

Abstract
Hyperactivation of β-catenin–T-cell-factor (TCF)-regulated gene transcription is a hallmark of colorectal cancer (CRC). The cell-neural adhesion molecule L1CAM (hereafter referred to as L1) is a target of β-catenin–TCF, exclusively expressed at the CRC invasive front in humans. L1 overexpression in CRC cells increases cell growth and motility, and promotes liver metastasis. Genes induced by L1 are also expressed in human CRC tissue but the mechanisms by which L1 confers metastasis are still unknown. We found that signaling by the nuclear factor κB (NF-κB) is essential, because inhibition of signaling by the inhibitor of κB super repressor (IκB-SR) blocked L1-mediated metastasis. Overexpression of the NF-κB p65 subunit was sufficient to increase CRC cell proliferation, motility and metastasis. Binding of the L1 cytodomain to ezrin – a cytoskeleton-crosslinking protein – is necessary for metastasis because when binding to L1 was interrupted or ezrin gene expression was suppressed with specific shRNA, metastasis did not occur. L1 and ezrin bound to and mediated the phosphorylation of IκB. We also observed a complex containing IκB, L1 and ezrin in the juxtamembrane region of CRC cells. Furthermore, we found that L1, ezrin and phosphorylated p65 are co-expressed at the invasive front in human CRC tissue, indicating that L1-mediated activation of NF-κB signaling involving ezrin is a major route of CRC progression.
Key words: Colon cancer, L1, Metastasis, NF-κB, IκB, Ezrin
Introduction
Studies of inherited and sporadic colorectal cancer (CRC) revealed the Wnt/β-catenin pathway (Kinzler and Vogelstein, 1996) as the primary mutation target, and mutations that lead to the hyperactivation of genes regulated by β-catenin–T-cell-factor (TCF) are among the first genetic changes and also a major cause for CRC development and progression (Clevers, 2006; Conacci-Sorrell et al., 2002a; Polakis, 2007). An important question in β-catenin-mediated oncogenesis is the identification and mode of action of genes targeted by β-catenin–TCF that are induced during invasive CRC formation. In addition to the β-catenin target genes that confer a growth advantage in cancer cells – such as cyclin D1 (Shtutman et al., 1999; Tetsu and McCormick, 1999) and MYC (He et al., 1998) – the accumulation of β-catenin in the nuclei of cancer cells continues with disease progression and is most evident at the invasive edge of CRC tissue (Brabletz et al., 1998; Brabletz et al., 2001). This indicates that β-catenin signaling also activates genes that are involved in later stages of CRC development. The changes in molecular and cellular properties that occur at the invasive front of CRC tissue cells are reminiscent of epithelial-to-mesenchymal transition (EMT) (Brabletz et al., 2005; Conacci-Sorrell et al., 2003; Gavert and Ben-Ze'ev, 2008; Thiery, 2002; Thiery and Sleeman, 2006), but the mechanisms driving these changes remain largely unknown. In recent studies, we identified genes encoding members of the neuronal L1-CAM family of immunoglobulin-like cell-adhesion receptors (the neural cell-adhesion molecule L1CAM – hereafter referred to as L1, and the neuron-glia-related cell-adhesion molecule NRCAM – hereafter referred to as Nr-CAM) as targets of β-catenin–TCF signaling in CRC cells (Conacci-Sorrell et al., 2002b; Gavert et al., 2005). We detected L1 in a small population of CRC cells at the invasive tumor front that displayed nuclear β-catenin (Gavert et al., 2005). Furthermore, forced expression of L1 in human CRC cells promotes their motility in vitro and metastatic capacity to the liver when injected into the spleen of nude mice (Gavert et al., 2007). However, the mechanisms by which L1 confers these dramatic changes in the behavior of CRC cells remain unclear.
The nuclear factor κB (NF-κB), known for its capacity to activate genes involved in immune response and inflammation but also in controlling oncogenesis (Bassères and Baldwin, 2006; Karin et al., 2002; Naugler and Karin, 2008), was recently implicated in both EMT and metastasis (Huber et al., 2004; Julien et al., 2007; Min et al., 2008; Solanas et al., 2008). In this study, we asked whether signaling by NF-κB is involved in eliciting the cellular changes induced by L1 that lead to invasive CRC development. We found that L1 induces NF-κB signaling in CRC cells and show that, by inhibiting NF-κB signaling in L1-expressing CRC cells, it is possible to block the metastatic capacity of these cells. We also found that this process requires the cytoskeletal crosslinking protein ezrin. Ezrin is a member of the ERM (ezrin, radixin, moesin) family of actin-associated proteins that have a key role in the formation of microvilli and other membrane protrusions (Bretscher et al., 2002). Several studies implicated the involvement of ezrin in the metastatic spread of various neoplasms (Khanna et al., 2004; Yu et al., 2004). We found that ezrin forms a complex with L1 and IκB in the juxtamembrane region, and that suppression of ezrin blocks the metastatic capacity of CRC cells. We demonstrate the presence of ezrin, L1 and the more active phosphorylated subunit p65 (p65-P) of NF-κB in human CRC tissue cells at the invasive front of tumors, and suggest that induction of NF-κB signaling and ezrin are key components in the mechanism(s) by which L1 promotes invasive metastatic colon cancer development.
Results
Expression of L1 in CRC cells induces NF-κB signaling by enhancing IκB phosphorylation
To determine whether L1 expression in CRC cells enhances signaling by NF-κB, a NF-κB-responsive reporter plasmid containing three copies of the NF-κB DNA-binding sequence linked to the luciferase gene (3xκB.luc) was introduced into Ls174T control cells and Ls174T cells stably transfected with L1. Levels of luciferase activity in these cells were then measured. Two independently derived L1-expressing cell clones (Gavert et al., 2007) displayed higher levels of NF-κB transactivation compared with control Ls174T cells that lacked L1 (Fig. 1A). L1 also induced a dramatic increase in NF-κB activity in HEK293T cells (Fig. 1B), indicating that its capacity to induce NF-κB signaling is not restricted to a single cell type. We also compared NF-κB signaling in SW620 CRC cells (Fig. 1C), which express L1endogenously, before and after suppressing L1 levels with specific small hairpin RNA (shRNA; Fig. 1D) and found that NF-κB signaling was reduced when L1 levels were suppressed (Fig. 1C). SW620 cells whose L1 levels were suppressed also displayed decreased tumorigenic capacity when injected subcutaneously into nude mice (supplementary material Fig. S1).
Fig. 1.
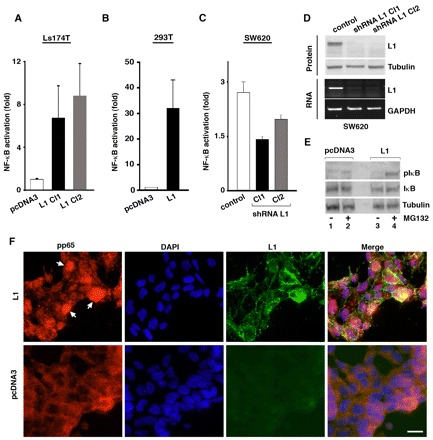
L1 induces NF-κB signaling and the phosphorylation of IκB in colon cancer cells. (A) The NF-κB-responsive reporter plasmid 3xκB.luc was transfected together with pSV β-galactosidase control vector (for transfection efficiency normalization) into human Ls174T CRC cells that do not express L1, and into two individually selected clones stably transfected to express L1 (L1 Cl1 and Cl2) and a control clone (pcDNA3). Fold NF-κB activation was determined after dividing luciferase activity by the values obtained with an empty reporter plasmid. (B) 293T cells were transfected as in A with either an empty pcDNA3 plasmid or with the same plasmid containing L1. Luciferase activity was then determined. (C) Human SW620 CRC cells that express endogenous L1, two individually selected clones stably transfected with shRNA targeting L1 (shRNA L1 Cl1 and Cl2) and a control clone (control) were transfected as in A, and luciferase activity was determined. (D) The cells described in C were analyzed for L1 RNA and protein levels by PCR and western blotting (bottom and top blots, respectively. (E) Ls174T cells stably transfected with the empty pcDNA3 vector or vector expressing L1 were either left untreated (lanes 1,3) or were treated with the proteasome inhibitor MG132 for 3 hours before harvesting (lanes 2,4). Levels of IκB total (IκB) and phosphorylated IκB (IκB-P) were determined by western blotting. Tubulin served as loading control. (F) Ls174T cells stably transfected with either pcDNA3 or vector expressing L1 were immunostained with anti-p65-P and anti-L1 antibodies (red and green staining, respectively). Nuclei were stained with DAPI (blue). Scale bar: 20 μm. pIκB is IκB-P and pp65 is p65-P.
A key step in the activation of NF-κB is the phosphorylation of the inhibitor of κB (IκB) leading to the release of NF-κB for nuclear translocation, and the polyubiquitylation and degradation of pIκB by the proteasome (Bassères and Baldwin, 2006; Karin and Ben-Neriah, 2008). In accordance with this mechanism, we detected in L1-expressing cells an increase in IκB phosphorylation (Fig. 1E; IκB-P, lane 4). Naturally, pIκB could only be detected when its degradation by the proteasome was inhibited by MG132 (Fig. 1E, lane 4). Employing immunofluorescence microscopy, we also observed the nuclear accumulation of p65-P, a phosphorylated (more active form) of the p65 NF-κB subunit (Perkins, 2006), in L1 expressing CRC cells (Fig. 1F, arrowheads), but not in control cells displaying diffuse cytoplasmic p65-P distribution. These results suggest that L1 expression induced nuclear translocation and increased activity of NF-κB signaling by a mechanism involving the classic pathway of IκB phosphorylation.
IκB-SR binds to and colocalizes with L1, thereby inhibiting CRC cell proliferation, motility and L1-mediated metastasis
L1 could be affecting NF-κB signaling by binding to IκB and releasing NF-κB for signaling in the nucleus. We wished to determine whether L1 binds to IκB thereby affecting NF-κB signaling. We isolated clones of CRC cells that stably expressed both L1 and the IκB super repressor (IκB-SR) – a stable point mutant of IκB (Fig. 2A, lanes 4,5) – and found that in such cells the L1-mediated induction of NF-κB signaling was suppressed (Fig. 2B). We then examined the localization of L1 and IκB-SR in CRC cells that stably expressed both proteins. Whereas IκB-SR was only localized in cytoplasmic aggregates in cells that expressed IκB-SR alone (Fig. 2C, left panel), cells that expressed both L1 and IκB-SR, showed IκB-SR localization also in the juxtamembrane region together with L1 (Fig. 2C, right panel and merged image). In addition, in co-immunoprecipitation experiments (using anti-L1 antibody), we identified a complex containing both IκB-SR and L1 (Fig. 2D, lane 8), and also wild-type (wt) IκB in complex with L1 (Fig. 4E), suggesting that L1 can interact with IκB and thereby affects NF-κB signaling. We determined the consequences of blocking NF-κB signaling in L1-expressing CRC cells (by IκB-SR) on several cellular properties that are altered by L1 (Gavert et al., 2005; Gavert et al., 2007). We found that proliferation of cells grown in 0.5% serum (Fig. 2E) and cell motility (Fig. 2F) (n=18, P<0.0001) are both inhibited in L1-expressing cells when NF-κB signaling was blocked by IκB-SR.
Fig. 2.
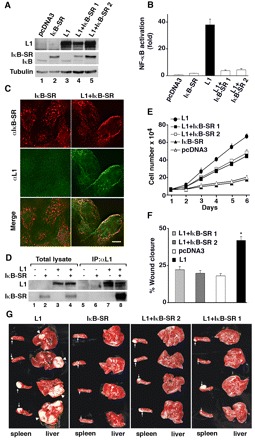
IκB-SR binds to and colocalizes with L1, inhibiting CRC cell proliferation, motility and L1-mediated metastasis of CRC cells to the liver. (A) pcDNA3 empty plasmid or L1-transfected Ls174T cell clones, were further transfected with the puror plasmid or the puror plasmid containing IκB-SR. Western blotting of these cell lines identified two cell clones that expressed both L1 and IκB-SR (lanes 4,5). (B) NF-κB activation in the various cell clones was determined as described for Fig. 1A. (C) The subcellular localization of cells stably expressing L1, IκB-SR or L1 and IκB-SR together was determined by double immunofluorescence microscopy with antibodies against L1 (green, αL1) and IκB (red, αIkB-SR). Scale bar: 20 μm. (D) Lysates of 293T cells transfected with L1, IκB-SR or both were immunoprecipitated (IP) with antibodies against L1. The immunoprecipitated proteins were analyzed by western blotting with antibodies against L1 and IκB. (E) The proliferation of Ls174T CRC cell clones (described in A) in the presence of 0.5% serum was determined in quadruplicate during 6 days. (F) An artificial wound was introduced into confluent monolayers of Ls174T cell clones (described in A), and the extent of wound closure in four different wounds for each cell clone was determined after 18 hours. (G) The CRC cell clones (described in A) were injected into the spleen of nude mice, and tumor growth at the site of injection (spleen) and formation of metastases (liver) were determined. Arrows point to tumors formed at the site of injection in the spleen and arrowheads to large macrometastases in the liver of mice.
Fig. 4.
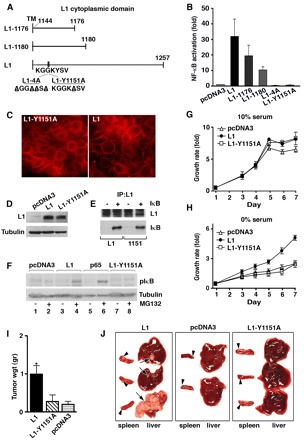
The juxtamembrane L1 cytodomain contains a tyrosine residue at position 1151 that is necessary for L1-mediated cancer progression. (A) Diagram of the L1 cytoplasmic domain in wt and mutant molecules. (B) The NF-κB-responsive reporter plasmid 3xκB.luc was transfected into 293T cells together with an empty pcDNA3 plasmid (control), or with the same plasmid expressing wt L1 or mutant L1 in the cytodomain. Constructs L1-1176 and L1-1180 express truncated versions of L1, whereas constructs L1-4A and L1-Y1151A express L1 point mutants. (C) Cells stably transfected with either pcDNA3 or L1-Y1151A were immunostained with anti-L1 antibody, and their levels of expression were determined by western blotting with antibodies against L1 and IκB (D). (E) Lysates of 293T cells transfected to express L1 or L1-Y1151A together with IκB-SR were immunoprecipitated (IP) with antibodies against L1 and analyzed by western blotting with antibodies against L1 and IκB. (F) Ls174T cells stably transfected with empty pcDNA3 vector, or plasmids expressing L1, L1-Y1151A or the p65 NF-κB subunit were either left untreated (lanes 1,3,5,7) or were treated with the proteasome inhibitor MG132 for 3 hours before cell harvesting (lanes 2,4,6,8), and levels of phosphorylated IκB (IκB-P) were determined. (G) Proliferation of CRC cell clones described in C in the presence of 10% or 0% serum (H) was determined during 7 days in quadruplicate. (I) Cell clones described in C were injected into the flanks of nude mice – on one side L1-expressing cells and on the other side of the same mouse L1-Y1151A-expressing cells or cells expressing the empty pcDNA3 vector (control). Tumor size was determined 14 days after injection. (J) The metastatic capacity of the CRC cell clones described in C was determined as described for Fig. 2G. IκB is IκB-P.
To examine whether NF-κB signaling is required for the induction of metastasis conferred by L1 in CRC cells, we injected into the spleen of nude mice control cells, Ls174T cells that expressed IκB-SR alone, or L1 either alone or in combination with IκB-SR, and determined their ability to grow at the site of injection and to form metastases in the liver. Whereas the injection of L1-expressing cells resulted in the formation of large liver metastases in all the injected animals (100%, n=9, P<0.05), co-expression of IκB-SR in two independently derived L1-expressing cell clones completely blocked their metastasis (100%; n=13, P<0.001) (Fig. 2G). Cells expressing IκB-SR alone (Fig. 2A, lane 2) did not form metastases (Fig. 2G), similar to control Ls174T cells. By contrast, all cell clones formed tumors to varying extents at the site of injection in the spleen, irrespective of their metastatic potential; a finding that has previously been reported (Gavert et al., 2007). We conclude that signaling by NF-κB is essential for the ability of L1 to confer a variety of cellular properties associated with CRC cell invasion and metastasis.
Overexpression of the p65 NF-κB subunit in CRC cells confers enhanced proliferation, increased motility and induces liver metastasis
To more closely link the tumorigenic and metastatic capacities conferred by L1 in CRC cells to NF-κB signaling, we asked whether this downstream activation of the NF-κB pathway (in the absence of L1) is sufficient to induce the cellular characteristics seen when L1 is expressed. We stably transfected the NF-κB p65 subunit in Ls174T cells (that lack L1) (Gavert et al., 2005; Gavert et al., 2007) and isolated two independent cell clones that overexpressed the p65 subunit (Fig. 3A, left panel). These cells displayed strong NF-κB signaling (Fig. 3A, right panel). Similar to L1-transfected cells (Fig. 2E), the p65-overexpressing cells grew better in 0.5% serum than control cells (Fig. 3B). Expression of p65 in these cells dramatically enhanced their capacity to close an artificial wound introduced in a confluent monolayer; cells were also more motile than control Ls174T cells (Fig. 3C,D) (n=9, P<0.0001). This increase was similar to that observed after L1 transfection (Fig. 2F). The injection of p65-overexpressing Ls174T cells into the spleen of nude mice resulted in the formation of metastases (Fig. 3E; 50%, n=15, P<0.08), but these were markedly smaller than those seen in L1-transfected cells. This might have been the result of insufficient levels of the p52 subunit, which is needed for the optimal activity of the p65-p52 NF-κB heterodimer. Histological sections showed that the gross morphology of the metastatic tissue formed in p65-transfected cells was indistinguishable from that formed in L1-overexpressing cells (Fig. 3F).
Fig. 3.
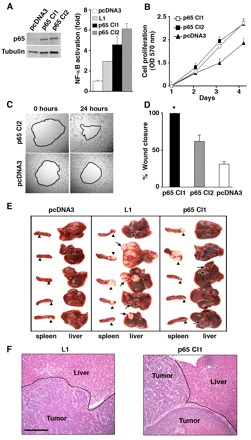
Overexpression of the NF-κB p65 subunit promotes cell proliferation and motility, and confers metastasis in human CRC cells. (A) Two cell clones that stably expressed p65 (p65 Cl1 and Cl2) or pcDNA3 (western blot, left panel) and also L1-transferred cells (Fig. 2A, lane 3) were transiently transfected with 3xκB.luc and β-galactosidase together. Activation of NF-κB was determined (bar graph, right panel) as described for Fig. 1A. (B) Proliferation (in 0.5% of serum) of pcDNA3-transfected cells and the two p65-transfected cell clones was compared. (C) The capacity of a p65-transfected cell clone to close an artificial wound was compared with that of control Ls174T cells after 24 hours. The same areas were photographed immediately after wounding (0 hours) and 24 hours later. (D) The motility of two p65 overexpressing cell clones was compared with that of control Ls174T cells. Wound closure was determined simultaneously in four wounds for each cell line. (E) The metastatic capacity of Ls174T cells stably transfected with pcDNA3, L1 or p65 was determined as described for Fig. 2G. (F) Sections of livers stained with hematoxylin and eosin show tumor metastases in mice that had been injected with either L1- or p65-expressing CRC cells. Scale bar: 250 μm.
A point mutation at Tyr1151 within the L1 cytodomain abolished its capacity to induce NF-κB signaling, cell proliferation, tumorigenesis and metastasis
To begin to unravel the molecular pathways through which L1 mediates the activation of NF-κB signaling, we employed several deletion and point mutations in the L1 cytodomain sequence, which is known to interact with various partners (Fig. 4A) (Cheng et al., 2005). The capacity of these L1 mutants to induce NF-κB activation was determined in 293T cells (Fig. 4B). The results showed that large deletions starting at amino acids (aa) 1176 or 1180 in the L1 cytodomain (L1-1176 or L1-1180, respectively) (Fig. 4A) reduced the capacity of L1 to activate NF-κB. However, four point mutations of lysine, lysine, tyrosine and valine to alanine within the sequence KGGKYSV in the juxtamembrane L1 domain (L1-4A) were sufficient to completely block NF-κB activation (Fig. 4B, L1-4A). Moreover, one single point mutation that of Tyr1151Ala (L1-Y1151A), suppressed the ability of L1 to activate NF-κB signaling in 293T cells (Fig. 4B). We, therefore, isolated CRC cells that stably expressed the L1-Y1151A mutant (Fig. 4C,D) and found that the L1-Y1151A mutant protein is expressed in the cell membrane at levels that are similarly to those in wt L1 (Fig. 4C). Like wtL1, L1L1-Y1151 still formed a complex with wt IκB, (Fig. 4E), but unlike wt L1 and p65, L1-Y1151A did not induce the phosphorylation of IκB (Fig. 4F, lane 8), a finding that is consistent with the inability of L1-Y1151A to activate NF-κB signaling (Fig. 4B). Moreover, unlike cells that express wt L1, CRC cells that express the mutant L1-Y1151A were unable to proliferate without serum (Fig. 4H), had a low tumorigenic capacity (similar to control Ls174T cells; Fig. 4I; n=23, P=0.0417) and were unable to form liver metastases (Fig. 4J; n=20, P<0.001). These results suggested that Tyr1151 in the L1 cytodomain is essential for NF-κB activation and to confer growth, tumorigenic and metastatic capacities in CRC cells.
The interaction of ezrin with L1 requires Tyr1151 and is necessary for the L1-mediated increase in cell motility and the induction of liver metastasis
Previous studies pointed to the importance of Tyr1151 in the L1 cytodomain (Cheng et al., 2005; Sakurai et al., 2008) and a close homologue of L1 (Schlatter et al., 2008) for the interaction with the actin-cytoskeleton adaptor protein ezrin (Bretscher et al., 2002; Fievet et al., 2008). We wished to determine whether ezrin forms a molecular complex with L1 and whether the L1-Y1151A mutant is defective in this capacity. In 293T cells transfected with wt and the L1-Y1151A mutant we found that wt L1 forms a complex that co-immunoprecipitates with ezrin (Fig. 5A, lane 7), whereas the L1-Y1151A mutant has a reduced capacity to bind ezrin (Fig. 5A, lane 8). Next, we wished to determine whether this interaction of L1 with ezrin is an essential part of the L1-mediated response we see in CRC cells. We isolated individual clones of CRC cells expressing L1, in which the level of ezrin was suppressed by shRNA (Fig. 5B, lanes 4,5). Cells that expressed shRNA targeting ezrin lost the increased capacity to grow in the absence of serum conferred by L1 (Fig. 5C), and their ability to close a wound in a monolayer was reduced to that of control Ls174T cells lacking L1 (Fig. 5D,E) (n=24, P<0.0001). Moreover, the metastatic capacity of L1-transfected cells with reduced ezrin levels was blocked (Fig. 5F; n=20, P<0.001), indicating that ezrin is essential for L1-mediated metastasis. The induction of NF-κB signaling through an increase in IκB phosphorylation displayed by L1- or p65-overexpressing CRC cells (Fig. 5G, lanes 6,8) was also inhibited when ezrin levels were suppressed (Fig. 5G, lane 10). This suggested that NF-κB activation and the metastatic ability conferred by L1 require ezrin. Similar results were obtained by the expression of dominant-negative ezrin (which lacks the actin-binding C-terminus; supplementary material Fig. S2), indicating that the actin-binding function of ezrin is required for the properties conferred to CRC cells by L1.
Fig. 5.
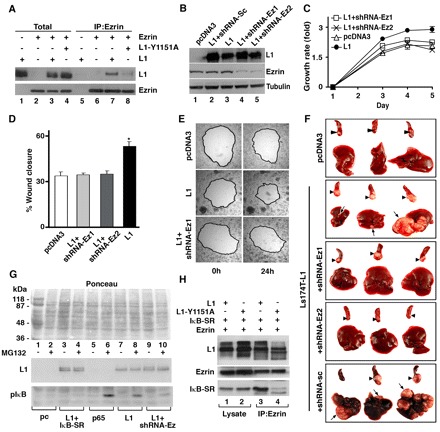
Ezrin is required for L1-mediated metastasis, and L1, IκB and ezrin form a complex in CRC cells. (A) Lysates of 293T cells transfected with different combinations of ezrin, L1 and L1-Y1151A, were immunoprecipitated (IP) with antibodies against ezrin. Samples of cell lysates (Total) and immunoprecipitated proteins (IP:Ezrin) were analyzed by western blotting with antibodies against L1 and ezrin. (B) L1-expressing Ls174T cell clones were transfected with shRNA targeting ezrin or a scrambled siRNA sequence (L1+siRNA-Sc). In two independently isolated cell clones (lanes 4,5) the expression of ezrin was suppressed. (C) The proliferation of CRC cell clones (described in B) in the presence of 0.5% serum was determined during 5 days. (D,E) An artificial wound was introduced into confluent monolayers of the cell clones described in B, and the extent of wound closure in four different wounds for each cell clone was determined after 24 hours (an image of one representative wound is shown in E). (F) The metastatic capacity of the CRC cell clones described in B was determined as described for Fig. 2G. (G) Cells stably transfected with either the empty pcDNA3 vector, the construct expressing L1 alone or together with shRNA targeting ezrin, or with the construct expressing the p65 subunit of NF-κB were either left untreated (−) or were treated with the proteasome inhibitor MG132 (+) for 3 hours before cell harvesting. Protein levels of L1 and IκB-P were determined. Ponceau staining shows the protein gel loading. (H) 293T cells were transfected with different combinations of L1, L1-Y1151A, ezrin and IκB-SR, and cell lysates were immunoprecipitated (IP) with antibodies against ezrin. Immunoprecipitated proteins were analyzed by western blotting with antibodies against L1, IκB and ezrin. Ezrin co-precipitates with IκB and wt L1 (lanes 1,3), but to a much lesser extent with L1-Y1151A and IκB. pIκB is IκB-P.
Since our results showed that L1 interacts with both IκB (Fig. 4E) and ezrin (Fig. 5A), we wished to determine whether a ternary complex containing L1, IκB and ezrin is formed. The formation of such ternary complex between L1, ezrin and IκB-SR was examined in 293T cells transfected with cDNAs encoding these molecules, and compared with the capacity of the L1-Y1151A mutant to form such a complex. Wt L1 can be found in a ternary complex containing both IκB and ezrin (Fig. 5H, lane 3), but much less IκB-SR was precipitated with ezrin when L1-Y1151A was employed (Fig. 5H, lane 4). We conclude that ezrin acts as a scaffold molecule that promotes the assembly of a multi-molecular complex containing L1 and IκB, and that enhances IκB phosphorylation and NF-κB signaling.
We then wished to determine the subcellular location of the L1-ezrin complex and found that, normally, endogenous ezrin is mostly detected in filopodia of Ls174T cells when they are lacking L1 (Fig. 6E,F). However, when L1 was expressed in such cells, ezrin was recruited to membrane areas of cell adhesion containing L1 (Fig. 6B,C), in a manner similar to that of sequestering IκB-SR to L1-containing cell adhesions in CRC cells following L1 expression (Fig. 2C). We examined the localization of endogenous ezrin and L1 in SW620 CRC cells, before and after suppressing L1 levels with L1-specific shRNA. As in Ls174T cells transfected with L1, endogenous ezrin and L1 colocalized at cell-cell contact sites in SW620 cells (Fig. 6M-O). Suppression of L1 (Fig. 1D) led to the redistribution of ezrin, mainly to filopodia but also to the cytoplasm (Fig. 6K,L, insert). Furthermore, when we looked at the localization of ezrin in cells that expressed the L1-Y1151A mutant, we found that more ezrin was found in filopodia and less in L1-containing cell-cell contacts (Fig. 6G-I) when compared with wt L1 (Fig. 6A-C). This is in accordance with the co-immmunoprecipitation results (Fig. 5A, lanes 7,8). Together with the results of Fig. 2C,D and Fig. 5H, these findings indicate that L1 recruits ezrin and IκB to a submembranal complex that promotes IκB phosphorylation and NF-κB signaling.
Fig. 6.
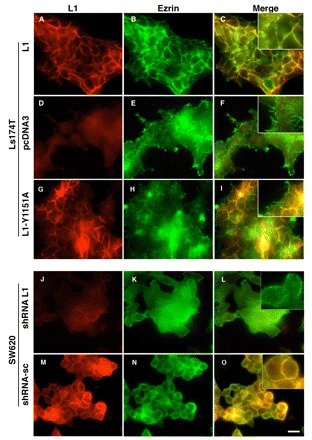
Endogenous wt L1 recruits ezrin from filopodia to cell-cell contact areas. The localization of L1 and ezrin in Ls174T cells stably transfected with either empty pcDNA3 vector, or the constructs expressing L1 wt or L1-Y1151A mutant, was determined by double immunofluorescence microscopy with antibodies against L1 (red, A,D,G) and ezrin (green, B,E,H). Merged images are shown in (C,F,I). The expression of endogenous L1 was stably suppressed in SW620 CRC cells with shRNA targeting L1, and the localization of endogenous L1 and ezrin was determined using antibodies against L1 (red, J,M) and ezrin (green, K,N). (L,O) Merged images. Scale bar: 20 μm for all panels.
Ezrin, activated NF-κB and L1 colocalize at the invasive front of human CRC tissue
We have previously reported on the specific localization of L1 at the invading edge of CRC tumors (Gavert et al., 2005). We now wished to determine whether ezrin is enriched in cells of human CRC tissue samples and whether NF-κB is activated in the nuclei of these cells. Analysis of human CRC tissue samples by immunohistochemistry on serial sections using anti-L1 and anti-p65-P antibodies, revealed that 75% (n=20) of the samples expressed L1 in invading tumor cells (Fig. 7A). In 80% (n=15) of L1-expressing samples of tumor tissue, the immunostaining of serial sections revealed coexpression of L1 in the membrane of invasive CRC cells (Fig. 7A,B) with activated NF-κB p65-P staining in their nuclei (Fig. 7D,E). The presence of p65-P was also evident in some cells localized in the central more differentiated areas of the tumor, but these were primarily mitotic cells (Fig. 7F, arrows) that did not express L1 (Fig. 7C). Sequential staining of CRC tissue for ezrin (Fig. 7J,K) and L1 (Fig. 7G,H) indicated strong expression and colocalization of ezrin and L1 at the invasive tumor front, and only weak ezrin staining in differentiated areas of the tumor (Fig. 7L). Normal colonic epithelial tissue neither expressed L1 (Fig. 7M), p65-P (Fig. 7N) nor ezrin (Fig. 7O) at detectable levels, and only weak occasional staining for these molecules was observed in the stroma. These results strongly suggest that activation of NF-κB signaling by L1 and the involvement of ezrin at the invasive CRC tumor front is an important route for the development of invasive colon cancer.
Fig. 7.
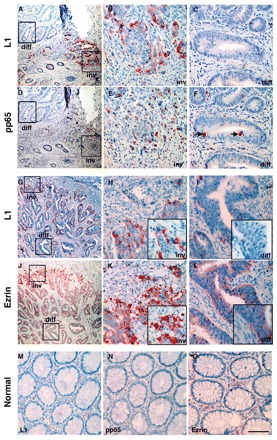
NF-κB, L1 and ezrin are preferentially co-expressed at the invading edge of human CRC tissue. Immunohistochemical staining for L1 (red; A-C,G-I), p65-P (D-F) and ezrin (J-L) was conducted in sequential paraffin sections of human colorectal adenocarcinoma tissue with differentiated (diff) central regions indicated by tubular structures and undifferentiated invading tumor cells (inv). Boxed areas in the first column of images indicate respective areas at higher magnifications that are shown in the middle and right column. Note that L1-expressing invading tumor cells (inv) contain nuclear p65-P (B,E); these invading tumor cells also coexpress ezrin (H,K). By contrast, tumor cells in central tumor regions lack L1 (diff), and p65-P is only detected in some mitotic cells (diff, arrows) (D,F). Ezrin staining in more central regions of the tumor is weak (J,L). (M-O) Staining of normal colon tissue. Scale bar: 400 μm (A,D,G,J), 75 μm (all other panels). In the magnification boxes seen in panels (H,I,K,L) the bar represent 40 μm. pp65 is p65-P.
Discussion
Here, we demonstrated that NF-κB signaling and ezrin are essential for the capacity of L1 to induce metastasis in CRC cells because by inhibiting NF-κB transactivation or by suppressing ezrin levels, or by using an L1 mutant with an altered ezrin-binding domain, the ability of L1 to induce liver metastasis by CRC cells could be blocked. We further showed that L1 recruits both IκB and ezrin into a juxtamembrane complex in CRC cells, and that ezrin is found in a ternary complex together with L1 and IκB in cells. Ezrin is required for the elevated IκB phosphorylation that leads to NF-κB signaling. We also found that L1, ezrin and the phosphorylated p65 NF-κB subunit are present in the same population of invading human CRC tissue cells. Together, these results strongly suggest a major route by which L1 confers an invasive metastatic potential in human CRC cells, by activating the NF-κB signaling pathway through a mechanism involving the cytoskeletal protein ezrin as indicated in the proposed model (Fig. 8). To our knowledge, this is the first indication of the involvement of this signaling complex in human CRC metastasis.
Fig. 8.

Scheme for L1-mediated activation of NF-κB that involves ezrin. (A) NF-κB is inactive when bound to IκB. Phosphorylation of IκB releases NF-κB, and allows its translocation to the nucleus and the subsequent activation of target genes. L1 can form a complex with IκB and can promote the phosphorylation of IκB by a process involving the binding of ezrin to L1 at the juxtamembrane domain. (B) When Tyr1151 in L1is mutated to alanine (L1-Y1151A) in the juxtamembrane domain, ezrin can no longer bind to L1 and, although L1 can still form a complex with IκB, the L1-1Y1151A mutant no longer promotes IκB phosphorylation. pIκB is IκB-P.
The essential function of ezrin in metastasis of CRC cell that we have detected is in agreement with studies on the involvement of ezrin in the metastasis of pediatric cancers (osteosarcoma and rhabdomyosarcoma) (Khanna et al., 2004; Yu et al., 2004). A recent study also reported on elevated ezrin expression in human CRC tissue (Wang et al., 2009). Our findings on the presence of p65-P in invasive CRC tissue cells in L1-expressing cells are supported by a recent study that reported on higher p65 immunoreactivity in CRC tissue compared with normal intestinal mucosa, and an increase in p65 that correlated with histological tumor progression (Aranha et al., 2007). In a CRC cell-culture system that mimicks the invasive versus the more differentiated areas of the tumor by varying the density of cells in culture (Brabletz et al., 2001; Conacci-Sorrell et al., 2003), we found much higher levels of L1 in less dense cell cultures, which also displayed higher nuclear β-catenin signaling – reminiscent of invasive CRC cells at the tumor edge (Gavert et al., 2005). Such sparse cultures also displayed a more substantial nuclear localization of NF-κB (Aranha et al., 2007). Together, these studies supports a link between the activation of β-catenin–TCF-regulated transcription (which induces L1) with the induction of NF-κB signaling leading to increased cell motility and the acquisition of invasive capacity by CRC cells.
These changes in the cellular phenotype of CRC also include the downregulation of E-cadherin (Brabletz et al., 2001; Conacci-Sorrell et al., 2003) through induction of the major EMT regulators Slug and ZEB1 (Spaderna et al., 2008), in both the cell-culture models of CRC cells and at the invasive front of CRC tissue, suggesting that L1 is a key mediator of an EMT-like phenotypic shift. In a recent study that used MCF7 breast cancer cells, the introduction of L1 into these cells resulted in the downregulation of E-cadherin levels and an increase in the invasive motility of these cells (Shtutman et al., 2006). EMT and NF-κB activation were also found to be essential for metastasis of breast cancer cells (Huber et al., 2004) and in the process of mesenchymal gene induction during EMT (Chua et al., 2007; Julien et al., 2007; Solanas et al., 2008). It is noteworthy that the expression of the homeoprotein Six1 that expands the stem cell pool in mouse mammary tumors and induces EMT and tumorigenesis (McCoy et al., 2009) also activates the promoter of the ezrin gene and promotes metastasis of rhabdomyosarcoma (Yu et al., 2006).
By which mechanism(s) does L1 activate the NF-κB signaling pathway? Previous studies indicated that L1 interacts with tyrosine kinase growth-factor receptors, thereby modulating the activation of MAPK, AKT and other signaling pathways (Gavert et al., 2008). These pathways were reported to activate the IκB kinase (IKK) complex upstream of the IκB-NF-κB complex, thereby inducing NF-κB signaling (Bassères and Baldwin, 2006). Another possibility, on the basis of the results of this study, is the formation of a submembrane complex between IκB, ezrin and L1 that leads to increased phosphorylation and p65-P degradation, and then to the release of NF-κB for transactivation in the nucleus. This notion is supported by a recent study demonstrating the presence of a submembrane complex between E-cadherin and the IκB-NF-κB complex, whose disruption during EMT (driven by the downregulation of E-cadherin) led to the release of NF-κB from this submembrane pool and its activation of mesenchymal genes (Solanas et al., 2008).
The most obvious mechanism by which NF-κB signaling contributes to cancer development is through suppression of apoptosis, by inducing the transcription of Bcl2 family members (Naugler and Karin, 2008; Van Antwerp et al., 1996); this was demonstrated in breast cancer cells (Wang et al., 2007) and other cancer cell types (Bassères and Baldwin, 2006). Our microarray analysis of gene expression in L1-transfected CRC cells, however, did not reveal the induction of a Bcl2 gene expression signature or of other anti-apoptotic genes (Gavert et al., 2007). Moreover, a recent study of apoptosis and Bcl2 expression in CRC tissue revealed a higher level of Bcl2 in normal mucosa compared with cancer tissue and Bcl2 protein levels inversely correlated with apoptosis – with more extensive apoptosis observed in the tumor tissue that increased from the adenoma to carcinoma stage, concluding that NF-κB does not exert an anti-apoptotic function through Bcl2 activation in CRC tissue (Aranha et al., 2007). Another means by which NF-κB often contributes to cancer progression is the induction of angiogenesis through the coordinated activation of inflammatory cytokines (Bassères and Baldwin, 2006; Naugler and Karin, 2008). Our gene-array studies of L1-induced genes in CRC cells did not support this notion either (Gavert et al., 2007), because we observed a coordinated downregulation of inflammatory cytokine RNAs in L1-transfected CRC cells (data not shown). Future studies should focus on the molecular details of the link between L1 and the IκB–NF-κB complex, and on the analysis of gene-expression patterns of CRC cells that overexpress L1 genes whose levels are reduced by IκB or ezrin shRNA (in L1-overexpressing cells). Such studies will hopefully narrow down the number and provide information on the genes whose activation by NF-κB is essential for conferring invasive metastatic capacities by L1 in CRC.
Materials and Methods
Cell culture, transfection and transactivation
293T, Ls174T and SW620 cells were grown as described (Gavert et al., 2005; Gavert et al., 2007). For cell growth, 5×103 cells per well were plated into 24-well dishes and their number was determined in triplicate every 24 hours for 5-7 days. Transient transfection of 293T cells was performed using the calcium-phosphate method. Ls174T and SW620 cells were transfected using LipofectamineTM 2000 (Invitrogen, Carlsbad, CA). In transactivation assays, 0.25 μg of β-galactosidase was co-transfected with 1 μg of a plasmid containing three copies of an NF-κB-responsive promoter sequence linked to luciferase (3xκB.luc) and 1 μg of the empty pGL3 plasmid. Cells were plated in triplicate, lysed after 48 hours, and luciferase and β-galactosidase levels determined by using enzyme assay kits from Promega (Madison, WI). Co-transfection with human IκB super repressor (IκB-SR; with mutations in Ser32Ala and Ser36Ala) stabilized against degradation (Zhang et al., 2000) was followed by selection with puromycin (10 μg/ml). SW620 cells stably expressing L1shRNA were co-transfected with four plasmids containing different shRNAs, followed by selection with puromycin. Ls174T cells stably expressing ezrin shRNA were established by co-transfecting cells with four plasmids containing different ezrin shRNA, followed by selection with zeocine (500 μg/ml).
Plasmids
The L1 cDNA and L1 mutants have been described previously (Hlavin and Lemmon, 1991). The IκB-SR cDNA, the NF-κB responsive reporter plasmid (3xκB.luc) and the cDNA for human p65 NF-κB subunit were from David Wallach and Andrew Kovalenko (Weizmann Institute of Science, Rehovot, Israel) (Zhang et al., 2000). shRNA against ezrin and unspecific control shRNA were from Monique Arpin (Curie Institute, Paris). The ezrin target sequences were: 5′-GATTGGCTTTCCTTGGAGTGA-3′, 5′-GAATCCTTAGCGATGAGATCT-3′. The scrambled ezrin sequence was: 5′-GATATGTGCGTACCTAGCAT-3′. L1 shRNA was prepared in pSuper according to the manufacturer's instructions (pSuper RNAi System, OligoEngine, Seattle, WA) using the target sequences 5′-GGATGGTGTCCACTTCAAA-3′, 5′-GAGAAGGGTGGGCTTCCC-3′, 5′-AGACCAGAAGTACCGGATA-3′ and the scrambled sequence 5′-AGCGCGCTTTGTAGGATTC-3′.
RT-PCR
RNA was isolated from cells using EZ-RNA kit (Biological Industries, Israel). PCR was performed using the sequences for L1: forward 5′-ACGGGCAACAACAGCAAC-3′, reverse 5′-CGGCTTCCTGCTAATCATG-3′.
Immunofluorescence and immunoblotting
Cells cultured on glass coverslips were permeabilized with 0.5% Triton X-100 and fixed with 3% PFA. Staining for p65-P was conducted with cells fixed in methanol for 5 minutes at 4°C. Polyclonal and monoclonal antibodies (pAb and mAb, respectively) against L1, recognizing both the extracellular and intracellular domains, have been described previously (Cheng and Lemmon, 2004). Other antibodies were, mAb IκBα/MAD-3 (BD Biosciences, Franklin Lakes, NJ), goat Ab against NF-κB p65 (sc-109, Santa Cruz Biotechnology, Inc. Santa Cruz, CA), rAb phosphospecific pS529 NF-κB p65 (Rockland, Gilbertsville, PA), and mAb ezrin (Sigma, St Louis, MI). The secondary antibodies were Alexa-488-conjugated goat anti-mouse, anti-rabbit IgG (Invitrogen, Carlsbad, CA) and Cy3-labeled goat anti-mouse or anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA). Images were acquired by using Eclipse E1000, Nikon objectives 60×/1.4 NA with a camera (ORCA-ER; Hamamatsu) and Volocity acquisition software (Improvision). Western blots were developed by using the ECL method (Amersham Biosciences, UK) with the antibodies described above and mouse anti-tubulin (Sigma-Israel).
Immunohistochemistry
Immunohistochemistry was carried out on 25 paraffin-embedded human colorectal adenocarcinomas as described (Brabletz et al., 1999). For L1 and p65-P detection, polyclonal rabbit anti-L1 antibody (described above; diluted 1:1000), rabbit antiserum against p65-P (no. 3037, Cell Signaling, Frankfurt, Germany; diluted 1:50), and mouse anti-ezrin antibody (described above) were used. The streptomycin/AB system was used for detection of Ab binding according to the manufacturer's protocol (Dako, Hamburg, Germany). Sections were counterstained with hemalaun (Merck, Darmstadt, Germany).
Artificial wound healing
A round wound was introduced into a confluent cell monolayer with a micropipette tip by using suction to remove the cells. Fresh culture medium with 40 μg/ml mitomycin C was added to inhibit cell proliferation. Cells were placed in a 37°C chamber above the microscope, the wounds were photographed in quadruplicate for 18-24 hours and percentage wound closure was calculated using the Photoshop CS3 analyzer to measure the open wound area compared with the wound area at the beginning of the experiment.
Metastasis assays
Groups of five mice were injected with 106 cells into the distal tip of the spleen; after 4-6 weeks their spleen and livers were removed as described (Gavert et al., 2007). Tissue histology was performed on 3-μm serial sections of formalin-fixed, paraffin-embedded livers that were stained with hematoxylin-eosin. Tumor growth was induced by injecting 2×106 cells into the flanks of nude mice (five mice per group). Control cells were injected into the opposite flank of the same mouse and tumors were removed and compared after 2 weeks.
Statistics
Statistical significance was determined by Fisher's exact test (Langsrud et al., 2007) for mouse metastasis experiments. Tumor mass was compared and significance determined by ANOVA. In wound closure, significance was determined using non-paired Student's t-test. A P value of <0.05 was considered significant.
Supplementary Material
Acknowledgements
These studies were supported by grants from Israel Science Foundation, Israel Cancer Research Fund, Yad Abraham Center for Cancer Diagnosis and Therapy, the YY fellowship from UICC (to A.B.-Z.). Studies in the laboratory of T.B. were supported by the Deutsche Forschungsgemeinschaft (no. BR 1399/4-3 and 5-1), the European Union (MCSCS contract no. 037297) and the Walter G. Ross Foundation.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/135/12/2135/DC1
References
- Aranha M. M., Borralho P. M., Ravasco P., Moreira da Silva I. B., Correia L., Fernandes A., Rodrigues C. M., Camilo M. (2007). NF-κB and apoptosis in colorectal tumourigenesis. Eur. J. Clin. Invest. 37, 416-424. [DOI] [PubMed] [Google Scholar]
- Bassères D. S., Baldwin A. S. (2006). Nuclear factor-κB and inhibitor of κB kinase pathways in oncogenic initiation and progression. Oncogene 25, 6817-6830. [DOI] [PubMed] [Google Scholar]
- Brabletz T., Jung A., Hermann K., Günther K., Hohenberger W., Kirchner T. (1998). Nuclear overexpression of the oncoprotein β-catenin in colorectal cancer is localized predominantly at the invasion front. Pathol. Res. Pract. 194, 701-704. [DOI] [PubMed] [Google Scholar]
- Brabletz T., Jung A., Dag S., Hlubek F., Kirchner T. (1999). β-catenin regulates the expression of the matrix metalloproteinase-7 in human colorectal cancer. Am. J. Pathol. 155, 1033-1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T., Jung A., Reu S., Porzner M., Hlubek F., Kunz-Schughart L. A., Knuechel R., Kirchner T. (2001). Variable β-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc. Natl. Acad. Sci. USA 98, 10356-10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T., Hlubek F., Spaderna S., Schmalhofer O., Hiendlmeyer E., Jung A., Kirchner T. (2005). Invasion and metastasis in colorectal cancer: epithelial-mesenchymal transition, mesenchymal-epithelial transition, stem cells and β-catenin. Cells Tissues Organs 179, 56-65. [DOI] [PubMed] [Google Scholar]
- Bretscher A., Edwards K., Fehon R. (2002). ERM proteins and merlin: integrators at the cell cortex. Nat. Rev. Mol. Cell Biol. 3, 586-599. [DOI] [PubMed] [Google Scholar]
- Cheng L., Lemmon V. (2004). Pathological missense mutations of neural cell adhesion molecule L1 affect neurite outgrowth and branching on an L1 substrate. Mol. Cell Neurosci. 27, 522-530. [DOI] [PubMed] [Google Scholar]
- Cheng L., Itoh K., Lemmon V. (2005). L1-mediated branching is regulated by two ezrin-radixin-moesin (ERM)-binding sites, the RSLE region and a novel juxtamembrane ERM-binding region. J. Neurosci. 25, 395-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua H. L., Bhat-Nakshatri P., Clare S. E., Morimiya A., Badve S., Nakshatri H. (2007). NF-κB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene 26, 711-724. [DOI] [PubMed] [Google Scholar]
- Clevers H. (2006). Wnt/β-catenin signaling in development and disease. Cell 3, 469-480. [DOI] [PubMed] [Google Scholar]
- Conacci-Sorrell M., Ben-Yedidia T., Shtutman M., Feinstein E., Einat P., Ben-Ze'ev A. (2002a). Nr-CAM is a target gene of the β-catenin/LEF-1 pathway in melanoma and colon cancer and its expression enhances motility and confers tumorigenesis. Genes Dev. 16, 2058-2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conacci-Sorrell M., Zhurinsky J., Ben-Ze'ev A. (2002b). The cadherin-catenin adhesion system in signaling and cancer. J. Clin. Invest. 109, 987-991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conacci-Sorrell M., Simcha I., Ben-Yedidia T., Blechman J., Savagner P., Ben-Ze'ev A. (2003). Autoregulation of E-cadherin expression by cadherin-cadherin interactions: the roles of β-catenin signaling, Slug, and MAPK. J. Cell Biol. 163, 847-857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fievet B., Louvard D., Arpin M. (2007). ERM proteins in epithelial cell organization and functions. Biochim. Biophys. Acta 1773, 653-660. [DOI] [PubMed] [Google Scholar]
- Gavert N., Ben-Ze'ev A. (2008). Epithelial-mesenchymal transition and the invasive potential of tumors. Trends Mol. Med. 14, 199-209. [DOI] [PubMed] [Google Scholar]
- Gavert N., Conacci-Sorrell M., Gast D., Schneider A., Altevogt P., Brabletz T., Ben-Ze'ev A. (2005). L1, a novel target of β-catenin signaling, transforms cells and is expressed at the invasive front of colon cancers. J. Cell Biol. 168, 633-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavert N., Sheffer M., Raveh S., Spaderna S., Shtutman M., Brabletz T., Barany F., Paty P., Notterman D., Domany E., et al. (2007). Expression of L1-CAM and ADAM10 in human colon cancer cells induces metastasis. Cancer Res. 67, 7703-7712. [DOI] [PubMed] [Google Scholar]
- Gavert N., Ben-Shmuel A., Raveh S., Ben-Ze'ev A. (2008). L1-CAM in cancerous tissues. Exp. Opin. Biol. Ther. 8, 1749-1757. [DOI] [PubMed] [Google Scholar]
- He T. C., Sparks A. B., Rago C., Hermeking H., Zawel L., da Costa L. T., Morin P. J., Vogelstein B., Kinzler K. W. (1998). Identification of c-MYC as a target of the APC pathway. Science 281, 1509-1512. [DOI] [PubMed] [Google Scholar]
- Hlavin M. L., Lemmon V. (1991). Molecular structure and functional testing of human L1CAM: an interspecies comparison. Genomics 11, 416-423. [DOI] [PubMed] [Google Scholar]
- Huber M. A., Azoitei N., Baumann B., Grünert S., Sommer A., Pehamberger H., Kraut N., Beug H., Wirth T. (2004). NF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J. Clin. Invest. 114, 569-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien S., Puig I., Caretti E., Bonaventure J., Nelles L., van Roy F., Dargemont C., de Herreros A. G., Bellacosa A., Larue L. (2007). Activation of NF-κB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene 26, 7445-7456. [DOI] [PubMed] [Google Scholar]
- Karin M., Ben-Neriah Y. (2008). Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol. 18, 621-663. [DOI] [PubMed] [Google Scholar]
- Karin M., Cao Y., Greten F. R., Li Z. W. (2002). NF-κB in cancer: from innocent bystander to major culprit. Nat. Rev. Cancer 2, 301-310. [DOI] [PubMed] [Google Scholar]
- Khanna C., Wan X., Bose S., Cassaday R., Olomu O., Mendoza A., Yeung C., Gorlick R., Hewitt S. M., Helman L. J. (2004). The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nature Med. 10, 182-186. [DOI] [PubMed] [Google Scholar]
- Kinzler K., Vogelstein B. (1996). Lessons from hereditary colon cancer. Cell 87, 159-170. [DOI] [PubMed] [Google Scholar]
- Langsrud Ø., Jørgensen K., Ragni Ofstad R., Næs T. (2007). Analyzing designed experiments with multiple responses. J. Appl. Stat. 34, 1275-1296. [Google Scholar]
- McCoy E., Iwanaga R., Jedlicka P., Abbey N.-S., Chodosh L., Heichman K., Welm A. L., Ford H. L. (2009). Six1 expands the mouse mammary epithelial stem/progenitor cell pool and induces mammary tumors that undergo epithelial-mesenchymal transition. J. Clin. Invest. 119, 2663-2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min C., Eddy S. F., Sherr D. H., Sonenshein G. E. (2008). NF-κB and epithelial to mesenchymal transition of cancer. J. Cell Biochem. 104, 733-744. [DOI] [PubMed] [Google Scholar]
- Naugler W. E., Karin M. (2008). NF-κB and cancer-identifying targets and mechanisms. Curr. Opin. Genet. Dev. 18, 19-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins N. D. (2006). Post-translational modifications regulating the activity and function of the nuclear factor κB pathway. Oncogene 25, 6717-6730. [DOI] [PubMed] [Google Scholar]
- Polakis P. (2007). The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 17, 45-51. [DOI] [PubMed] [Google Scholar]
- Sakurai T., Gil O. D., Whittard J. D., Gazdoiu M., Joseph T., Wu J., Waksman A., Benson D. L., Salton S. R., Felsenfeld D. P. (2008). Interactions between the L1 cell adhesion molecule and ezrin support traction-force generation and can be regulated by tyrosine phosphorylation. J. Neurosci. Res. 86, 2602-2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlatter M., Buhusi M., Wright A., Maness P. (2008). CHL1 promotes Sema3A-induced growth cone collapse and neurite elaboration through a motif required for recruitment of ERM proteins to the plasma membrane. J. Neurochem. 104, 731-744. [DOI] [PubMed] [Google Scholar]
- Shtutman M., Zhurinsky J., Simcha I., Albanese C., D'Amico M., Pestell R., Ben-Ze'ev A. (1999). The cyclin D1 gene is a target of the β-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. USA 96, 5522-5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtutman M., Levina E., Ohouo P., Baig M., Roninson I. B. (2006). Cell adhesion molecule L1 disrupts E-cadherin-containing adherens junctions and increases scattering and motility of MCF7 breast carcinoma cells. Cancer Res. 66, 11370-11380. [DOI] [PubMed] [Google Scholar]
- Solanas G., Porta-de-la-Riva M., Agustí C., Casagolda D., Sánchez-Aguilera F., Larriba M. J., Pons F., Peiró S., Escrivà M., Muñoz A., et al. (2008). E-cadherin controls β-catenin and NF-κB transcriptional activity in mesenchymal gene expression. J. Cell Sci. 121, 2224-2234. [DOI] [PubMed] [Google Scholar]
- Spaderna S., Schmalhofer O., Wahlbuhl M., Dimmler A., Bauer K., Sultan A., Hlubek F., Jung A., Strand D., Eger A., et al. (2008). The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res. 68, 537-544. [DOI] [PubMed] [Google Scholar]
- Tetsu O., McCormick F. (1999). β-Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398, 422-426. [DOI] [PubMed] [Google Scholar]
- Thiery J. P. (2002). Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2, 442-454. [DOI] [PubMed] [Google Scholar]
- Thiery J. P., Sleeman J. P. (2006). Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 7, 131-142. [DOI] [PubMed] [Google Scholar]
- Van Antwerp D. J., Martin S. J., Kafri T., Green D. R., Verma I. M. (1996). Suppression of TNF-α-induced apoptosis by NF-κB. Science 274, 787-789. [DOI] [PubMed] [Google Scholar]
- Wang H.-J., Zhu J.-S., Zhang Q., Sun Q., Guo H. (2009). High levels of ezrin expression in colorectal cancer tissue is closely related to tumor malignancy. World J. Gastroenterol. 15, 2016-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Belguise K., Kersual N., Kirsch K. H., Mineva N. D., Galtier F., Galtier F., Chalbos D., Sonenshein G. E. (2007). Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat. Cell Biol. 9, 470-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y., Khan J., Khanna C., Helman L., Meltzer P., Merlino G. (2004). Expression profiling identifies the cytoskeletal organizer ezrin and the developmental homeoprotein Six-1 as key metastatic regulators. Nature Med. 10, 175-181. [DOI] [PubMed] [Google Scholar]
- Yu Y., Davicioni E., Triche T., Merlino G. (2006). The homeoprotein Six1 transcriptionally activates multiple protumorigenic genes but requires ezrin to promote metastasis. Cancer Res. 66, 1982-1989. [DOI] [PubMed] [Google Scholar]
- Zhang S. Q., Kovalenko A., Cantarella G., Wallach D. (2000). Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKγ) upon receptor stimulation. Immunity 12, 301-311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}