

Abstract
Background. An association between GB virus C (GBV-C) and improved outcomes of human immunodeficiency virus (HIV) infection has been reported in HIV-positive individuals with active GBV-C coinfection. This study provides insights into the immune mechanisms underlying the protective role of GBV-C in HIV-infected patients.
Methods. The concentrations of 64 cytokines and chemokines were measured in plasma samples obtained from the Viral Activation Transfusion Study cohort before transfusion and longitudinally from 30 patients positive for both HIV and GBV-C (hereafter, “cases”) and 30 patients positive for HIV and negative for GBV-C (hereafter, “controls”).
Results. Cases had lower HIV viral loads and higher CD4 T-cell counts than controls after acquisition of GBV-C infection. Most of the modulated cytokines and chemokines were reduced after GBV-C detection, including many proinflammatory cytokines, suggesting an overall antiinflammatory effect of GBV-C in HIV-positive subjects. Most pathways and functions of the measured cytokines were downregulated in cases, except cell death pathways, which were upregulated in various cell subsets in the 3 months after GBV-C detection.
Conclusions. GBV-C has a protective effect, in part through a competition mechanism leading to decreased inflammation and improved HIV disease outcome in cases. Further studies are necessary to establish whether GBV-C may have deleterious effects on the host at the cellular level, including depleting the cells that are the targets of HIV.
Keywords: GBV-C, transfusion-transmission, HIV coinfection, cytokine and chemokine responses, HIV disease progression markers, HIV disease outcome
GB virus C (GBV-C), also known as human pegivirus, is an RNA virus within the Pegivirus genus in the Flaviviridae family [1, 2] and is found within T and B lymphocytes, natural killer (NK) cells, and monocytes [3, 4]. GBV-C can cause persistent human infection, especially in immunosuppressed individuals [5]; however, 60%–75% of immune competent individuals resolve viremia, coincident with the development of anti–GBV-C E2 antibodies [6]. GBV-C is transmitted sexually, vertically, and parentally through infected blood products [7–11]. GBV-C is common in healthy blood donors in developed countries, with 1%–4% testing positive for GBV-C RNA and 17% having viral envelope protein E2 (E2) antibodies [12–14]. Studies have failed to demonstrate an association with any particular disease [15–17], with the potential exception of an association with non-Hodgkin lymphoma [18–20]. Further research is needed to determine whether this association is causally related to GBV-C infection. Therefore, blood products are not routinely screened for the presence of GBV-C RNA [6, 21]. GBV-C RNA prevalence was 7% in the Viral Activation Transfusion Study (VATS) cohort and individuals with advanced human immunodeficiency virus (HIV) infection [22]. E2 antibody–negative, transfusion-naive VATS subjects developed GBV-C viremia within 120 days after transfusion, with a 9% incident infection rate per unit transfused [22].
Reports have shown an association between GBV-C and prolonged survival in HIV-infected individuals with active GBV-C coinfection [12]. GBV-C viremia is associated with slower HIV disease progression, and coinfected subjects have lower HIV viral loads and higher CD4+ T-cell than HIV-1–monoinfected patients [8, 23, 24]. We recently reported that HIV-infected people acquiring incident GBV-C infection following transfusion have longer survival durations than HIV-infected people who underwent transfusion but did not acquire GBV-C infection [25]. The findings suggested that the intentional infection of HIV-positive individuals with GBV-C could represent a therapeutic approach for HIV infection [26].
The host immunological response underlying GBV-C and HIV coinfection that may contribute to reduced HIV replication and CD4+ T-cell loss and, consequently, to better survival are not well characterized. Prior studies found reduced lymphocyte, monocyte, and NK cell markers of activation in patients with HIV and GBV-C coinfection, compared with those with HIV monoinfection, suggesting that GBV-C infection may modulate host inflammation, thus reducing HIV replication and pathogenesis [27–31]. Furthermore, in vitro studies suggest that E2 and virus particles interfere with T-cell activation and proliferation [32–35].
Here, VATS plasma samples were evaluated for cytokine and chemokine levels during acute GBV-C viremia following transfusion-associated transmission in HIV-infected patients. With HIV disease progression markers, treatment data, and GBV-C infection parameters, this longitudinal study provided a unique opportunity to characterize the patterns of cytokines and chemokines during incident GBV-C coinfection and provides insight into the immune mechanisms underlying the protective role of GBV-C coinfection in HIV-infected patients.
METHODS
The VATS (1995–1999) was a multicenter clinical trial that randomized 531 transfusion-naive HIV-positive patients with anemia to receive either a filtered leuko-reduced or standard non–leuko-reduced blood, funded by the National Heart Lung and Blood Institute (NHLBI) [36, 37]. VATS plasma samples were collected before transfusion (baseline) and during follow-up visits, weekly for 1 month and quarterly up to 45 months after transfusion, and stored at −70°C at the NHLBI-funded Biologic Specimen and Data Repository Information Coordinating Center (BioLINCC). A limited-access VATS public use data set with demographic characteristics and clinical parameters (HIV viral load, CD4+ T-cell counts, and CD8+CD38+ T-cell frequencies) of the patients is also available at BioLINCC.
Approval from the University of California–San Francisco and University of Iowa institutional review boards were obtained for this study. All VATS patients provided written informed consent for HIV and transfusion outcomes research.
Subject Selection and Sample Accession
Of the 531 HIV-positive VATS subjects, 489 had paired pretransfusion and final samples available for GBV-C evaluation. All paired serum/plasma samples were previously tested for GBV-C E2 antibody, using the anti-GBenv uplate enzyme immunoassay, and for RNA, using the quantitative GBV-RNA reverse transcription–polymerase chain reaction assay (both from Roche Diagnostics, Penzberg, Germany) [22]. To examine the effect of incident GBV-C infection on immunological parameters, a subset of 294 HIV-positive subjects who were GBV-C RNA and E2 antibody negative before transfusion was evaluated [22]. Plasma samples (not previously thawed) were requested from the BioLINCC for confirmation of GBV-C status and additional cytokine/chemokine testing.
Based on availability, 870 pretransfusion and posttransfusion samples from 143 subjects were selected for GBV-C RNA analysis.
GBV-C RNA Detection and Case Definition
Selected plasma samples were tested for GBV-C RNA at the University of Iowa [35, 38]. RNA extraction and amplification was performed as previously described [35, 38]. Four negative control wells (2 blanks and 2 water controls) were included with each set of samples, and a standard curve was generated using synthetically transcribed GBV-C RNA [35, 39, 40]. These samples consisted of 322 GBV-C RNA–positive samples; 358 GBV-C antibody–negative, GBV-C RNA–negative samples; and 190 GBV-C antibody–positive, GBV-C RNA–negative samples.
Cases were defined as subjects with incident GBV-C infection, based on the presence of at 3 posttransfusion samples that were positive for GBV-C RNA or anti-E2 antibodies within a 3-month period (eg, 2 RNA and one serology) and at least 1 of samples was positive for GBV-C RNA. Controls were defined as subjects who remained GBV-C RNA and GBV-C antibody negative at all time points after transfusion. We identified 30 cases and 30 controls for analysis on the basis of sample availability and comparable sampling time points from baseline.
Definition of Treatment
VATS data on treatment were available from baseline and quarterly visits. Highly active antiretroviral therapy (HAART) was defined as ≥2 nucleoside reverse-transcriptase inhibitors (NRTIs) in combination with at least 1 protease inhibitor or 1 nonnucleoside reverse-transcriptase inhibitor (NNRTI), consistent with available medications when VATS was active. Antiretroviral therapy (ART) was defined as the use of only 1 NRTI, at least 1 protease inhibitor, and/or at least 1 NNRTI.
Multiple Cytokine and Chemokine Analysis
Specimens from the 30 cases and 30 controls underwent cytokine and chemokine analysis. The concentrations of 64 analytes were measured in plasma samples, using an array of Luminex-based assays (EMD Millipore, Hayward, California): the Milliplex MAP Human Cytokine/Chemokine Magnetic Bead Panel (HCYTMAG-60K-PX29; standard curve range, 3.2–1 × 104 pg/mL), the cytokine/chemokine panel II (HCP2MAG-62K-PX23; standard curve range, 0.98–1 × 106 pg/mL), and the cytokine/chemokine panel III (HCP3MAG-63K-PX11; standard curve range, 1.95–2 × 105pg/mL); the high-sensitivity cytokine/chemokine panel (HSCYTO-60SPMX13; standard curve range, 3.2–1 × 104 pg/mL) was used to measure the concentration of 13 cytokines also assessed by the HCYTMAG-60K-PX29 assay. Subjects' plasma samples were assayed according to the manufacturer's protocols. Samples and standard curves were run in duplicate. Fluorescence signals were detected using the multiplex array reader Luminex 200 system (Invitrogen) and analyzed using Bio-Plex manager 6.1 software (Bio-Rad). For each of the analytes in the panels, results that were out of range low of the standard curve were set as half the lowest standard point on the curve for that analyte. The values that were out of range high were set as 2 times the highest standard point on the curve.
Statistical Analysis
Baseline variables were contrasted between cases and controls, using t tests for continuous variables and Fisher exact or χ2 tests for categorical variables. Baseline cytokine and viral load values were log-transformed before analysis, owing to nonnormal distribution of the data.
The analysis period spanned the subset of visits selected for cytokine testing, from baseline up to approximately 15 months (450 days) after transfusion. Percentage changes in cytokine concentrations from the pre-HAART period to the post-HAART period were assessed in subjects with at least 1 sample in both periods. Absolute changes in cytokine concentrations and viral load values were assessed in cases between baseline and time points closest to 6 months (approximately 200 days) and 15 months (approximately 400 days) after transfusion, corresponding to samples obtained closest to 100 and 300 days after GBV-C detection and were assessed in controls between baseline and the samples obtained closest to 400 days after transfusion. Because of nonnormal distribution of data, change values were analyzed using nonparametric Wilcoxon signed rank tests.
Least-squares means, adjusted for time-varying HAART status and individual subject variation, were used to illustrate trends in GBV-C and HIV viral load since GBV-C detection. Adjusted mixed modeling also was used to assess the impact of GBV-C infection on log-transformed cytokine concentrations over time, with GBV-C positivity, days from baseline, HAART treatment status, and HIV viral load considered as time-varying covariates.
P values were adjusted for multiple comparisons into false discovery rates (FDRs) by the Benjamini and Hochberg controlling procedure; statistical significance was defined as a P value of < .05 and a FDR of ≤ 0.2. All statistical analysis was performed using SAS/STAT software (SAS Institute, Cary, North Carolina).
A heat map of cytokine modulations was generated using R and its gplots package. Pathway analysis (Qiagen Ingenuity Pathway Analysis) was performed to help predict effects the observed cytokine changes might have on the host immune system.
RESULTS
Transfusion-Transmitted GBV-C Improves HIV Disease Outcome
At baseline, there was no significant difference between cases and controls for demographics, HIV viral load, and CD4+ T-cell count (Table 1). However, a significant decrease in HIV viral load was observed in cases, from a mean log10 HIV viral load of 4.33 copies/mL at baseline down to 3.24 copies/mL at 100 days after GBV-C detection (P < .01) and maintained at 3.39 copies/mL at 300 days after GBV-C detection (P = .02). In contrast, controls maintained a higher mean log10 HIV viral load, from 4.54 copies/mL at baseline to 4.32 copies/mL at the last visit (P = .49; Figure 1A). The significant decrease of HIV viral load in cases was also accompanied by an increase in CD4+ T-cell count from 77 cell/µL to 125 cells/µL at 100 days (P = .09) and to 196 cells/µL at 300 days after GBV-C detection (P < .01), while there was no significant change in CD4+ T-cell count in the controls (Figure 1A). HIV viral load fell in the first year after GBV-C viremia developed (P = .07), after adjustment for HAART status, time elapsed, and subject (Figure 1B). There was an inverse correlation (P = .003) between GBV-C and HIV viral load during the first year after GBV-C diagnosis, after adjustment for HAART status, time elapsed, and subject.
Table 1.
Demographic and Clinical Characteristics at Baseline and During Follow-up Among Human Immunodeficiency Virus (HIV)-Infected Case and Control Subjects in the Viral Activation Transfusion Study
| Characteristic | Cases |
Controls |
P Valuea | ||
|---|---|---|---|---|---|
| Subjects, No. | Value | Subjects, No. | Value | ||
| Age at baseline, y | 30 | 37.7 ± 6.9 | 30 | 36.9 ± 7.3 | .67 |
| Male sex | 30 | 23 (76.7) | 30 | 21 (70.0) | .77 |
| Race/ethnicity | 30 | 30 | .48 | ||
| White, non-Hispanic | 21 (70.0) | 16 (53.3) | |||
| Black, non-Hispanic | 6 (20.0) | 9 (30.0) | |||
| Other | 3 (10.0) | 5 (16.7) | |||
| History of CD4+ T-cell count <50 cells/µL | 30 | 30 | .11 | ||
| Yes | 22 (73.3) | 20 (66.7) | |||
| No | 5 (16.7) | 10 (33.3) | |||
| Don't know | 3 (10.0) | 0 (0.0) | |||
| History of CMV disease | 30 | 7 (23.3) | 30 | 2 (6.7) | .15 |
| Treatment at baseline | 30 | 30 | .54 | ||
| ART | 16 (53.3) | 13 (43.3) | |||
| HAART | 3 (10.0) | 6 (20.0) | |||
| Neither | 11 (36.7) | 11 (36.7) | |||
| Treatment at any visit | 30 | 30 | .08 | ||
| ART only | 5 (16.7) | 12 (40.0) | |||
| HAART | 25 (83.3) | 18 (60.0) | |||
| Neither | 0 (0.0) | 0 (0.0) | |||
| Visits involving HAART receipt, % | 30 | 40.3 ± 33.9 | 30 | 33.2 ± 27.9 | .38 |
| HIV viral load at baseline, log10 copies/mL | 30 | 4.4 ± 1.2 | 30 | 4.5 ± 1.2 | .66 |
| CD4+ T-cell count at baseline, cells/µL | 28 | 63.9 ± 75.4 | 29 | 87.6 ± 161.6 | .48 |
| Bright CD8+ T cells expressing CD38 at baseline, % | 25 | 47.8 ± 16.6 | 24 | 41.5 ± 12.0 | .14 |
Data are mean value ± SD or no. (%) of subjects. Case subjects were positive for both GB virus C (GBV-C) and HIV RNA, and control subjects were positive for HIV RNA and negative for GBV-C antibody and GBV-C RNA.
Abbreviations: ART, antiretroviral therapy; HAART, highly active antiretroviral therapy; SD, standard deviation.
a The t test was used for continuous variables, and the Fisher exact test was used for categorical variables.
Figure 1.

Human immunodeficiency virus (HIV) disease progression markers in subjects positive for both HIV and GB virus C (GBV-C; cases) and subjects positive for HIV and negative for GBV-C (controls). A, Mean HIV disease progression markers (HIV viral load and CD4+ T-cell counts) across time for cases and controls. Symbols are for significant differences in mean change from baseline. *P < .05, **P < .01. B, Least-squares means of GBV-C and HIV viral loads for cases during follow-up, adjusted for subject and time-varying highly active antiretroviral therapy status.
Changes in Cytokines and Chemokines Within and Between Cases and Controls Receiving HIV Treatment During VATS
The concentrations of cytokines and chemokines were measured at baseline and at 2 weeks (14 days), 3 months (approximately 100 days), and 15 months (approximately 400 days) for controls; concentrations were measured at the same time points, as well as at 6 months (approximately 200 days) for cases, to capture both long-term and short-term effects of GBV-C infection (Supplementary Figure 1). At baseline, there was no significant difference between cases and controls in cytokine/chemokine levels (data not shown). Only 3 cases and 6 controls were receiving HAART at baseline, while others were either receiving ART or no treatment (Table 1). All cases and controls at some point received treatment in the form of ART, for 17% of the cases and 40% of the controls, or HAART, for 83% of the cases and 60% of the controls (Table 1).
For subjects who initiated HAART while in the VATS, cytokine and chemokine concentrations were averaged over the period pre-HAART and post-HAART (Figure 2A), and significant changes from the pre-HAART period to the post-HAART period were evaluated for cases and controls and between cases and controls (Figure 2B). Concentrations of 9 cytokines and chemokines were significantly decreased after HAART in cases, including interleukin 10 (IL-10; P < .001 and FDR = 0.002), interleukin 6 (IL-6; P = .01 and FDR = 0.06), tumor necrosis factor α (TNF-α; P = .004 and FDR = 0.03), granulocyte macrophage colony-stimulating factor (GM-CSF; P = .02 and FDR = 0.09), interferon α2 (IFN-α2; P < .001 and FDR = 0.004), interleukin 7 (IL-7; P = .004 and FDR = 0.03), interferon-inducible T-cell α chemoattractant (I-TAC; P = .001 and FDR = 0.02), I-309 (P = .01 and FDR = 0.04), and granulocyte colony-stimulating factor (G-CSF; P = .001 and FDR = 0.01). In contrast, levels of only 2 cytokines and chemokines, eotaxin 2 (P = .001 and FDR = 0.002) and thymus- and activation-regulation chemokine (TARC; P = .003 and FDR = 0.03), were significantly increased after HAART in cases. Similar trends were observed in controls, except for concentrations of interleukin 20 (IL-20) and interferon λ1, which significantly decreased (P = .04 and 0.03, respectively), and of leukemia inhibitory factor (LIF), which exhibited a decreasing trend. After correction for multiple comparisons, differences between cases and controls for interleukin 8 (IL-8), IL-20, and LIF concentrations were not significant.
Figure 2.

Impact of human immunodeficiency virus (HIV) treatment on cytokine and chemokine levels in subjects positive for both HIV and GB virus C (GBV-C; cases) and subjects positive for HIV and negative for GBV-C (controls). A, For the 25 cases and 18 controls who initiated highly active antiretroviral therapy (HAART) during the Viral Activation Transfusion Study, visit spacing was reorganized around the first day of HAART use. Red dots are for the first visit with a GBV-C RN–positive sample. B, Median percentage changes in cytokine/chemokine levels after initiation of HAART are represented for cases and controls.*P < .05, **P < .1, for comparison of cases to controls. Abbreviations: G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte macrophage colony-stimulating factor; IFN-α2, interferon α2; IFN-λ1, interferon λ1; IL-6, interleukin 6; IL-7, interleukin 7; IL-8, interleukin 8; IL-10, interleukin 10; IL-19, interleukin 19; IL-20, interleukin 20; I-TAC, interferon-inducible T-cell α chemoattractant; LIF, leukemia inhibitory factor; TARC, thymus- and activation-regulation chemokine; TNF-α, tumor necrosis factor α.
Significant Modulation of 27 Cytokines and Chemokines Over the Year Following GBV-C Infection
Cytokine and chemokine concentrations in the 23 cases for whom longitudinal samples were available after GBV-C detection (excluding the 7 cases for whom GBV-C was detected in the last tested sample) were determined at baseline and, on average, 100 and 300 days after GBV-C detection to evaluate the early and late effect, respectively, of GBV-C infection (Figure 3). Significant changes over time were found for concentrations of 27 cytokines and chemokines, with most of them decreasing, including those of 2 antiinflammatory cytokines (IL-10 and interleukin 1 receptor α chain [IL-1Rα]), 12 proinflammatory cytokines (GM-CSF, interferon γ [IFN-γ], IFN-α2, interleukin 12p40, interleukin 12p70 [IL-12p70], interleukin 15, interleukin 1α [IL-1α], interleukin 2 [IL-2], IL-7, IL-8, TNF-α, and IL-6), 8 chemokines (interferon γ–inducible protein 10 [IP-10], monocyte chemoattractant protein 1, eotaxin 2, 6Ckine, I-TAC, granulocyte chemoattractant protein 2 [GCP-2], macrophage inflammatory protein 1α [MIP-1α], and monokine induced by interferon γ), and 5 growth factors (epidermal growth factor, G-CSF, macrophage colony-stimulating factor [M-CSF], thrombopoietin, and vascular endothelial growth factor 1; Supplementary Table 1). The level of only 1 parameter, the chemoattractant eotaxin 2, significantly increased over time after GBV-C detection.
Figure 3.

Changes in cytokine and chemokine concentrations over time after transfusion among subjects positive for both human immunodeficiency virus (HIV) and GB virus C (GBV-C; cases) and subjects positive for HIV and negative for GBV-C (controls). Changes in cytokine and chemokine concentrations over time after transfusion in cases and controls. Changes in cytokine and chemokine concentrations are represented across 3 time points for 23 cases (baseline and approximately 100 and 300 days after GBV-C detection) and across 2 time points for controls (baseline and approximately 400 days from first visit). Cytokine and chemokine mean concentrations with significant changes are displayed in solid lines, and those with nonsignificant changes over time are displayed in dashed lines. Cytokines and chemokines are only represented in these graphs if a significant change is observed within either cases or controls. Abbreviations: EGF, epidermal growth factor; GCP-2, granulocyte chemoattractant protein 2; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte macrophage colony-stimulating factor; IFN-α2, interferon α2; IFN-γ, interferon γ; IL-1Rα, interleukin 1 receptor α chain; IL-1α, interleukin 1α; IL-2, interleukin 2; IL-6, interleukin 6; IL-7, interleukin 7; IL-8, interleukin 8; IL-10, interleukin 10; IL-12p40, interleukin 12p40; IL-12p70, interleukin 12p70; IL-15, interleukin 15; IP-10, interferon γ–inducible protein 10; I-TAC, interferon-inducible T-cell α chemoattractant; MCP-1, monocyte chemoattractant protein 1; M-CSF, macrophage colony-stimulating factor; MIG, monokine induced by interferon γ; MIP-1α, macrophage inflammatory protein 1α; TARC, thymus- and activation-regulation chemokine; TNF-α, tumor necrosis factor α; TPO, thrombopoietin; VEGF, vascular endothelial growth factor.
In contrast, concentrations of only 4 cytokines and chemokines were significantly altered in the controls on the last visit (approximately 400 days after transfusion), compared with their baseline values (with levels of 1 antiinflammatory cytokine decreasing [IL-10, P = .0001 and FDR = 0.008], 1 proinflammatory cytokine increasing [IL-6, P = .002 and FDR = 0.03], and 2 chemoattractants increasing [TARC, P = .002 and FDR = 0.03; and eotaxin 2, P = .001 and FDR = 0.03]; Figure 3). Heat map representation of the x-fold changes for each individual within each group highlights the cytokine and chemokine modulation over the acute phase of GBV-C infection and the year after GBV-C detection, for cases, and from baseline to the last visit, for controls (Supplementary Figure 2). A significant change was observed within proinflammatory cytokines, with higher enrichment of upregulation observed in controls, compared with cases, in the acute phase (100 days after GBV-C diagnosis) and beyond the acute phase (300 days after GBV-C diagnosis) of GBV-C infection. There was a 2-fold upregulation in 20% of the controls, compared with 11% and 10% of the cases at 100 and 300 days, respectively, after GBV-C detection (P < .001, by the Fisher exact test).
Pathway analysis was performed to help predict the effects that the observed cytokine changes might have on the host immune system. This analysis showed a decrease in pathways associated with chemoattractants for blood cells, leukocytes, phagocytes, and myeloid cells; in the differentiation and survival of hematopoietic cells and progenitors; and in the migration of monocyte-derived dendritic cells in controls. This was accompanied by an increase in pathways associated with cell death and apoptosis; in the migration and proliferation of eosinophils, myeloid cells, leukocytes, and endothelial cells; and in the function of antigen-presenting cells, generation of leukocytes, and infection of lymphocytes (Figure 4A). Cases had an increase in pathways associated with cell death and apoptosis of various cells and in the development of phagocytes and function of antigen-presenting cells and a decrease in binding, migration, and movement of cells. Similarly, 300 days after GBV-C infection diagnosis, there was a further decrease in cellular activation (peripheral blood mononuclear cells [PBMCs] and myeloid cells) and cellular trafficking, with an increase in the proliferation of myeloid progenitor cells and leukocyte infection (Figure 4B).
Figure 4.
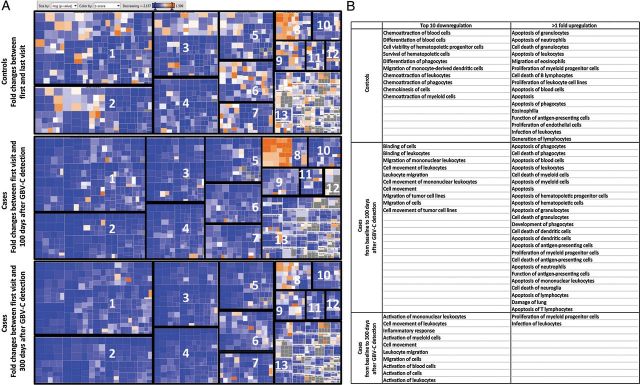
Impact of GB virus C (GBV-C) and human immunodeficiency virus (HIV) infection on cell functions and pathways. A, Fold changes (z scores) in cytokine and chemokine concentrations from baseline to 100 days and from baseline to 300 days after GBV-C detection, for subjects positive for both HIV and GBV-C (cases), and from baseline to the last visit, for subjects positive for HIV and negative for GBV-C (controls), were imputed into Qiagen Ingenuity Pathway Analysis (IPA) software. IPA generated a heat map in which cell functions and pathways are represented by squares sized by –log P value and colored by z score in blue, for negative scores, and in red, for positive scores. Cell functions and pathways are labeled 1–13 and defined as follows: 1, hematological system development and function; 2, cellular movement; 3, immune cell trafficking; 4, cell to cell signaling and interaction; 5, inflammatory response; 6, cellular growth and proliferation; 7, cellular development; 8, cell death and survival; 9, tissue development; 10, hematopoiesis; 11, cellular function; 12, cell-mediated immunity; and 13, others. B, The top 10 pathways and cell functions downregulated in controls and cases and those upregulated (>1-fold increase) are listed.
10 Cytokines and Chemokines Were Significantly Associated With GBV-C/HIV Coinfection, After Adjustment for Covariates
Multivariable mixed modeling adjusted for factors expected to impact the relationship between GBV-C status and cytokines and chemokines, including days from baseline, HAART status, and HIV viral load. After adjusted modeling, GBV-C infection was significantly associated with decreases in levels of 9 cytokines: 2 antiinflammatory cytokines (IL-10, P = .02; and IL-1Rα, P = .03), 2 proinflammatory cytokines (IL-6, P < .01; and IL-7, P = .02), 4 chemoattractants (MIP-1α, P = .02; 6Ckine, P < .01; I-TAC, P = .02; and GCP-2, P < .01), and the growth factor stem cell factor (SCF; P = .01; Table 2). GBV-C infection was associated with a significant increase of the chemoattractant eotaxin 2 (P = .04), independent of covariates. After correction for multiple comparisons, 7 cytokines remained significantly associated with GBV-C infection (Table 2, FDR ≤ 0.2).
Table 2.
Multivariable Analysis for the Association Between GB Virus C (GBV-C) and Cytokines, Adjusting for Days From Baseline, Highly Active Antiretroviral Therapy (HAART) Status, and Human Immunodeficiency Virus (HIV) Viral Load
| Cytokine | GBV-C Status |
HAART Status |
HIV Viral Load |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Slope | P Value | FDR | Slope | P Value | FDR | Slope | P Value | FDR | |
| Antiinflammatory | |||||||||
| IL-4 | 0.46 | .2 | 0.51 | 0.24 | .4 | 0.90 | −0.37 | .007 | 0.05 |
| IL-10a | −0.53 | .02 | 0.19 | −0.44 | .02 | 0.30 | 0.07 | .4 | 0.63 |
| IL-13a | −0.11 | .5 | 0.65 | 0.16 | .2 | 0.71 | 0.13 | .04 | 0.14 |
| IL-1Rα | −0.63 | .03 | 0.24 | −0.05 | .8 | 0.95 | −0.04 | .7 | 0.83 |
| TRAIL | 0.18 | .1 | 0.42 | 0.25 | .009 | 0.24 | 0.19 | <.0001 | 0.004 |
| Proinflammatory | |||||||||
| GM-CSF | −0.11 | .4 | 0.63 | −0.12 | .3 | 0.83 | −0.18 | .001 | 0.02 |
| IFN-γ | −0.02 | .9 | 0.95 | −0.10 | .5 | 0.90 | −0.13 | .03 | 0.14 |
| IL-12p70 | −0.08 | .6 | 0.78 | −0.01 | .9 | 0.97 | −0.18 | .005 | 0.04 |
| IL-1β | 0.04 | .8 | 0.93 | −0.06 | .7 | 0.91 | −0.25 | .001 | 0.01 |
| IL-6 | −0.65 | .003 | 0.13 | −0.11 | .6 | 0.91 | −0.08 | .3 | 0.56 |
| IL-15a | −0.16 | .4 | 0.59 | −0.29 | .046 | 0.48 | 0.06 | .4 | 0.58 |
| IL-1α | 0.22 | .3 | 0.57 | −0.24 | .2 | 0.70 | 0.24 | .005 | 0.04 |
| IL-7a | −0.38 | .02 | 0.19 | 0.06 | .7 | 0.91 | 0.00 | 1.0 | 0.99 |
| ENA-78 | −0.14 | .4 | 0.60 | 0.23 | .1 | 0.56 | −0.15 | .02 | 0.12 |
| IL-33 | 0.14 | .2 | 0.47 | 0.16 | .049 | 0.48 | 0.06 | .08 | 0.25 |
| Chemoattractant | |||||||||
| Eotaxin | 0.01 | .9 | 0.95 | 0.02 | .7 | 0.91 | −0.07 | .02 | 0.12 |
| IP-10 | −0.01 | .9 | 0.95 | −0.20 | .09 | 0.56 | 0.17 | .002 | 0.02 |
| MIP-1α | −0.36 | .02 | 0.20 | −0.21 | .1 | 0.56 | −0.08 | .2 | 0.34 |
| MIP-1β | −0.13 | .2 | 0.47 | −0.03 | .7 | 0.91 | −0.10 | .005 | 0.04 |
| BCA-1/CXCL-13 | 0.04 | .8 | 0.93 | −0.07 | .6 | 0.91 | 0.14 | .027 | 0.14 |
| Eotaxin-2 | 0.25 | .04 | 0.28 | 0.13 | .2 | 0.70 | −0.28 | <.0001 | 0.004 |
| MCP-2/CCL-8 | 0.00 | 1.0 | 0.99 | 0.05 | .6 | 0.91 | 0.13 | .001 | 0.01 |
| MCP-4 | −0.03 | .7 | 0.85 | 0.06 | .4 | 0.88 | −0.06 | .035 | 0.14 |
| TARC | −0.22 | .2 | 0.52 | 0.47 | .004 | 0.20 | −0.16 | .027 | 0.14 |
| 6Ckine | −0.25 | .004 | 0.13 | 0.11 | .1 | 0.56 | 0.03 | .4 | 0.56 |
| CCL-19/MIP-3β | −0.13 | .3 | 0.54 | 0.04 | .7 | 0.91 | 0.09 | .032 | 0.14 |
| CXCL-11/I-TAC | −0.32 | .02 | 0.19 | −0.10 | .4 | 0.88 | 0.11 | .030 | 0.14 |
| CXCL-6/GCP-2 | −0.26 | .007 | 0.13 | 0.00 | 1.0 | 0.98 | −0.05 | .2 | 0.31 |
| Growth factor | |||||||||
| SCF | −0.40 | .01 | 0.19 | 0.38 | .005 | 0.20 | 0.09 | .1 | 0.29 |
| M-CSF | −0.37 | .1 | 0.42 | 0.25 | .2 | 0.70 | 0.31 | .001 | 0.01 |
Each time-varying covariate's estimates were jointly adjusted for the presence of all other covariates. Each model was additionally adjusted for individual subject variation. All subjects were included in modeling. Visits following a case's first GBV-C–positive diagnosis were considered GBV-C positive, while visits for controls were coded as GBV-C negative. P values of < .05 and FDRs of ≤ 0.2 are statistically significant.
Abbreviations: BCA-1, B-cell–attracting chemokine 1; ENA-78, epithelial cell–derived neutrophil attractant 78; FDR, false discovery rate; GCP-2, granulocyte chemoattractant protein 2; GM-CSF, granulocyte macrophage colony-stimulating factor; IFN-γ, interferon γ; IL-1Rα, interleukin 1 receptor α chain; IL-1α, interleukin 1α; IL-1β, interleukin 1β; IL-4, interleukin 4; IL-6, interleukin 6; IL-7, interleukin 7; IL-10, interleukin 10; IL-12p70, interleukin 12p70; IL-13, interleukin 13; IL-15, interleukin 15; IL-33, interleukin 33; IP-10, interferon γ–inducible protein 10; I-TAC, interferon-inducible T-cell α chemoattractant; MCP-2, monocyte chemoattractant protein 2; MCP-4, monocyte chemoattractant protein 4; M-CSF, macrophage colony-stimulating factor; MIP-1α, macrophage inflammatory protein 1α; MIP-1β, macrophage inflammatory protein 1β; MIP-3β, macrophage inflammatory protein 3β; SCF, stem cell factor; TARC, thymus- and activation-regulation chemokine.
a Tested in duplicate in the HCYTMAG-60K-PX29 and HSCYTO-60SPMX13 assays with data obtained with the HCYTMAG-60K-PX29 assay displayed in this table.
The decrease in IL-10 level was associated with GBV-C infection, as well as with HAART use, while the SCF level was decreased in those with GBV-C infection and increased in response to HAART use (Figure 5). The eotaxin 2 level was inversely correlated with HIV viral load and significantly increased after GBV-C infection, while I-TAC was downregulated after GBV-C infection and correlated with HIV viral load. Independently from GBV-C infection, TRAIL was positively associated with HIV viral load but also with HAART use, while TARC had a negative relationship with HIV viral load and was positively associated with HAART use (Figure 5). HIV viral load was also significantly associated with decreased levels of interleukin 4, GM-CSF, IFN-γ, IL-12p70, interleukin 1β, epithelial cell–derived neutrophil attractant 78, eotaxin, macrophage inflammatory protein 1β (MIP-1β), and monocyte chemoattractant protein 4 and with increased levels of interleukin 13, IL-1α, IP-10, B-cell–attracting chemokine 1, monocyte chemoattractant protein 2, macrophage inflammatory protein 3β, and M-CSF.
Figure 5.

Proposed model for the impact of GB virus C (GBV-C) infection, human immunodeficiency virus (HIV) infection, and highly active antiretroviral therapy (HAART) on cytokines and chemokines. A multivariable mixed model adjusted for factors thought to impact the relationship between GBV-C status and cytokines and chemokines. After adjusted modeling, cytokines and chemokines impacted by GBV-C infection are shown in the orange circle, those modulated by HIV infection are shown in the blue rectangle, and those modulated by HAART are shown in the green circle. The upward-angled symbols are for upregulation, and the downward-angled symbols are for downregulation. The arrows are also colored to correspond to GBV-C (orange), HIV (blue), and HAART (green). Abbreviations: BCA-1, B-cell–attracting chemokine 1; ENA-78, epithelial cell–derived neutrophil attractant 78; GCP-2, granulocyte chemoattractant protein 2; GM-CSF, granulocyte macrophage colony-stimulating factor; IFN-γ, interferon γ; IL-1Rα, interleukin 1 receptor α chain; IL-1α, interleukin 1α; IL-1β, interleukin 1β; IL-4, interleukin 4; IL-6, interleukin 6; IL-7, interleukin 7; IL-10, interleukin 10; IL-12p70, interleukin 12p70; IL-13, interleukin 13; IL-15, interleukin 15; IL-33, interleukin 33; IP-10, interferon γ–inducible protein 10; I-TAC, interferon-inducible T-cell α chemoattractant; MCP-2, monocyte chemoattractant protein 2; MCP-4, monocyte chemoattractant protein 4; M-CSF, macrophage colony-stimulating factor; MIP-1α, macrophage inflammatory protein 1α; MIP-1β, macrophage inflammatory protein 1β; MIP-3β, macrophage inflammatory protein 3β; SCF, stem cell factor; TARC, thymus- and activation-regulation chemokine.
DISCUSSION
Accessing plasma samples collected before and at various time points after inadvertent transfusion-transmission of GBV-C allowed for a longitudinal investigation of the cytokine and chemokine responses through and beyond the acute phase of GBV-C infection in 30 cases and 30 controls. The analysis of HIV viral load and CD4+ T-cell count confirmed that cases had a better HIV disease outcome than controls, thus substantiating our previous report on the effect of GBV-C infection in the VATS cohort [25].
When immune parameter concentrations were evaluated longitudinally over time after transfusion, more cytokines and chemokines were altered by GBV-C infection in cases than in controls. Levels of most of the modulated cytokines and chemokines were reduced after GBV-C detection, including those of many proinflammatory cytokines, suggesting an overall antiinflammatory effect of GBV-C after coinfection in HIV-positive subjects. Additionally, there was an overall downregulation of immune pathways and functions 100 and 300 days following GBV-C detection, compared with baseline, and an increase in pathways associated with cell death in the cellular subsets that may support HIV replication. As proinflammatory cytokines such as type I interferons can prolong survival of activated T cells [41], it is perhaps not surprising that increased cell death is found to be associated with the decrease of proinflammatory cytokine levels in GBV-C–infected subjects, assuming that activated and not naive cells are dying.
The VATS enrolled late-stage, anemic, HIV-positive subjects with different HIV viral loads and treatment regimens at baseline and then followed subjects over time after transfusion, with heterogeneity in visit spacing. Recruitment for the VATS started before HAART was implemented and continued after HAART became available. Therefore, a multivariable analysis that adjusted for HIV viral load, time since baseline, HAART use, and subject allowed us to identify cytokines and chemokines independently associated with GBV-C infection. Significant associations between GBV-C infection and decreased levels of IL-1Rα, IL-6, IL-7, IL-10, MIP-1α, 6Ckine, SCF, I-TAC, and GCP-2 and increased level of eotaxin 2 were observed, independent of HAART use and HIV viral load.
The kinetics of acute GBV-C viremia and the immune response following infection or persistent GBV-C viremia are not well described. GBV-C RNA can be detected as early as 7 days after infection [13]. Of note, 29 of 30 cases sustained GBV-C viremia throughout the year after GBV-C infection and up to 2 years after GBV-C infection. Therefore, we are reporting the association between GBV-C and decreased levels of cytokines and chemokines in a group of HIV-positive individuals with sustained GBV-C viremia. Other studies have reported GBV-C RNA detection increases after the implementation of HAART [24, 42], months after potential transfusion-transmitted GBV-C infection in some cases. Our results are consistent with the observation that GBV-C viral load and HIV viral load are inversely correlated [43].
Several studies report in vitro and in vivo evidence of interaction between GBV-C and HIV. GBV-C replicates mostly in lymphocytes and could interfere with HIV replication by different possible mechanisms; GBV-C has been shown to downregulate the expression of HIV entry receptors CCR5 and CXCR4 by triggering the production of chemokines such as regulated upon activation, normal T-cell expressed (RANTES), MIP-1α, MIP-1β, and stromal cell–derived factor 1 (SDF-1) in GBV-C–infected PBMCs [12]. The binding of RANTES, MIP-1α, and MIP-1β to CCR5 and of SDF-1 to CXCR4 would prevent the binding of HIV to its coreceptors and its replication. Recent studies have investigated the effect of GBV-C coinfection on the host immune system [44]. An increase of naive T-cell frequency and a decrease of CD4+ and CD8+ T-cell activation has been demonstrated in HIV-infected individuals, irrespective of their ART exposure [27, 30]. GBV-C was associated with a reduced basal level of immune activation, T-cell activation, and proliferation markers in vivo in HIV-infected people [33] and in vitro [32]. It is also suggested that GBV-C infection induces non–T-cell immune responses and reduces NK-cell, monocyte, and B-cell activation markers [45]. Alteration of the T-helper type 1 (Th1)–Th2 cytokine profiles, interaction with T-cell activation, and blockage of IL-2–mediated CD4+ T-cell proliferation have also been suggested [46–48].
The findings in this study suggest an independent mechanism of GBV-C leading to decreased inflammation and improved HIV disease outcome in individuals coinfected with GBV-C and HIV, consistent with a protective role of GBV-C during HIV infection. The more dramatic changes in cytokine levels seen in the controls were dampened in many of the cases with decreases in both proinflammatory and antiinflammatory signals. A possible explanation is that the decrease in proinflammatory signals was followed by a decrease in antiinflammatory signals in a negative feedback loop.
Although pathway analyses revealed that most were downregulated in cases at 100 and 300 days after GBV-C infection, cell death pathways were upregulated in various cell subsets in the earlier time point. Further investigation will be needed to understand the cause and effect of cytokine-virus interactions. On one hand, it would be important to know whether GBV-C has a silent deleterious effect on the host at the cellular level. On the other hand, during acute GBV-C infection, the depletion of activated or proliferating T cells [30], which are potential targets for HIV, along with a decrease in T-cell activation [32], could decrease the targets for HIV infection and reduce the risk of acquisition of HIV infection and/or downstream viral dissemination and replication manifested in set point HIV viral load.
STUDY GROUP PARTICIPANTS
Participating REDS-III investigators are as follows: program office, Simone Glynn (National Heart, Lung, and Blood Institute, NIH, Bethesda, Maryland); data coordinating center, Don Brambilla and Susan Sullivan (RTI international, Rockville, Maryland); and central laboratory, Michael P. Busch and Phillip J. Norris (Blood Systems Research Institute, San Francisco, California). Additional participating investigators and centers are as follows: Marion C. Lanteri, Farnaz Vahidnia, John Heitman, Xutao Deng, Sheila M. Keating, and Brian Custer (Blood Systems Research Institute); Sylvia Tan (RTI International); and Jack T. Stapleton (Iowa City Veterans Affairs Hospital and University of Iowa Carver College of Medicine, Iowa City, Iowa).
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgment. We thank Donna Klinzman (Iowa City, Virginia), for technical assistance.
Financial support. This work was supported by the National Heart, Lung, and Blood Institute, National Institutes of Health (contract HHSN268201100001); and the Merit Review Award Program (grants BX000207 and CX000821 to J. T. S.).
Potential conflicts of interest. All authors: No reported conflicts.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
Contributor Information
Collaborators: for the NHLBI REDS III Study, Simone Glynn, Don Brambilla, Susan Sullivan, Michael P. Busch, Phillip J. Norris, Marion C. Lanteri, Farnaz Vahidnia, John Heitman, Xutao Deng, Sheila M. Keating, Brian Custer, Sylvia Tan, and Jack T. Stapleton
References
- 1.Adams MJ, King AM, Carstens EB. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses. Arch Virol 2013; 158:2023–30. [DOI] [PubMed] [Google Scholar]
- 2.Stapleton JT, Foung S, Muerhoff AS, Bukh J, Simmonds P. The GB viruses: a review and proposed classification of GBV-A, GBV-C (HGV), and GBV-D in genus Pegivirus within the family Flaviviridae. J Gen Virol 2011; 92:233–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chivero ET, Bhattarai N, Rydze RT, Winters MA, Holodniy M, Stapleton JT. Human pegivirus RNA is found in multiple blood mononuclear cells in vivo and serum-derived viral RNA-containing particles are infectious in vitro. J Gen Virol 2014; 95:1307–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.George SL, Varmaz D, Stapleton JT. GB virus C replicates in primary T and B lymphocytes. J Infect Dis 2006; 193:451–4. [DOI] [PubMed] [Google Scholar]
- 5.Williams CF, Klinzman D, Yamashita TE, et al. Persistent GB virus C infection and survival in HIV-infected men. N Engl J Med 2004; 350:981–90. [DOI] [PubMed] [Google Scholar]
- 6.Tillmann HL, Heringlake S, Trautwein C, et al. Antibodies against the GB virus C envelope 2 protein before liver transplantation protect against GB virus C de novo infection. Hepatology 1998; 28:379–84. [DOI] [PubMed] [Google Scholar]
- 7.Bhanich Supapol W, Remis RS, Raboud J, et al. Mother-to-child transmission of GB virus C in a cohort of women coinfected with GB virus C and HIV in Bangkok, Thailand. J Infect Dis 2009; 200:227–35. [DOI] [PubMed] [Google Scholar]
- 8.Lefrere JJ, Roudot-Thoraval F, Morand-Joubert L, et al. Prevalence of GB virus type C/hepatitis G virus RNA and of anti-E2 in individuals at high or low risk for blood-borne or sexually transmitted viruses: evidence of sexual and parenteral transmission. Transfusion 1999; 39:83–94. [DOI] [PubMed] [Google Scholar]
- 9.Scallan MF, Clutterbuck D, Jarvis LM, Scott GR, Simmonds P. Sexual transmission of GB virus C/hepatitis G virus. J Med Virol 1998; 55:203–8. [DOI] [PubMed] [Google Scholar]
- 10.Rey D, Vidinic-Moularde J, Meyer P, et al. High prevalence of GB virus C/hepatitis G virus RNA and antibodies in patients infected with human immunodeficiency virus type 1. Eur J Clin Microbiol Infect Dis 2000; 19:721–4. [DOI] [PubMed] [Google Scholar]
- 11.Bourlet T, Guglielminotti C, Evrard M, et al. Prevalence of GBV-C/hepatitis G virus RNA and E2 antibody among subjects infected with human immunodeficiency virus type 1 after parenteral or sexual exposure. J Med Virol 1999; 58:373–7. [PubMed] [Google Scholar]
- 12.Stapleton JT, Williams CF, Xiang J. GB virus type C: a beneficial infection? J Clin Microbiol 2004; 42:3915–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tacke M, Schmolke S, Schlueter V, et al. Humoral immune response to the E2 protein of hepatitis G virus is associated with long-term recovery from infection and reveals a high frequency of hepatitis G virus exposure among healthy blood donors. Hepatology 1997; 26:1626–33. [DOI] [PubMed] [Google Scholar]
- 14.Alter HJ. G-pers creepers, where'd you get those papers? A reassessment of the literature on the hepatitis G virus. Transfusion 1997; 37:569–72. [DOI] [PubMed] [Google Scholar]
- 15.Alter HJ. Emerging, re-emerging and submerging infectious threats to the blood supply. Vox Sang 2004; 87(suppl 2):56–61. [DOI] [PubMed] [Google Scholar]
- 16.Laskus T, Radkowski M, Wang LF, Vargas H, Rakela J. Detection of hepatitis G virus replication sites by using highly strand-specific Tth-based reverse transcriptase PCR. J Virol 1998; 72:3072–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linnen J, Wages J, Jr, Zhang-Keck ZY, et al. Molecular cloning and disease association of hepatitis G virus: a transfusion-transmissible agent. Science 1996; 271:505–8. [DOI] [PubMed] [Google Scholar]
- 18.Krajden M, Yu A, Braybrook H, et al. GBV-C/hepatitis G virus infection and non-Hodgkin lymphoma: a case control study. Int J Cancer 2010; 126:2885–92. [DOI] [PubMed] [Google Scholar]
- 19.Stapleton JT, Chaloner K. GB virus C infection and non-Hodgkin lymphoma: important to know but the jury is out. Int J Cancer 2010; 126:2759–61. [DOI] [PubMed] [Google Scholar]
- 20.Chang CM, Stapleton JT, Klinzman D, et al. GBV-C infection and risk of NHL among U.S. adults. Cancer Res 2014; 74:5553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heuft HG, Berg T, Schreier E, et al. Epidemiological and clinical aspects of hepatitis G virus infection in blood donors and immunocompromised recipients of HGV-contaminated blood. Vox Sang 1998; 74:161–7. [PubMed] [Google Scholar]
- 22.Vahidnia F, Petersen M, Rutherford G, et al. Transmission of GB virus type C via transfusion in a cohort of HIV-infected patients. J Infect Dis 2012; 205:1436–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spellberg B, Edwards JE., Jr Type 1/Type 2 immunity in infectious diseases. Clin Infect Dis 2001; 32:76–102. [DOI] [PubMed] [Google Scholar]
- 24.Tillmann HL, Heiken H, Knapik-Botor A, et al. Infection with GB virus C and reduced mortality among HIV-infected patients. N Engl J Med 2001; 345:715–24. [DOI] [PubMed] [Google Scholar]
- 25.Vahidnia F, Petersen M, Stapleton JT, Rutherford GW, Busch M, Custer B. Acquisition of GB virus type C and lower mortality in patients with advanced HIV disease. Clin Infect Dis 2012; 55:1012–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gretch D. Editorial commentary: advocating the concept of GB virus C biotherapy against AIDS. Clin Infect Dis 2012; 55:1020–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maidana-Giret MT, Silva TM, Sauer MM, et al. GB virus type C infection modulates T-cell activation independently of HIV-1 viral load. AIDS 2009; 23:2277–87. [DOI] [PubMed] [Google Scholar]
- 28.Nattermann J, Nischalke HD, Kupfer B, et al. Regulation of CC chemokine receptor 5 in hepatitis G virus infection. AIDS 2003; 17:1457–62. [DOI] [PubMed] [Google Scholar]
- 29.Schwarze-Zander C, Neibecker M, Othman S, et al. GB virus C coinfection in advanced HIV type-1 disease is associated with low CCR5 and CXCR4 surface expression on CD4(+) T-cells. Antivir Ther 2010; 15:745–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stapleton JT, Chaloner K, Martenson JA, et al. GB virus C infection is associated with altered lymphocyte subset distribution and reduced T cell activation and proliferation in HIV-infected individuals. PLoS One 2012; 7:e50563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stapleton JT, Martinson JA, Klinzman D, Xiang J, Desai SN, Landay A. GB virus C infection and B-cell, natural killer cell, and monocyte activation markers in HIV-infected individuals. AIDS 2013; 27:1829–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhattarai N, McLinden JH, Xiang J, Landay AL, Chivero ET, Stapleton JT. GB virus C particles inhibit T cell activation via envelope E2 protein-mediated inhibition of TCR signaling. J Immunol 2013; 190:6351–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhattarai N, Rydze RT, Chivero ET, Stapleton JT. GB virus C viremia is associated with higher levels of double-negative T cells and lower T-cell activation in HIV-infected individuals receiving antiretroviral therapy. J Infect Dis 2012; 206:1469–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McLinden JH, Stapleton JT, Klinzman D, et al. Chimpanzee GB virus C and GB virus A E2 envelope glycoproteins contain a peptide motif that inhibits human immunodeficiency virus type 1 replication in human CD4(+) T-cells. J Gen Virol 2013; 94:774–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rydze RT, Bhattarai N, Stapleton JT. GB virus C infection is associated with a reduced rate of reactivation of latent HIV and protection against activation-induced T-cell death. Antivir Ther 2012; 17:1271–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Busch MP, Collier A, Gernsheimer T, et al. The Viral Activation Transfusion Study (VATS): rationale, objectives, and design overview. Transfusion 1996; 36:854–9. [DOI] [PubMed] [Google Scholar]
- 37.Collier AC, Kalish LA, Busch MP, et al. Leukocyte-reduced red blood cell transfusions in patients with anemia and human immunodeficiency virus infection: the viral activation transfusion study: a randomized controlled trial. JAMA 2001; 285:1592–601. [DOI] [PubMed] [Google Scholar]
- 38.Souza IE, Allen JB, Xiang J, et al. Effect of primer selection on estimates of GB virus C (GBV-C) prevalence and response to antiretroviral therapy for optimal testing for GBV-C viremia. J Clin Microbiol 2006; 44:3105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stapleton JT, Chaloner K, Zhang J, et al. GBV-C viremia is associated with reduced CD4 expansion in HIV-infected people receiving HAART and interleukin-2 therapy. AIDS 2009; 23:605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiang J, Wunschmann S, Schmidt W, Shao J, Stapleton JT. Full-length GB virus C (Hepatitis G virus) RNA transcripts are infectious in primary CD4-positive T cells. J Virol 2000; 74:9125–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Starbeck-Miller GR, Xue HH, Harty JT. IL-12 and type I interferon prolong the division of activated CD8 T cells by maintaining high-affinity IL-2 signaling in vivo. J Exp Med 2014; 211:105–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bjorkman P, Flamholc L, Molnegren V, Marshall A, Guner N, Widell A. Enhanced and resumed GB virus C replication in HIV-1-infected individuals receiving HAART. AIDS 2007; 21:1641–3. [DOI] [PubMed] [Google Scholar]
- 43.Tillmann HL, Manns MP. GB virus-C infection in patients infected with the human immunodeficiency virus. Antiviral Res 2001; 52:83–90. [DOI] [PubMed] [Google Scholar]
- 44.Bhattarai N, Stapleton JT. GB virus C: the good boy virus? Trends Microbiol 2012; 20:124–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stapleton JT, Jeffrey A. Martinson, Donna Klinzman, Jinhua Xiang, Desai SN, Alan Landay. GB virus C infection and B-cell, natural killer cell, and monocyte activation markers in HIV-infected individuals. AIDS 2013; 27:1829–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhattarai N, McLinden JH, Xiang J, Kaufman TM, Stapleton JT. GB virus C envelope protein E2 inhibits TCR-induced IL-2 production and alters IL-2–signaling pathways. J Immunol 2012; 189:2211–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rydze RT, Xiang J, McLinden JH, Stapleton JT. GB virus type C infection polarizes T-cell cytokine gene expression toward a Th1 cytokine profile via NS5A protein expression. J Infect Dis 2012; 206:69–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nunnari G, Nigro L, Palermo F, et al. Slower progression of HIV-1 infection in persons with GB virus C co-infection correlates with an intact T-helper 1 cytokine profile. Ann Intern Med 2003; 139:26–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.