly attenuated both basal and PE-stimulated GATA4-luciferase activities, suggesting that ANKRD1 enhances GATA4 activity in cardiomyocytes.
Figure 1.

ANKRD1 regulates GATA4 DNA-binding and activity. (A) NRVM transfected with a GATA4-dependent luciferase reporter (black bars) or control (reporter with no GATA4 binding sites; white bars) and treated with 100 µM PE in the presence or absence of Ankrd1 siRNA. Values normalized to untreated non-silencing siRNA (NS) control and expressed as mean ± SEM, n = 4–6, *P < 0.05 vs. untreated NS control. (B) Electrophoretic mobility shift assay using NRVM nuclear extracts revealed a single band shift with an α-MHC probe which was specific for GATA4 as shown by co-incubation with anti-GATA4 antibody resulting in a supershift (SS), or loss of band with cold or mutant (Mut) α-MHC probe. Co-incubation with anti-ANKRD1 antibody or using nuclear extracts from NRVM treated with Ankrd1 siRNA resulted in depletion of the GATA4 band. Asterisks indicate non-specific bands. Representative image from three independent experiments.
To determine whether ANKRD1 alters GATA4-DNA binding affinity, we performed EMSA using an α-myosin heavy chain (αMHC) probe that contained two GATA4 DNA binding motifs.20 While non-specific bands were detectable in the assay, the αMHC probe produced a single slow migrating band shift (Figure 1B). This band shift was a distinct GATA4 DNA-binding protein complex as demonstrated by loss of the band shift with excess unlabelled αMHC probe or when using an αMHC probe with the GATA4 domains mutated, and by supershift in the presence of GATA4 antibody. In NRVM treated with Ankrd1 siRNA, the GATA4 band shift intensity was decreased suggesting that loss of ANKRD1 decreases GATA4-DNA binding. Co-incubation of NRVM nuclear extracts with ANKRD1 antibody resulted in depletion of the GATA4 band shift, suggesting that ANKRD1 is part of the GATA4-DNA binding complex.
3.3. ANKRD1 knockdown attenuates PE-induced cardiomyocyte hypertrophy
While the above data demonstrated that ANKRD1 enhanced GATA4 activity and DNA binding, ANKRD1 has no known kinase activity. In cardiomyocytes, PE is a potent activator of the MEK1/2-ERK1/2 kinase cascade which phosphorylates GATA4 at serine 105 to enhance transcriptional activation and DNA binding.16,24 Thus, one potential mechanism is that ANKRD1 indirectly enhances GATA4 phosphorylation via ERK1/2. To examine this, we transfected NRVMs with Ankrd1 siRNA then treated the cells with 100 µM PE for 10 min. Under basal conditions, Ankrd1 siRNA reduced the levels of phosphorylated ERK1/2 (pERK1/2) and GATA4 (pGATA4) (Figure 2A). PE resulted in a significant increase in pERK1/2 and pGATA4 levels, which were blunted in NRVM transfected with Ankrd1 siRNA. MEK1/2, the upstream activator of ERK1/2, was also significantly phosphorylated with PE treatment and this effect was again blunted in NRVMs transfected with Ankrd1 siRNA (Figure 2B). It should be noted that the relative increase in PE-induced phosphorylation of GATA4, ERK, and MEK in Ankrd1 siRNA transfected cells was comparable to the non-silencing control, suggesting that Ankrd1 knockdown does not prevent PE-induced phosphorylation of these proteins. Instead, Ankrd1 plays a role in scaffolding of these proteins thereby enhancing the phosphorylation cascade, as will be discussed later.
Figure 2.

ANKRD1 knockdown attenuates MEK1/2, ERK1/2, and GATA4 phosphorylation. (A) Immunoblots from NRVM treated with 100 µM PE showed induction of ERK1/2 and GATA4 phosphorylation, which was blunted in NRVM pre-transfected with Ankrd1 siRNA. (B) Immunoblots from NRVM treated with PE showed induction of MEK1/2 and p38 phosphorylation; Ankrd siRNA attenuated MEK1/2 but not p38 phosphorylation. Immunoblots were quantified and values expressed as mean ± SEM, n = 3–4, *P < 0.05 vs. non-silencing (NS) control, †P < 0.05 vs. PE-treated control.
Interestingly, p38 MAPK, part of a distinct MAPK signalling cascade also known to phosphorylate GATA4,25 was unaffected by Ankrd1 siRNA either under basal conditions or with PE treatment.
To examine whether ANKRD1 plays a role in cardiomyocyte hypertrophy, NRVM were treated with PE for 24 h in the presence or absence of Ankrd1 siRNA. PE treatment induced a hypertrophic phenotype in NRVM characterized by increased cell-surface area (Figure 3A) and robust accumulation of ANP (Figure 3B), which were both attenuated in NRVMs transfected with Ankrd1 siRNA.
Figure 3.
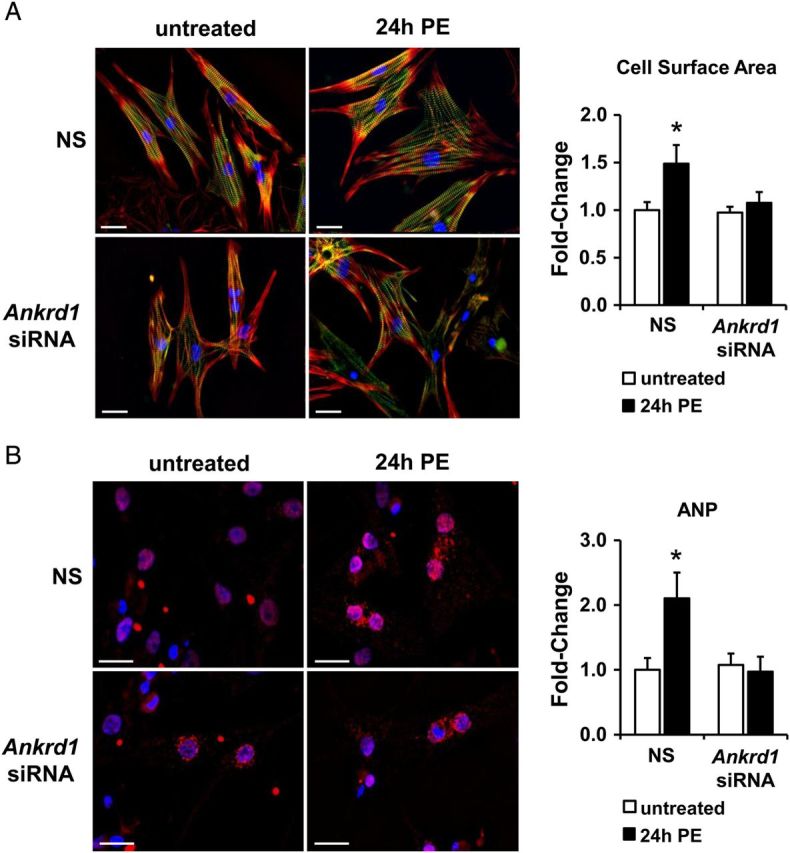
ANKRD1 knockdown attenuates PE-induced cardiomyocyte hypertrophy. (A) Representative images of NRVM showed an increase in cell area with 100 µM PE for 24 h, which was attenuated in cells pre-transfected with Ankrd1 siRNA. NRVM were immunostained for myomesin (green), DAPI (blue), and filamentous actin (red). Cell area was quantified and values expressed as mean ± SEM, n = 4–5, *P < 0.05 vs. untreated non-silencing (NS) control. Scale bar = 25 µm. (B) Representative images of cardiomyocytes treated with 100 µM PE for 24 h showed an increase in ANP levels (red), which is blunted in Ankrd1 siRNA cells. NRVM nuclei were stained with DAPI (blue) and ANP staining was quantified and values expressed as mean ± SEM, n = 4–5, *P < 0.05 vs. untreated non-silencing (NS) control. Scale bar = 20 µm.
3.4. ANKRD1 interacts with ERK and GATA4 at the sarcomere and nucleus
The detected interaction between ANKRD1 and GATA4 and the role of ANKRD1 in regulating ERK1/2 and GATA4 phosphorylation led us to hypothesize that ANKRD1 might serve as a scaffold protein for ERK1/2 and GATA4 to enhance the ability of activated ERK1/2 to phosphorylate GATA4. To first test whether ANKRD1 interacts with ERK1/2, NRVM whole extracts were subjected to IP using anti-pERK1/2 antibody followed by ANKRD1 western blot. Supplementary material online, Figure S2A showed that full-length ANKRD1 was detected in the immunoprecipitate as well as a faster migrating band (presumably the ANKRD1 cleavage product). IP using anti-ANKRD1 antibody followed by ERK1/2 western blot also showed an interaction between these two proteins, and PLA demonstrated cytoplasmic as well as nuclear protein interactions between ANKRD1 and ERK1/2 (see Supplementary material online, Figure S2B). IP using anti-ANKRD1 and anti-MEK1/2 antibodies also demonstrated an interaction between these two proteins (see Supplementary material online, Figure S2C).
Confocal immunofluorescence microscopy was used to assess the localization of ANKRD1, ERK, and GATA4 in NRVM. As previously reported,17 ANKRD1 localized in the nucleus and as a narrow doublet in the sarcomeric I-band (Figure 4). ERK1/2 and GATA4 were also nuclear and both co-localized as narrow doublets with ANKRD1. This observation together with the results from the PLA interactions suggests that ERK and GATA4 form a complex with ANKRD1 in the nucleus and in the sarcomeric I-band. To further confirm that GATA4 can be localized with sarcomeric ANKRD1, we expressed FLAG-tagged GATA4 in NRVM. Confocal imaging using a FLAG-antibody showed that GATA4 was striated and co-localized with sarcomeric ANKRD1 (see Supplementary material online, Figure S3).
Figure 4.
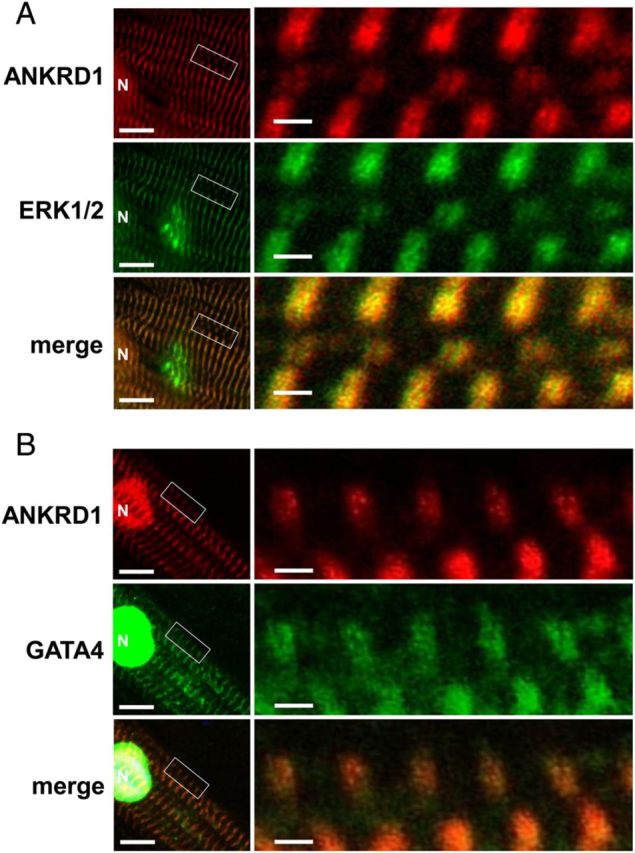
GATA4 and ERK1/2 co-localize with ANKRD1 in the sarcomere and nucleus. NRVM were double immunostained with antibodies against ANKRD1 (red) and ERK1/2 (green) (A) and ANKRD1 (red) and GATA4 (green) (B). Merged low magnification images on left panels show co-localization in the nucleus (N), scale bar = 10 µm. Boxed inset in left panels are shown as enlarged images on the right panels. Enlarged images show immunostaining as merged narrow doublets which is consistent with co-localization at the sarcomeric I-band, scale bar = 1 µm.
To determine whether ANKRD1 plays a role in anchoring ERK and/or GATA4 to the sarcomere, NRVMs were transfected with Ankrd1 siRNA followed by immunostaining for ERK and GATA4. NRVM transfection with a non-silencing control showed sarcomeric ERK1/2 (see Supplementary material online, Figure S4A) and GATA4 (see Supplementary material online, Figure S4B) immunostaining, while transfection with Ankrd1 siRNA resulted in diffuse cytoplasmic staining of both ERK1/2 and GATA4, The reverse experiments were also performed and while knockdown of Erk1/2 by siRNA seemed not to affect sarcomeric ANKRD1, Gata4 siRNA resulted in sarcomeric ANKRD1 disarray and loss of nuclear ANKRD1. This is consistent with our previous study showing that GATA4 regulates ANKDR1 gene expression and sarcomere homeostasis.17 Overall, these data suggest that ANKRD1 serves as a sarcomeric scaffolding protein for ERK1/2 and GATA4, presumably to enhance protein phosphorylation and augment hypertrophic signal transduction.
3.5. Sarcomeric ANKRD1/ERK/GATA4 complex translocates to the nucleus with PE stimulation
To elucidate the interactions between ANKRD1, ERK1/2, and GATA4 and their sub-cellular localization during hypertrophic signalling, NRVMs were treated for 3 h with 100 µM PE and processed for PLA. Untreated NRVM showed cytoplasmic and nuclear ERK1/2-GATA4, ANKRD1-ERK1/2, and ANKRD1-GATA4 PLA signals (Figure 5A). Based on the confocal images, we interpret the cytoplasmic PLA signals as interactions between ANKRD1, ERK1/2, and GATA4 at the sarcomeric I-band. PE stimulation significantly enhanced the nuclear PLA signal for all three protein interactions, suggesting nuclear translocation of an ANKRD1/ERK/GATA4 complex since total levels of these three proteins were unaltered with 3 h PE stimulation (data not shown). This nuclear translocation is dependent on MEK/ERK phosphorylation as pretreatment with MEK inhibitor, U0126, attenuated the PE-induced increase in nuclear PLA signals. ANKRD1 knockdown by siRNA attenuated the PE-induced increase in nuclear ERK1/2-GATA4 PLA signal (Figure 5B), thus confirming a role for ANKRD1 in ERK1/2-GATA4 nuclear translocation. It is interesting to note that we were still able to detect ERK-GATA PLA interactions with ANKRd1 knockdown. Based on the anchoring role of sarcomeric ANKRD1, we speculate that these ERK-GATA4 interactions are non-sarcomeric and therefore deficient in transducing a hypertrophic signal to the nucleus.
Figure 5.
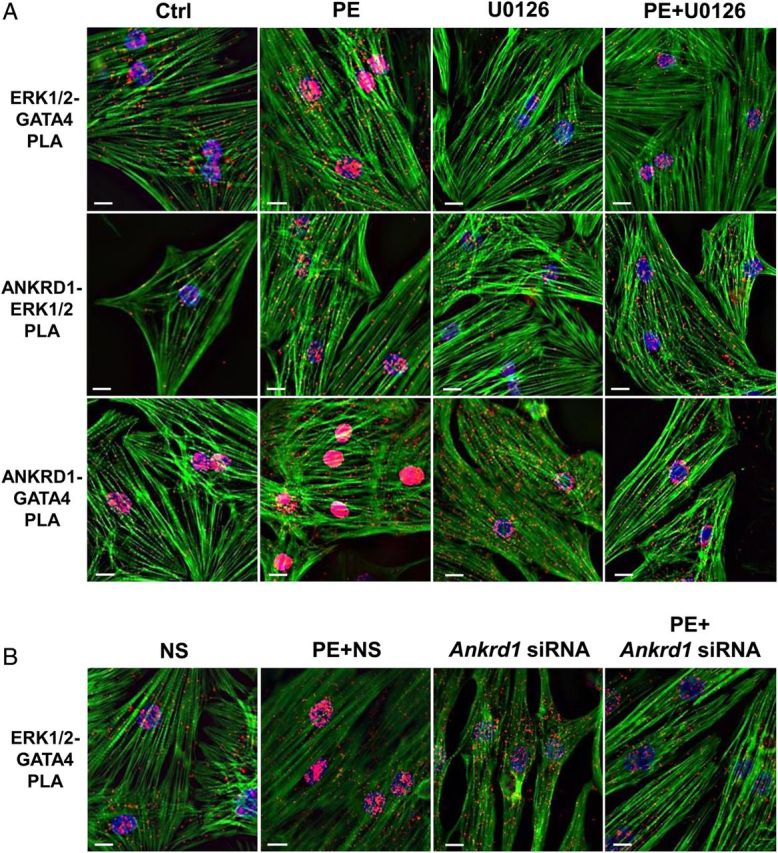
PE induces nuclear translocation of an ANKRD1/ERK/GATA4 complex. Representative images of paraformaldehyde fixed NRVM subjected to proximity ligation assay (PLA). (A) Untreated NRVM showed PLA interactions (red puncta) between ERK1/2 and GATA4 (upper panel), ANKRD1 and ERK1/2 (middle panel), and ANKRD1 and GATA4 (lower panel). NRVM treated with 100 µM PE showed significantly enhanced nuclear PLA signals for all three protein interactions, which were attenuated with MEK/ERK inhibitor (U0126). (B) NRVM transfected with non-silencing (NS) control show a PE-induced increase in nuclear ERK1/2-GATA4 PLA interactions, which was attenuated in NRVM transfected with Ankrd1 siRNA. NRVM were stained for filamentous actin (green) and DAPI (blue). Scale bar = 10 µm.
We examined the effect of PE-induced phosphorylation of ERK and GATA4 to further delineate the signalling interactions between ANKRD1, ERK1/2, and GATA4. In contrast to the robust PLA interactions observed with ANKRD1 and unphosphorylated ERK and GATA4 (Figure 5A), untreated NRVM showed rare pERK1/2-pGATA4, ANKRD1-pERK1/2, and ANKRD1-pGATA4 PLA signals (Figure 6A). PE induced an increase in pERK1/2-pGATA4 PLA signals that were primarily cytosolic, peaking at 1 h and returning to baseline levels at 3 h. ANKRD1-pERK1/2 PLA signals were also primarily cytosolic peaking at 1 h PE and returning to baseline levels at 3 h. ANKRD1-pGATA4 PLA signals were greatest in the cytosol with 1 h of PE treatment and immunofluorescence imaging confirmed that the PE-induced interaction between ANKRD1 and pGATA4 was localized at the sarcomere (see Supplementary material online, Figure S5). Interestingly, following 3 h PE, ANKRD1-pGATA4 PLA signals were localized almost exclusively in the nucleus. These data suggest that PE induces a transient sarcomeric signalling complex that includes ANKRD1 and phosphorylated forms of ERK and GATA4. To further test whether ANKRD1 acts as a scaffold protein for pERK1/2 and pGATA4, we performed PLA experiments in NRVMs that were pre-transfected with Ankrd1 siRNA. Knockdown of ANKRD1 in cells treated with PE for 1 h significantly decreased the pERK1/2-pGATA4 PLA signals (see Supplementary material online, Figure S6), suggesting that ANKRD1 facilitates the PE-induced interaction between pERK1/2 and pGATA4.
Figure 6.
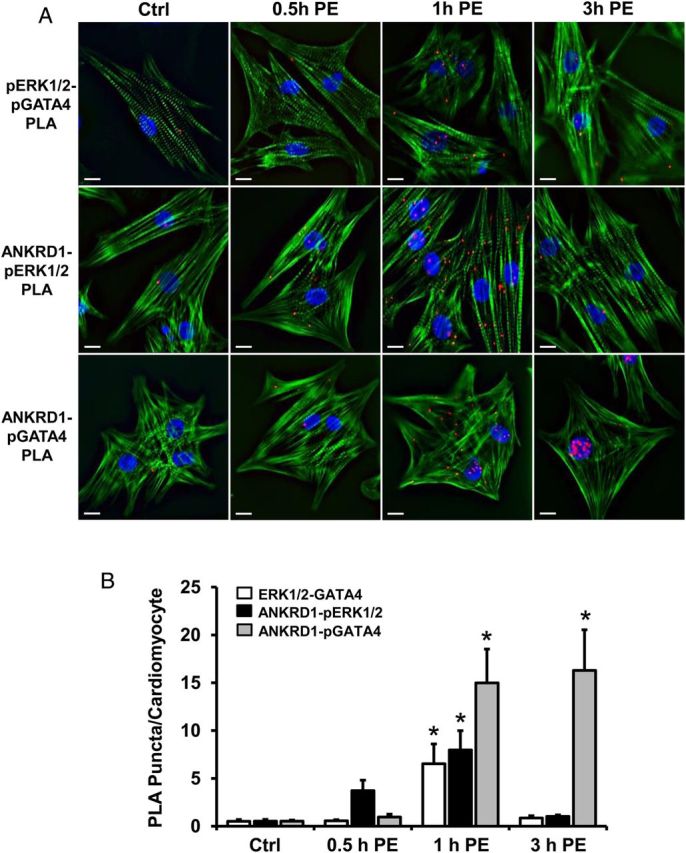
Spatiotemporal interactions between ANKRD1, pERK, and pGATA4 in PE-treated NRVM. (A) Representative images of NRVM treated with 100 µM PE at various time-points, showed PLA interactions between pERK1/2 and pGATA4 (upper panel), ANKRD1 and pERK1/2 (middle panel), and ANKRD1 and pGATA4 (lower panel). NRVM were stained for filamentous actin (green) and DAPI (blue). Scale bar = 10 µm. (B) PLA interactions were quantified for each time-point by determining the number of red puncta per cardiomyocyte. A total of 40–50 myocytes were averaged per time-point. Values expressed as mean ± SEM, n = 5, *P < 0.05 vs. untreated control (Ctrl).
3.6. ANKRD1 knockdown disrupts sarcomeric FHL-1 and FHL-2
We have shown that sarcomeric ANKRD1 interacts with and regulates GATA4 phosphorylation via pERK1/2, however, it remains unclear how ANKRD1 regulates ERK1/2 phosphorylation. Previous reports have shown that four-and-a-half-LIM domains 1 and 2, FHL-1 and FHL-2, both of which interact with the titin spring domain, also interact with each other and both regulate ERK1/2 phosphorylation.26,27 We hypothesized that ANKRD1 could regulate ERK phosphorylation via interaction with the sarcomeric FHL proteins. We detected an interaction between ANKRD1 and FHL-2 by IP and by PLA (see Supplementary material online, Figure S7A), and confirmed an interaction between FHL-1 and FHL-2 by PLA (see Supplementary material online, Figure S7B), but we were not able to detect an interaction between ANKRD1 and FHL-1 (data not shown). Interestingly, knockdown of ANKRD1 in NRVM resulted in loss of FHL-1 protein (see Supplementary material online, Figure S8A). Total FHL-2 protein levels were not affected by loss of ANKRD1, however, cells harvested using a mild lysis buffer showed that FHL-2 levels were increased in the soluble fraction (see Supplementary material online, Figure S8B), suggesting a disruption in binding of FHL2 to the sarcomere. Immunofluorescence imaging confirmed a sarcomeric co-localization of ANKRD1 and FHL-2, which became diffuse and non-sarcomeric in NRVMs transfected with Ankrd1 siRNA (see Supplementary material online, Figure S8C).
These results suggest that ANKRD1 may be part of a larger macro-molecular sarcomere complex that includes the FHL proteins, and that loss of ANKRD1 disrupts sarcomeric FHL proteins and ERK signalling.
3.7. Ankrd1−/− mice maintain a normal cardiac hypertrophic response to pressure overload
To examine the role of ANKRD1 in vivo, WT and Ankrd1−/− mice were subjected to transverse aortic constriction (TAC) to induce haemodynamic overload. Loss of Ankrd1 protein content was confirmed in hearts from Ankrd1−/− mice (see Supplementary material online, Figure S9A). Pressure overload (as assessed by the pressure gradient across the aortic constriction) was similar between Ankrd1−/− and WT mice and significantly higher compared with the corresponding sham controls (see Supplementary material online, Figure S9B). TAC for 4 weeks resulted in significant thickening of the LV posterior and septal walls with early signs of heart failure as evidenced by a decrease in cardiac function (reduced fractional shortening and ejection fraction) and increased LV chamber dilation (increased LV inner diameter) that was comparable between the WT and Ankrd1−/− groups (see Supplementary material online, Table S1). Moreover, TAC induced comparable hypertrophy in both WT and Ankrd1−/− groups as indicated by similar increases in the ratio of heart weight to body weight (see Supplementary material online, Table S1), heart weight to tibia length, and fetal gene markers (see Supplementary material online, Figure S8C and D). Thus, ANKRD1 does not appear to play a role in cardiac hypertrophy under conditions of haemodynamic pressure overload induced by TAC.
3.8. Ankrd1−/ − mice exhibit a blunted cardiac hypertrophic response to PE stimulation
To examine the in vivo role of ANKRD1 with PE-induced cardiac hypertrophy, Ankrd1−/− and WT mice were implanted with osmotic mini-pumps to continuously deliver PE over a 2-week period. At baseline, there were no differences in systolic blood pressure (WT = 104 ± 3 mmHg vs. Ankrd1−/− = 107 ± 2 mmHg; P = NS) or echocardiographic measures of myocardial structure and function (see Supplementary material online, Table S1). PE infusion resulted in similar increases in systolic blood pressure (WT = 121 ± 6 mmHg vs. Ankrd1−/− = 117 ± 8 mmHg; P = NS), however, significant thickening of the LV posterior wall was only observed in WT and not Ankrd1−/− mice, when compared with vehicle controls. Cardiac hypertrophy was confirmed in the WT group as measured by an increase in the ratio of heart weight to body weight (see Supplementary material online, Table S1) and heart weight to tibia length (Figure 7A) when compared with the vehicle control. In contrast, the ratio of heart weight to body weight or tibia length in the PE-treated Ankrd1−/− group was no different from the vehicle control. A blunted response to PE-induced cardiac hypertrophy in the Ankrd1−/− group was further indicated by the attenuated expression of phosphorylated GATA4 and fetal gene markers ANP, β-MHC, and skeletal α-actin, and attenuation of an increase in cardiomyocyte area (Figure 7B–D).
Figure 7.
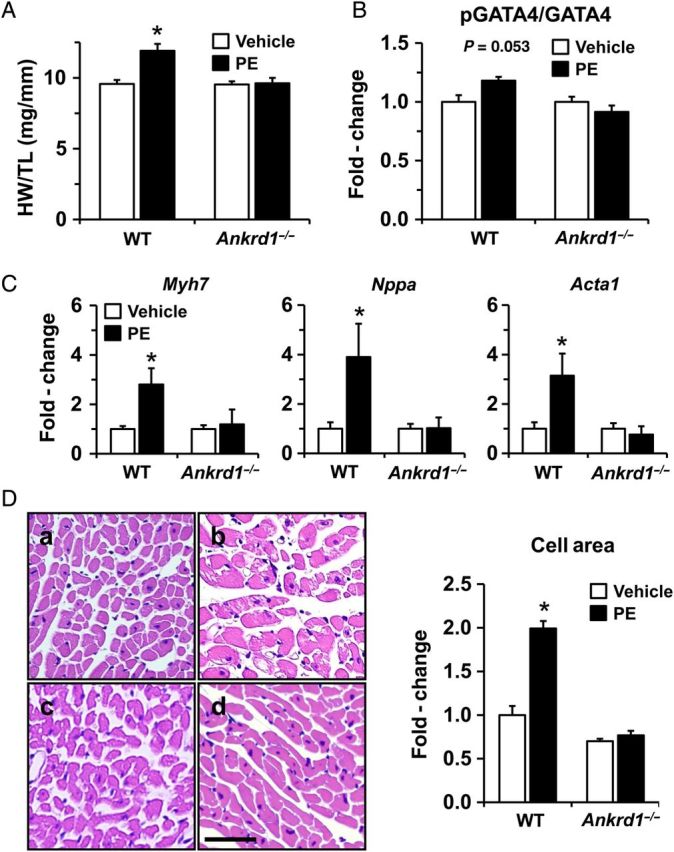
PE-induced cardiac hypertrophy is abrogated in Ankrd1−/− mice. (A) PE infusion for 2 weeks resulted in a significant increase in the ratio of heart weight to tibia length (HW/TL) when compared with vehicle controls in WT but not Ankrd1−/− mice (n = 6 in WT groups, n = 7 in Anrkd1−/− groups). (B) Ankrd1−/− mice exhibit a blunted response to PE-induced GATA4 phosphorylation, n = 3. (C) The PE-induced increase in mRNA levels for fetal gene markers Myh7, Nppa, Acta1 was attenuated in Ankrd1−/− mice, n = 5. Values expressed as mean ± SEM, *P < 0.05 vs. vehicle controls. (D) Histological sections stained with haematoxylin and eosin (H & E) are shown at ×40 magnification; a = WT vehicle, b = WT PE, c = Ankrd1−/− vehicle, d = Ankrd1−/− PE. Scale bar = 20 µm. Cardiomyocyte cross-sectional area was quantified and fold-change values expressed as mean ± SEM, n = 3, *P < 0.05 vs. vehicle control.
To confirm the localization of ERK1/2 and GATA4 observed in vitro, we performed immunohistochemistry in mouse hearts. We were able to verify sarcomeric localization of ERK1/2 and GATA4 in WT hearts (see Supplementary material online, Figure S10A and B). Furthermore, ERK1/2 and GATA4 immunostaining appeared diffuse in Ankrd1−/− hearts (see Supplementary material online, Figure S10B), further confirming our in vitro observations (see Supplementary material online, Figure S4). Finally, FHL-1 levels were significantly decreased in the Ankrd1−/− hearts (see Supplementary material online, Figure S10C), further supporting a role for ANKRD1 as an important stabilizing component of a larger I-band macro-molecular complex that includes FHL-1.
4. Discussion
The sarcomere is increasingly being recognized as an important signalosome that integrates and conveys signals from the contractile machinery to the nucleus to induce gene expression. In this report, we demonstrate that ANKRD1, ERK, and GATA4 are part of a sarcomere to nuclear transitional complex that plays a pivotal role in phenylephrine-induced cardiac hypertrophy.
Cardiac titin is a gigantic elastic protein that forms an intricate filament network in the sarcomere and is the principal determinant of cardiomyocyte passive stiffness. While the viscoelastic properties of the titin spring have been well-characterized, there is increasing evidence that this spring region also serves as an important regulatory node in integrating and coordinating signalling pathways important in cardiac hypertrophy and mechanosensing.28,29
ANKRD1 interacts with the N2A segment of the titin spring and Miller et al.5 reported that ANKRD1 translocates to the nucleus upon static stretch in rat fetal cardiomyocytes, though the physiological significance of this in vitro ANKRD1 translocation remains to be elucidated. Our data strongly support a role for ANKRD1 in mediating sarcomeric MEK-ERK-GATA4 signal transduction. To our knowledge, this is also the first report of a transcription factor, GATA4, found to localize to the sarcomere. Previous studies have shown that ERK1/2 phosphorylates GATA4 at S105 which is necessary for GATA4 DNA binding and stress-induced cardiac hypertrophy.16,24 Our study specifically identifies a role for ANKRD1 in mediating MEK/ERK-induced GATA4 phosphorylation at the level of the sarcomere, suggesting a more localized upstream activation of GATA4 than previously thought. This is distinct from other sarcomere signalling proteins such as FHL-1/FHL-2, MLP, MURF, and calcineurin which are believed to signal and activate transcription factors more downstream in the nucleus.27,30,31 It should be pointed out that GATA4 is subject to a host of post-translational modifications by diverse enzymes including p38 MAPK and PKA (phosphorylation), p300 (acetylation), Hdac2 and Hopx (deacytelation), Sumo (sumoylation), and PRC2 (methylation).32 Localizing GATA4 to the ANKRD1 sarcomere complex could serve as a way to restrict GATA4 phosphorylation to ERK signalling.
In our model, TAC for 4 weeks resulted in a significant dilation of the LV chamber where the titin spring is presumed to be under high pathological strain. Our results, however, show that the hypertrophic response was comparable between Ankrd1−/− and WT mice, and therefore argue against a role for ANKRD1 in mechanosensation, at least in the context of TAC-induced pressure overload. These results are consistent with a recent report in triple (Ankrd1/Ankrd2/Ankrd23) MARP knockout mice showing no abnormal cardiac phenotype in response to TAC.33 The authors concluded that the MARPs are not essential for cardiac function in response to pressure overload, however, the response to chronic agonist stimulation was not examined. That ANKRD1 might act as a mechanosensor under different mechanical stimuli such as volume overload or exercise hypertrophy warrants further investigation. Our findings are reminiscent of FHL-2 which interacts with the unique N2B sequence (N2B-Us) of the titin spring, in that FHL-2 plays no role in pressure overload-induced hypertrophy but modifies the hypertrophic response to β-adrenergic stimulation.31,34,35 Our results appear to contradict a recent study in transgenic ANKRD1-overexpressing mice reporting that ANKRD1 attenuates both TAC and isoproterenol-induced cardiac hypertrophy via suppression of ERK signalling.36 This apparent discrepancy could be due to the supra-physiological levels of ANKRD1 in the transgenic mice leading to off-target or dominant-negative effects.
Our data suggest that PE-stimulation induces sarcomeric translocation of ANKRD1, ERK1/2, and GATA4 to the nucleus, presumably to induce gene expression. Our finding that ANKRD1 interacts and co-localizes with GATA4 and ERK1/2 in the nucleus suggests that they shuttle as a complex to the nucleus. ANKRD1 was initially identified as a co-repressor of YB-1 and a negative regulator of cardiac-specific genes including atrial natriuretic factor, myosin light chain-2v, and cardiac troponin C.3,6 More recently, ANKRD1 was shown to be a co-activator of p53 in a myoblast cell line and was suggested to play a role in myogenic differentiation.21 We have previously shown that GATA4 is upstream of ANKRD1 and that both GATA4 and ANKRD1 co-regulate sarcomere gene expression.17 Our current study suggests that with PE stimulation, ANKRD1 in a positive feedback loop with GATA4 amplifies a mutually reinforcing signalling circuit to augment cardiac gene expression. GATA4 is also known to be activated under conditions of cardiac hypertrophy induced by pressure overload,16 and it is important to consider the role of GATA4 in this context. As previously discussed, multiple other signalling pathways are able to post-translationally modify GATA4 and regulate its transcriptional activity.32 While PE stimulation may activate a specific pathway that involves ANKRD1-ERK-GATA4 signalling, pressure overload may evoke a more global response by recruiting diverse hypertrophic signalling pathways that converge to enhance GATA4 transcriptional activity; thus, compensating for loss of ANKRD1 signalling in the Ankrd1−/− mouse. While beyond the scope of this study, it would be important to determine whether PE-induced ANKRD1 signalling extends to other Gq-coupled receptor agonists such as angiotension II and endothelin-1.
Our study shows an interaction between ANKRD1 and FHL-2, and ANKRD1 knockdown in vitro and in vivo results in partial loss or disruption of FHL-1 and FHL-2. FHL-1 has some striking parallels with ANKRD1 in that it interacts with the titin N2B-Us spring and has been shown to form a sarcomeric complex by recruitment of Raf/MEK/ERK to enhance phosphorylation of ERK2 to induce a downstream hypertrophic response.27 Mice deficient in FHL-1 show loss of ERK1/2 phosphorylation and a blunted pathological hypertrophic response to TAC-induced pressure overload.27 Given that FHL-1 and FHL-2 interact with each other and with ERK2 at titin N2B-Us,26,37 we speculate that sarcomeric ANKRD1 is part of a larger macro-molecular complex that helps anchor FHL-1 and FHL-2 to regulate sarcomeric Raf/MEK/ERK signalling. We believe that loss of ANKRD1 disrupts this signalling pathway and ultimately perturbs ERK-induced GATA4 phosphorylation and gene transcription. Adding to the complexity of ANKRD1 signalling is myopalladin which interacts with ANKRD1 at its N-terminus and α-actinin at its C-terminus,7 in essence bridging ANKRD1 to another major sarcomeric signalosome, i.e. the Z-line.38 It is interesting to note that a recent study reported that ANKRD1 is phosphorylated by PKA and PKC, potentially altering its structure and function.39 Whether ANKRD1 phosphorylation occurs with hypertrophy and/or plays a role in sarcomere signalling or mechanosensing merits investigation.
Human mutations in ANKRD1 have recently been shown to be associated with hypertrophic and dilated cardiomyopathy.11–13 In vitro studies indicate that a mutation associated with hypertrophic cardiomyopathy enhanced ANKRD1 stability while a mutation associated with dilated cardiomyopathy disrupted the ANKRD1 interaction with FHL2.13,40 Stabilized ANKRD1 could augment sarcomeric ANKRD1-GATA4 signalling resulting in hypertrophic cardiomyopathy, while loss of ANKRD1-FHL2 interaction could impair sarcomeric signalling leading to dilated cardiomyopathy.
Taken together, we propose a model whereby ANKRD1 plays an important role in phenylephrine-induced hypertrophy by serving as a sarcomere scaffolding protein to induce ERK-GATA4 phosphorylation; this is followed by translocation of ANKRD1, ERK, and GATA4 to the nucleus with subsequent GATA4 DNA binding and gene transcription. It is increasingly being recognized that the development of cardiac hypertrophy involves an intricate and complex interplay of biomechanical and neurohormonal factors that elicit a myriad of sub-cellular signalling complexes and pathways that lead to cell growth.1 A detailed dissection of the signalling mechanisms governing cardiac hypertrophy raises the prospect of designing more effective therapeutic strategies tailored to specific forms of hypertrophic cardiomyopathy.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This study was supported in part by NIH grant RO1DK065656 and the US Department of Veterans Affairs support to J.M.D., Vanderbilt University Stahlman grant and RO1HL095813 to C.C.L.
Acknowledgements
We thank Douglas B. Sawyer for stimulating discussions and valuable feedback. We acknowledge the support of the VUMC Cell Imaging Shared Resource and Mouse Metabolic Phenotyping Center (MMPC, NIH grant U24 DK0596).
Conflict of interest: none declared.
References
- 1.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med 2008;358:1370–1380. [DOI] [PubMed] [Google Scholar]
- 2.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol 2006;7:589–600. [DOI] [PubMed] [Google Scholar]
- 3.Jeyaseelan R, Poizat C, Baker RK, Abdishoo S, Isterabadi LB, Lyons GE, Kedes L. A novel cardiac-restricted target for doxorubicin. CARP, a nuclear modulator of gene expression in cardiac progenitor cells and cardiomyocytes. J Biol Chem 1997;272:22800–22808. [DOI] [PubMed] [Google Scholar]
- 4.Kemp TJ, Sadusky TJ, Saltisi F, Carey N, Moss J, Yang SY, Sassoon DA, Goldspink G, Coulton GR. Identification of Ankrd2, a novel skeletal muscle gene coding for a stretch-responsive ankyrin-repeat protein. Genomics 2000;66:229–241. [DOI] [PubMed] [Google Scholar]
- 5.Miller MK, Bang ML, Witt CC, Labeit D, Trombitas C, Watanabe K, Granzier H, McElhinny AS, Gregorio CC, Labeit S. The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. J Mol Biol 2003;333:951–964. [DOI] [PubMed] [Google Scholar]
- 6.Zou Y, Evans S, Chen J, Kuo HC, Harvey RP, Chien KR. CARP, a cardiac ankyrin repeat protein, is downstream in the Nkx2–5 homeobox gene pathway. Development 1997;124:793–804. [DOI] [PubMed] [Google Scholar]
- 7.Bang ML, Mudry RE, McElhinny AS, Trombitas K, Geach AJ, Yamasaki R, Sorimachi H, Granzier H, Gregorio CC, Labeit S. Myopalladin, a novel 145-kilodalton sarcomeric protein with multiple roles in Z-disc and I-band protein assemblies. J Cell Biol 2001;153:413–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aihara Y, Kurabayashi M, Saito Y, Ohyama Y, Tanaka T, Takeda S, Tomaru K, Sekiguchi K, Arai M, Nakamura T, Nagai R. Cardiac ankyrin repeat protein is a novel marker of cardiac hypertrophy: role of M-CAT element within the promoter. Hypertension 2000;36:48–53. [DOI] [PubMed] [Google Scholar]
- 9.Nagueh SF, Shah G, Wu Y, Torre-Amione G, King NM, Lahmers S, Witt CC, Becker K, Labeit S, Granzier HL. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 2004;110:155–162. [DOI] [PubMed] [Google Scholar]
- 10.Zolk O, Marx M, Jackel E, El-Armouche A, Eschenhagen T. Beta-adrenergic stimulation induces cardiac ankyrin repeat protein expression: involvement of protein kinase A and calmodulin-dependent kinase. Cardiovasc Res 2003;59:563–572. [DOI] [PubMed] [Google Scholar]
- 11.Arimura T, Bos JM, Sato A, Kubo T, Okamoto H, Nishi H, Harada H, Koga Y, Moulik M, Doi YL, Towbin JA, Ackerman MJ, Kimura A. Cardiac ankyrin repeat protein gene (ANKRD1) mutations in hypertrophic cardiomyopathy. J Am Coll Cardiol 2009;54:334–342. [DOI] [PubMed] [Google Scholar]
- 12.Duboscq-Bidot L, Charron P, Ruppert V, Fauchier L, Richter A, Tavazzi L, Arbustini E, Wichter T, Maisch B, Komajda M, Isnard R, Villard E. Mutations in the ANKRD1 gene encoding CARP are responsible for human dilated cardiomyopathy. Eur Heart J 2009;30:2128–2136. [DOI] [PubMed] [Google Scholar]
- 13.Moulik M, Vatta M, Witt SH, Arola AM, Murphy RT, McKenna WJ, Boriek AM, Oka K, Labeit S, Bowles NE, Arimura T, Kimura A, Towbin JA. ANKRD1, the gene encoding cardiac ankyrin repeat protein, is a novel dilated cardiomyopathy gene. J Am Coll Cardiol 2009;54:325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuo CT, Morrisey EE, Anandappa R, Sigrist K, Lu MM, Parmacek MS, Soudais C, Leiden JM. GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev 1997;11:1048–1060. [DOI] [PubMed] [Google Scholar]
- 15.Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res 2006;98:837–845. [DOI] [PubMed] [Google Scholar]
- 16.van Berlo JH, Elrod JW, Aronow BJ, Pu WT, Molkentin JD. Serine 105 phosphorylation of transcription factor GATA4 is necessary for stress-induced cardiac hypertrophy in vivo. Proc Natl Acad Sci USA 2011;108:12331–12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen B, Zhong L, Roush SF, Pentassuglia L, Peng X, Samaras S, Davidson JM, Sawyer DB, Lim CC. Disruption of a GATA4/Ankrd1 signaling axis in cardiomyocytes leads to sarcomere disarray: implications for anthracycline cardiomyopathy. PLoS One 2012;7:e35743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. doi: 10.1016/j.ajpath.2014.09.018. Samaras SE, Almodovar-Garcia K, Wu N, Yu F, Davidson JM. Global deletion of Ankrd1 results in a wound-healing phenotype associated with dermal fibroblast dysfunction. American J of Pathol 2015;185:96–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim CC, Zuppinger C, Guo X, Kuster GM, Helmes M, Eppenberger HM, Suter TM, Liao R, Sawyer DB. Anthracyclines induce calpain-dependent titin proteolysis and necrosis in cardiomyocytes. J Biol Chem 2004;279:8290–8299. [DOI] [PubMed] [Google Scholar]
- 20.Molkentin JD, Kalvakolanu DV, Markham BE. Transcription factor GATA-4 regulates cardiac muscle-specific expression of the alpha-myosin heavy-chain gene. Mol Cell Biol 1994;14:4947–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kojic S, Nestorovic A, Rakicevic L, Belgrano A, Stankovic M, Divac A, Faulkner G. A novel role for cardiac ankyrin repeat protein Ankrd1/CARP as a co-activator of the p53 tumor suppressor protein. Arch Biochem Biophys 2010;502:60–67. [DOI] [PubMed] [Google Scholar]
- 22.Almodovar-Garcia K, Kwon M, Samaras SE, Davidson JM. ANKRD1 acts as a transcriptional repressor of MMP13 via the AP-1 site. Mol Cell Biol 2014;34:1500–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laure L, Suel L, Roudaut C, Bourg N, Ouali A, Bartoli M, Richard I, Daniele N. Cardiac ankyrin repeat protein is a marker of skeletal muscle pathological remodelling. FEBS J 2009;276:669–684. [DOI] [PubMed] [Google Scholar]
- 24.Liang Q, Wiese RJ, Bueno OF, Dai YS, Markham BE, Molkentin JD. The transcription factor GATA4 is activated by extracellular signal-regulated kinase 1- and 2-mediated phosphorylation of serine 105 in cardiomyocytes. Mol Cell Biol 2001;21:7460–7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Charron F, Tsimiklis G, Arcand M, Robitaille L, Liang Q, Molkentin JD, Meloche S, Nemer M. Tissue-specific GATA factors are transcriptional effectors of the small GTPase RhoA. Genes Dev 2001;15:2702–2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Purcell NH, Darwis D, Bueno OF, Muller JM, Schule R, Molkentin JD. Extracellular signal-regulated kinase 2 interacts with and is negatively regulated by the LIM-only protein FHL2 in cardiomyocytes. Mol Cell Biol 2004;24:1081–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sheikh F, Raskin A, Chu PH, Lange S, Domenighetti AA, Zheng M, Liang X, Zhang T, Yajima T, Gu Y, Dalton ND, Mahata SK, Dorn GW, II, Brown JH, Peterson KL, Omens JH, McCulloch AD, Chen J. An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. J Clin Invest 2008;118:3870–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Granzier HL, Labeit S. The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circ Res 2004;94:284–295. [DOI] [PubMed] [Google Scholar]
- 29.Linke WA, Hamdani N. Gigantic business: titin properties and function through thick and thin. Circ Res 2014;114:1052–1068. [DOI] [PubMed] [Google Scholar]
- 30.Frank D, Kuhn C, Katus HA, Frey N. The sarcomeric Z-disc: a nodal point in signalling and disease. J Mol Med (Berl) 2006;84:446–468. [DOI] [PubMed] [Google Scholar]
- 31.Hojayev B, Rothermel BA, Gillette TG, Hill JA. FHL2 binds calcineurin and represses pathological cardiac growth. Mol Cell Biol 2012;32:4025–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou P, He A, Pu WT. Regulation of GATA4 transcriptional activity in cardiovascular development and disease. Curr Top Dev Biol 2012;100:143–169. [DOI] [PubMed] [Google Scholar]
- 33.Bang ML, Gu Y, Dalton ND, Peterson KL, Chien KR, Chen J. The muscle ankyrin repeat proteins CARP, Ankrd2, and DARP are not essential for normal cardiac development and function at basal conditions and in response to pressure overload. PLoS One 2014;9:e93638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chu PH, Bardwell WM, Gu Y, Ross J, Jr, Chen J. FHL2 (SLIM3) is not essential for cardiac development and function. Mol Cell Biol 2000;20:7460–7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kong Y, Shelton JM, Rothermel B, Li X, Richardson JA, Bassel-Duby R, Williams RS. Cardiac-specific LIM protein FHL2 modifies the hypertrophic response to beta-adrenergic stimulation. Circulation 2001;103:2731–2738. [DOI] [PubMed] [Google Scholar]
- 36.Song Y, Xu J, Li Y, Jia C, Ma X, Zhang L, Xie X, Zhang Y, Gao X, Zhang Y, Zhu D. Cardiac ankyrin repeat protein attenuates cardiac hypertrophy by inhibition of ERK1/2 and TGF-beta signaling pathways. PLoS One 2012;7:e50436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lange S, Auerbach D, McLoughlin P, Perriard E, Schafer BW, Perriard JC, Ehler E. Subcellular targeting of metabolic enzymes to titin in heart muscle may be mediated by DRAL/FHL-2. J Cell Sci 2002;115:4925–4936. [DOI] [PubMed] [Google Scholar]
- 38.Pyle WG, Solaro RJ. At the crossroads of myocardial signaling: the role of Z-discs in intracellular signaling and cardiac function. Circ Res 2004;94:296–305. [DOI] [PubMed] [Google Scholar]
- 39.Lun AS, Chen J, Lange S. Probing muscle ankyrin-repeat protein (MARP) structure and function. Anat Rec (Hoboken) 2014;297:1615–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crocini C, Arimura T, Reischmann S, Eder A, Braren I, Hansen A, Eschenhagen T, Kimura A, Carrier L. Impact of ANKRD1 mutations associated with hypertrophic cardiomyopathy on contraction parameters of engineered heart tissue. Basic Res Cardiol 2013;108:349. [DOI] [PubMed] [Google Scholar]