

Abstract
Although ribosomal proteins are known for playing an essential role in ribosome assembly and protein translation, their ribosome-independent functions have also been greatly appreciated. Over the past decade, more than a dozen of ribosomal proteins have been found to activate the tumor suppressor p53 pathway in response to ribosomal stress. In addition, these ribosomal proteins are involved in various physiological and pathological processes. This review is composed to overview the current understanding of how ribosomal stress provokes the accumulation of ribosome-free ribosomal proteins, as well as the ribosome-independent functions of ribosomal proteins in tumorigenesis, immune signaling, and development. We also propose the potential of applying these pieces of knowledge to the development of ribosomal stress-based cancer therapeutics.
Keywords: ribosomal protein, ribosomal stress, p53, MDM2, cancer, immunity, ribosomopathy
Introduction
The eukaryotic ribosome is the cellular translational machinery primarily responsible for protein synthesis from messenger RNAs (mRNA) and consists of four ribosomal RNA (rRNA) species and 79 ribosomal proteins (RPs). The production of this machinery called ribosome biogenesis is an extraordinarily complex process involving all three RNA polymerases and >150 non-ribosomal factors that are required for the synthesis, processing, transportation, and assembly of pre-ribosomes (Fatica and Tollervey, 2002; Tschochner and Hurt, 2003). In principle, 47S/45S pre-rRNA synthesized by RNA Polymerase I (RNA Pol I), 5S rRNA generated by RNA Polymerase III (RNA Pol III), and RP-encoding mRNAs produced by RNA Polymerase II (RNA Pol II), along with non-ribosomal factors and small nucleolar RNAs (snoRNA), are assembled to yield the 90S pre-ribosomes in the nucleolus, which undergo multiple modifications and subsequent separation into pre-60S and pre-40S particles. During their transport from the nucleolus to the cytoplasm, these pre-ribosomes are dissociated from most of their non-ribosomal factors and then matured to 60S and 40S subunits for protein translation (Fatica and Tollervey, 2002; Tschochner and Hurt, 2003). Protein translation is also a highly organized event in the rough endoplasmic reticulum (RER) where the ribosomes form a 80S translational machinery, which decodes mRNA to produce an amino acid chain or polypeptide facilitated by transfer RNAs (tRNA) that directly bind to the mRNA template through their complementary anticodon sequences (Dai and Lu, 2008).
Ribosome biogenesis and protein translation are finely coordinated with and essential for cell growth, proliferation, differentiation, and animal development. Impairment of any of these two cellular processes can severely retard cell growth and perturb animal development. This has been demonstrated in almost all experimental eukaryote systems tested. For example, a genetic study that analyzes the complete set of yeast strains with deletion of each of the ∼6000 yeast genes reveals that depletion of Sfp1, a transcription factor important for controlling expression of over 60 genes involved in ribosome assembly, results in the smallest cell size (Jorgensen et al., 2002). RP gene insufficiency that reduces the number of ribosomes and protein synthesis leads to the classic minute phenotype in Drosophila, including delayed larval development, small body size, short thin bristles, female sterility, malformation of wings and eyes, and recessive lethality (Lambertsson, 1998; Saeboe-Larssen et al., 1998). In mammals, conditional deletion of RPS6 in mice blocks liver cell proliferation after partial hepatectomy (Volarevic et al., 2000). Homozygous RPL29-knockout mice, although viable, display lower body weight at birth and reduced postnatal viability due to global skeletal growth defects. RPL29-null MEFs show decreased cell proliferation and protein synthesis (Kirn-Safran et al., 2007). Additionally, the embryonic lethality resulting from complete loss of RPS19 (Matsson et al., 2004) or Sbds (Zhang et al., 2006), a nucleolar protein necessary for rRNA processing and ribosome biogenesis (Austin et al., 2005; Ganapathi et al., 2007; Menne et al., 2007), consolidates the crucially important role of well-executed ribosome biogenesis in cell growth and development, although the effect is also partially attributed to abnormal activation of tumor suppressor p53 (Zhang and Lu, 2009; Zhou et al., 2012).
It has been known that the rate of cell proliferation is finely tuned to match the rate of protein synthesis (Ruggero and Pandolfi, 2003). Several tumor suppressors inhibit tumor cell growth and proliferation by interfering with ribosome biogenesis and thus restricting global protein synthesis. For instance, the members of the retinoblastoma proteins (RB) family, such as RB and p130, have been shown to attenuate cell growth and proliferation by reducing rRNA production via a mechanism of direct association with and inactivation of UBF that is required for RNA Pol I activity (Cavanaugh et al., 1995; Hannan et al., 2000; Ciarmatori et al., 2001). The tumor suppressor p53 can also dampen RNA Pol I-catalyzed transcription by preventing the interaction between SL1 and UBF (Zhai and Comai, 2000). Moreover, RB and p53 have also been implicated in regulation of RNA Pol III-catalyzed transcription (White et al., 1996; Cairns and White, 1998; Sutcliffe et al., 2000). RNA Pol III is responsible for transcription of key components in ribosome biogenesis and translation, such as 5S rRNA and tRNAs. RB and p53 can bind to TF-IIIB, a core initiation factor of RNA Pol III, and perturb its interactions with RNA Pol III and other components, such as TF-IIIC2, thus markedly inhibiting RNA Pol III-mediated transcription (White et al., 1996; Cairns and White, 1998; Sutcliffe et al., 2000). Hence, inactivation of either RB or p53 in human cancers results in uncontrolled ribosome biogenesis and protein synthesis, leading to elevated cell proliferation (Cordon-Cardo and Reuter, 1997). By contrast, oncogenic signals often promote cell growth and proliferation by boosting ribosome biogenesis and global protein synthesis (van Riggelen et al., 2010). A delicate study by crossing c-Myc-transgenic mice that contain overexpressed ectopic c-Myc in B-cells with RPL24-haploinsufficient mice validates that c-Myc oncogenic activity requires robust ribosome biogenesis and protein translation (Barna et al., 2008). Remarkably, upregulation of total protein synthesis was well correlated with the increased cell growth and proliferation in the Eμ-Myc/+-transgenic mice. Consistently, when the upregulated protein synthesis was reduced to normal level by deleting one allele of RPL24, the oncogenic potential of c-Myc was markedly abrogated (Barna et al., 2008). These studies demonstrate that the increased rates of ribosomal biogenesis and protein synthesis are tightly bound with the elevated cell growth and proliferation during tumor development and also highly regulated by both tumor suppressors and oncoproteins.
In addition to their essential house-keeping roles in ribosome biogenesis and protein production during cell survival and animal development, RPs have been believed to possess ribosome-independent functions since their discovery. Over the past decade, a great progress has been made in understanding the biochemical, cellular, and physiological roles of RPs independent of the ribosome machinery. In this review, we attempt to offer an overview of the progress in this research area with emphasis on the roles of several interesting ribosomal proteins in tumorigenesis, immune signaling, and development (Table 1). For further reading, we would like to refer readers to other reviews on the related subjects (Warner and McIntosh, 2009; Zhang and Lu, 2009; Boulon et al., 2010; Zhou et al., 2012; de Las Heras-Rubio et al., 2014).
Table 1.
Extraribosomal functions of ribosomal proteins.
aMay not include all the related references.
bNF-κB is involved in both tumorigenesis and immune signal.
Ribosomal stress leads to accumulation of ribosome-free ribosomal proteins
As discussed above, ribosome biogenesis is a tightly organized multistep process, during which RPs are synthesized in the cytoplasm and immediately imported to the nucleolus where they are assembled into the pre-ribosome with rRNA. Also, it has been shown that newly synthesized, but unassembled, RPs are very unstable and immediately cleaned up by the cellular proteasome system in the nucleoplasm (Lam et al., 2007). Then, is it possible for some of the RPs to escape from the tight regulation and take other cellular jobs beyond the functions of the ribosome in translation? Over the past decade, more and more studies using biochemical, molecular biology, cellular biology, and genetic tools and based on different model systems have demonstrated that this is indeed the case. It has been shown that disturbance of any single step in the process of ribosome biogenesis by distinct extracellular and/or intracellular stimulations results in ribosomal stress (RS, also called nucleolar stress), leading to accumulation of ribosome-free form of RPs (Zhang and Lu, 2009; Zhou et al., 2012). These RS-causing stimuli can be categorized into at least three groups: (i) chemical agents or radiation that perturbs rRNA production or mediates RP degradation, (ii) lack of nutrients, including serum or glucose starvation and hypoxia, and (iii) gene deregulation, e.g. malfunction of genes required for ribosome biogenesis resulting from genetic alterations or experimental manipulation (Figure 1). Several tested RS-causing reagents and conditions are briefly described below.
Figure 1.
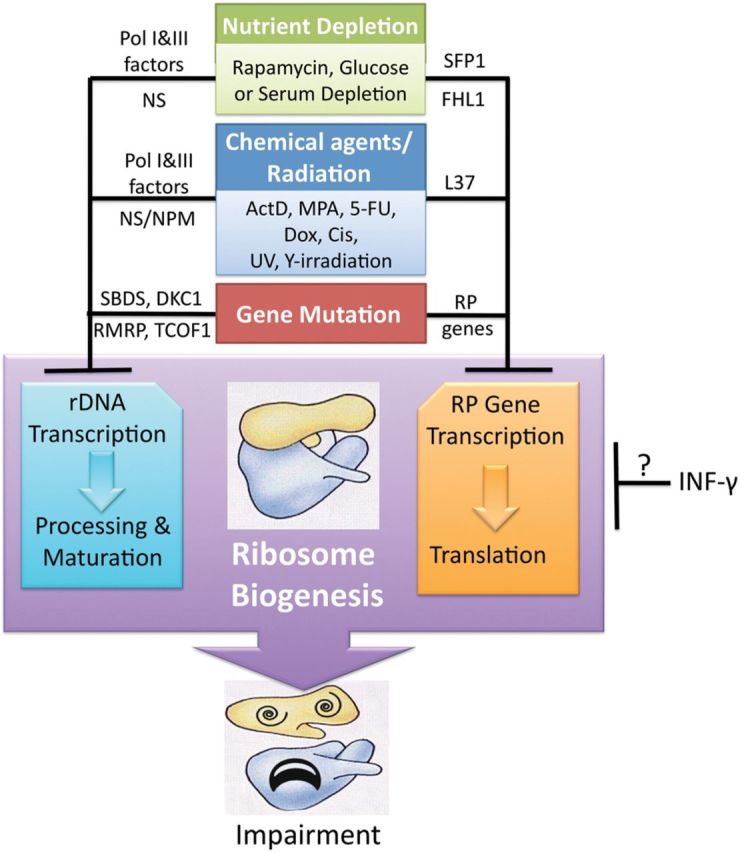
Numerous external or genetic stimuli, including chemical agents or radiation, nutrient depletion, and gene mutation, impair ribosome biogenesis and trigger RS. Several chemical agents or radiation are shown to inhibit RNA Pol I and III-associated factors and rRNA processing-associated factors, or mediate degradation of RPL37 (Llanos and Serrano, 2010). Nutrient depletion is also found to deregulate RNA Pol I and III-associated factors and rRNA processing-associated factors as well as SFP1 and FHL1 that are required for RP gene transcription. Genetic mutations of several rRNA synthesis-associated genes (SBDS, DKC1, RMRP, and TCOF1) (Narla and Ebert, 2010) and RP-encoding genes also cause RS.
Actinomycin D (Act D), a widely used anti-cancer drug, is a prominent example of RS-inducing chemical agents. An extremely low dose of Act D (<10 nM) has been shown to exclusively repress RNA Pol I activity, leading to impaired rRNA transcription (Perry and Kelley, 1970; Iapalucci-Espinoza and Franze-Fernandez, 1979). Recently, a new activity of triggering RS has also been unveiled for several traditional drugs. For example, mycophenolic acid (MPA) is an immunosuppressant drug commonly used for the prevention of organ transplant rejection (Allison and Eugui, 2005). Interestingly, treatment of human cells with MPA makes a nucleolar protein, nucleophosmin (NPM, also called B23), shuttling out of the nucleolus, resulting in the inhibition of rRNA processing (Sun et al., 2008). MPA also reduces cellular GTP levels by inhibiting IMPDH2, a rate limiting enzyme important for guanine nucleotide synthesis, resulting in the decrease of another nucleolar protein called nucleostemin (NS), a GTP-binding protein crucial for rRNA processing (Tsai and McKay, 2005; Romanova et al., 2009), and consequently provoking RS as well (Dai et al., 2008). In addition, several traditional DNA damage-inducing drugs, e.g. 5-Fluorouracil (5-Fu) (Ghoshal and Jacob, 1994; Rubbi and Milner, 2003; Sun et al., 2007), cisplatin (Jordan and Carmo-Fonseca, 1998; Rubbi and Milner, 2003), doxorubicin (Burger et al., 2010), and UV (Zatsepina et al., 1989; Rubbi and Milner, 2003) or γ-irradiation (Kruhlak et al., 2007), have been found to impede either rRNA transcription or rRNA processing, and thus being able to trigger RS as well.
Efficient ribosome biogenesis consumes >60% of cellular energy (ATP) and thus is tightly coupled with the energy status of a cell. This feature makes the nucleolar process highly sensitive to nutrient deprivation (Zhou et al., 2012), as demonstrated by several recent studies on the target of rapamycin (TOR) signaling that has been shown to play a central role in linking the cellular nutrient status to ribosome biogenesis. This signaling pathway promotes ribosome biogenesis by regulating the production of RPs and rRNAs through three different mechanisms. First, studies using yeast showed that inhibition of TOR globally represses the transcription of most RP-encoding genes (Cardenas et al., 1999). Consistently, several transcription factors, such as SFP1 (Jorgensen et al., 2004; Marion et al., 2004) and FHL1 (Martin et al., 2004; Schawalder et al., 2004; Rudra et al., 2005), were found to directly associate with RP gene promoters, activating their transcription under growth-favorable conditions in a TOR-dependent manner, whereas these two transcription factors were inactivated upon nutrient deficiency or TOR inhibition, leading to the downregulation of RP gene expression. Also, TOR can boost the translation of RP-encoding genes. Because the translation of RP-coding mRNAs is negatively controlled by the 5′ TOP (terminal oligopyrimidines or track of pyrimidines) sequence at their 5′ transcriptional start site (Meyuhas, 2000; Fumagalli et al., 2009), TOR could promote 5′ TOP mRNA translation by phosphorylating and activating S6K1, leading to elevated ribosome production and cell growth (Mayer and Grummt, 2006). Furthermore, TOR is required for the rapid and sustained serum-induced rDNA transcription in a S6K1-dependent manner (Hannan et al., 2003). Later on, TOR was also found to bolster rDNA transcription by sequestering TIF-IA in the nucleus and facilitating the formation of the RNA Pol I initiation complex (Claypool et al., 2004; Mayer et al., 2004). Rapamycin has also been shown to reduce the activity of RNA Pol III and its specific transcription factor TF-IIIB, repressing the transcription of the 5S rRNA gene (Zaragoza et al., 1998). Therefore, inhibition of the TOR signaling by nutrient depletion perturbs ribosome biogenesis at different levels. In line with this, a downstream target of the TOR pathway, AMP-activated protein kinase (AMPK), can be activated upon glucose deprivation to phosphorylate and inactivate TIF-IA, leading to the repression of RNA Pol I-mediated transcription (Hoppe et al., 2009). Interestingly, it was found that RPs and the ribosome itself could be degraded in response to nutrient starvation (Kraft et al., 2008), leading to the severe impairment of ribosome assembly. Hence, ribosomal biogenesis is highly sensitive to the energy status of cells, and abnormal nutrient levels could cause RS by disrupting this process.
Mutations of RP-encoding genes or ribosome biogenesis-associated genes can also perturb ribosome biogenesis, thus causing RS. These mutations are often highly associated with genetic diseases. One of these inherited diseases is Diamond–Blackfan anemia (DBA), also known as inherited erythroblastopenia. DBA is a congenital erythroid aplasia that is highly associated with mutations of several RP-encoding genes, including RPS19, RPS24, RPS17, RPL11, RPL5, and RPL35A (Narla and Ebert, 2010). Dysfunction of any of these RP genes leads to abnormal ribosome biogenesis resulting in growth arrest of erythroid progenitor cells (Narla and Ebert, 2010). The other disease is 5q-syndrome, which is an acquired DBA-like genetic disease predominantly found in females of advanced age and caused by an isolated interstitial deletion of the long arm of chromosome 5. One of the genes within this lost 5q chromosome region is the RPS14-encoding gene, whose deletion has been identified to be responsible for the development of this disease through a mechanism similar to that for DBA (Ebert et al., 2008; Barlow et al., 2010). Remarkably, RS that is caused by dysfunction of these RP genes can also be manipulated in vitro, such as impairment of 40S by RPS6 depletion (Fumagalli et al., 2009) or by RPS14 or RPS19 depletion (Zhou et al., 2013a; Wang et al., 2014), or disruption of 60S by RPL29 or RPL30 depletion (Sun et al., 2010) in cultured cells.
Altogether, ribosome biogenesis can be compromised via multiple mechanisms in response to a variety of stress signals, and, under such circumstances, some of the RPs free from the ribosome are accumulated in the nucleoplasm to execute their enchanting ribosome-independent functions (Figure 2) as further described below.
Figure 2.

Functions of ribosome-free RPs in response to RS. RS unleashes many free forms of RPs that are involved in tumorigenesis or immune response by regulating multiple signaling pathways. Additionally, impairment of ribosomal biogenesis through mutation or deregulation of RP-encoding genes leads to ribosomopathies and developmental defects.
Ribosomal proteins in cancers: oncoprotein or tumor suppressor
Constantly growing and highly proliferating cancer cells demand a huge amount of proteins and thus acquire increased protein synthesis. This means that cancer cells need a lot of more highly efficient ribosome translational machineries than do normal cells. Consistent with this notion is that a number of tumor suppressors and oncogenic proteins often control the progression of cancer cells by regulating ribosome biogenesis and global protein synthesis (Ruggero and Pandolfi, 2003; Silvera et al., 2010). Interestingly, besides the importance of ribosome to the growth and proliferation of cancer cells, individual ribosome-free RPs could also play a role in tumorigenesis. This statement is supported by the following lines of evidence. First, numerous RPs have been found to be up-regulated at either mRNA or protein level in various human tumors (Shuda et al., 2000; Kondoh et al., 2001; Artero-Castro et al., 2011; Chen et al., 2014). Also, overexpression of RPS3A promoted transformation of NIH-3T3 cells and tumor growth in nude mice (Naora et al., 1998). In addition, RPS13, which is highly expressed in multidrug-resistant gastric cancer cells, has been shown to prevent drug-induced apoptosis and to promote gastric cancer cell proliferation (Shi et al., 2004; Guo et al., 2011). The fact that some individual RPs are selectively upregulated in cancers implies that some RPs possess oncogenic activity independent of the translational machinery.
By contrast to the oncogenic functions of some RPs as briefed above, other ribosome-free RPs have been shown to play a role in suppressing tumorigenesis by either activating other tumor suppressors or inactivating oncoproteins (de Las Heras-Rubio et al., 2014). Over the past decade, more than a dozen of RPs, including RPL5 (Dai and Lu, 2004), RPL6 (Bai et al., 2014), RPL11 (Lohrum et al., 2003; Zhang et al., 2003), RPL23 (Dai et al., 2004; Jin et al., 2004), RPL26 (Zhang et al., 2010), RPL37 (Daftuar et al., 2013), RPS3 (Yadavilli et al., 2009), RPS7 (Chen et al., 2007; Zhu et al., 2009), RPS14 (Zhou et al., 2013a), RPS15 (Daftuar et al., 2013), RPS20 (Daftuar et al., 2013), RPS25 (Zhang et al., 2013b), RPS26 (Cui et al., 2014), RPS27 (Xiong et al., 2011), RPS27A (Sun et al., 2011), and RPS27L (Xiong et al., 2011, 2014), have been identified to regulate the MDM2/MDMX–p53 cascade, consequently suppressing tumor cell proliferation. The first interaction between RP and MDM2 was revealed in 1994 (Marechal et al., 1994). However, the physiological relevance of this RP–MDM2 interaction had remained elusive for almost 10 years until RPL11, RPL5, and RPL23 were reported to regulate the MDM2–p53 feedback loop (Lohrum et al., 2003; Zhang et al., 2003; Dai and Lu, 2004; Dai et al., 2004; Jin et al., 2004). When cells are under RS triggered by chemical agents or RP depletion, these three RPs were prevented from ribosome assembly or released from pre-ribosome to the nucleoplasm, where they bind to MDM2 and inhibit MDM2-mediated p53 ubiquitination and degradation, consequently leading to cell cycle and proliferation arrest. Although most of the RPs interact with MDM2 directly, some of them, such as RPS7 (Zhu et al., 2009), RPS15 (Daftuar et al., 2013), RPS20 (Daftuar et al., 2013), RPS25 (Zhang et al., 2013b), and RPL37 (Daftuar et al., 2013), have also been shown to bind to MDMX, the homologue and partner of MDM2. RPL26 is one of the most enchanting RPs because it not only interacts with MDM2, but also associates with p53 mRNA and enhances its translation (Takagi et al., 2005), and this enhancement can be interfered with by oncogenic signals, such as MDM2 (Ofir-Rosenfeld et al., 2008) and TGF-β1 (Lopez-Diaz et al., 2013). RPS3 is another interesting MDM2-interacting RP that was also shown to induce apoptosis by collaborating with E2F1. However, Akt-mediated phosphorylation of RPS3 attenuates apoptosis by abrogating the RPS3–E2F1 interaction in response to DNA damage (Lee et al., 2010). These results imply that Akt phosphorylation of RPS3 may play an oncogenic role. Indeed, Phospho-RPS3 was shown to translocate into the nucleus and upregulate pro-survival gene expression via association with NF-κB in non-small cell lung cancer cells (Wan et al., 2007; Yang et al., 2013a). More studies have underscored the notion that the MDM2- or MDMX-binding RPs, particularly RPL5 and RPL11, are potentially key tumor suppressors by modulating p53 activity. It has been found that RPL5 and RPL11 can associate with each other via 5S rRNA, and this preribosomal complex is essential for p53 activation upon impairment of ribosome biogenesis (Horn and Vousden, 2008; Donati et al., 2013). Intriguingly, a recent study has shown that only RPL5 and RPL11, but not other RPs (e.g. RPS7 and RPL23), are required for RS-elicited p53 activation (Fumagalli et al., 2012). The anti-tumor functions of RPL5 and RPL11 could be regulated by some oncoproteins. On the one hand, RPL11 can be inactivated by PICT1, which is encoded by a gene that resides on human chromosome 19q13.32. Although this is a so-called tumor suppressive region, as its frequent mutations are highly associated with human cancers, a later study using Pict1−/− mice showed that PICT1 actually acts as an oncoprotein (Sasaki et al., 2011). Pict1−/− mice died at E3.5 largely due to p53-induced apoptosis, whereas overexpression of PICT1 in cancer cells can sequester RPL11 in the nucleolus and prevent this RP from association with MDM2, leading to p53 inactivation (Sasaki et al., 2011). On the other hand, another oncoprotein, splicing factor SRSF1, was shown to stabilize p53 as an indispensable component of the RPL5–MDM2 complex and to trigger oncogene-induced and p53-dependent senescence (Fregoso et al., 2013). An elegant mouse genetic study further supports the role of RPs in preventing tumorigenesis (Macias et al., 2010). This study showed that RS fails to activate p53 in a knock-in mouse line that carries a cancer-associated Mdm2 C305F mutation, which disrupts the interaction of MDM2 with RPL5 and RPL11 (Lindstrom et al., 2007). When crossing this line with a c-Myc-transgenic mouse line, this MDM2 mutant accelerated Myc-induced lymphomagenesis (Macias et al., 2010), indicating that the RS–RPL11–RPL5–MDM2–p53 pathway plays a physiological role in protecting mammalian cells from tumorigenesis. Interestingly, a latest study generating a Rps27l knockout mouse model has demonstrated that Rps27l can either positively or negatively regulate p53 signals in different TP53 genetic backgrounds (Xiong et al., 2014). It was found that depletion of Rps27 l, in the p53+/+ background, triggers RS and thus promotes MDM2-mediated MDMX degradation, consequently leading to the impairment of the E3 activity of MDM2/MDMX complexes toward p53, whereas in the p53+/− background, Rps27 l acts as a tumor suppressor for its depletion induces genomic instability and spontaneous lymphoma (Xiong et al., 2014). Recently, we also found that TAp73, the p53 homologue, can be activated by RPL5, RPL11, and RPS14. Although these RPs have never been shown to bind to p53, they can directly bind to TAp73 and circumvent MDM2 inhibition of this transcription factor, leading to TAp73 activation and p73-dependent apoptosis (Zhou et al., 2014).
In addition to activating tumor suppressors, several RPs can also inactivate oncoproteins, such as c-Myc. Initially, our group showed that RPL11 specifically binds to the Myc box II (MB II) domain of c-Myc oncoprotein and inhibits its transcriptional activity by preventing the recruitment of its coactivator TRRAP on their target gene promoters (Dai et al., 2007). Later on, RPL11 was shown to not only suppress c-Myc activity, but also promote miR-24/miRISC-mediated c-Myc mRNA degradation (Challagundla et al., 2011). Likewise, RPS14 and RPL5 have also been found to suppress c-Myc activity by directly binding to this transcription factor and regulating its mRNA turnover (Liao et al., 2013; Zhou et al., 2013b). Another RP, RPL41, was shown to be able to facilitate the shuttling of activating transcription factor 4 (ATF4), a major regulator of tumor cell survival and a potential therapeutic target of human cancers, from the nucleus to the cytoplasm for degradation, consequently sensitizing tumor cells to chemotherapy (Wang et al., 2011). Recent identification of mutations of some RP-encoding genes, such as RPL5, RPL10, and RPL22, in cancer patients by genomic deep sequencing further consolidates the notion that these RPs can function as tumor suppressors (De Keersmaecker et al., 2013; Kandoth et al., 2013).
The tumor suppressive function of RPs may also be attributed to their inhibitory effect on angiogenesis, which is essential for solid tumor progression, as newly formed blood vessels can efficiently facilitate supplies of nutrients and oxygen to and disposal of waste metabolites from constantly growing tumors. Blood vessels are also the channels for tumor cells to metastasize to other organs, and metastasis is the ultimate cause of cancer deaths. Intriguingly, one of the RPs, RPL17, was identified by comparing the transcriptomes of two mouse strains, C3H/F (resistant to intima formation) and SJL (with increased intima formation), to play a potential role in vascular smooth muscle cells (VSMC) growth, as RPL17 expression was inversely correlated with VSMC growth, and RPL17 depletion promotes VSMC proliferation (Smolock et al., 2012). This study suggests that RPL17 might play a role in suppressing angiogenesis, though the underlying mechanism remains to be elucidated.
Based on these studies, ribosome-free RPs can act as either oncoproteins or tumor suppressors (Table 1). However, it remains to be further investigated how their differential functions are finely balanced or well-coordinated with ribosome biogenesis and protein production during normal cell growth and proliferation.
Roles of ribosomal proteins in immune signaling
Beyond their roles in oncogenesis, RPs have also been shown by several studies to participate in the innate immune response. One of the prominent examples is RPL13A, which was reported to engage the interferon-γ (IFN-γ)-mediated inflammatory response by selectively modulating gene expression. Inflammation is a self-protective process, through which the body attempts to remove pathogens and cell debris. Because this process is detrimental to both intruding organisms and host tissues, it needs to be restrictively controlled. IFN-γ is a type II pro- and anti-inflammatory cytokine and has been shown to fine tune inflammation process by coordinating the expression of inflammation-related genes. It has been shown that IFN-γ treatment elevates mRNA levels of a group of inflammatory genes, including Ceruloplasmin (CP) (Mazumder et al., 2003), VEGF-A (Ray and Fox, 2007), and those encoding chemokine ligand and receptors (Vyas et al., 2009), up to 12 h; however, translation of these mRNAs were suppressed by the IFN-γ-activated inhibitor of translation (GAIT) complex formed at about 12–16 h of treatment (Mukhopadhyay et al., 2009). Importantly, RPL13A is a key component of the GAIT complex and required for selective translational inhibition (Mazumder et al., 2003). Mechanistically, IFN-γ, through the DAPK1–ZIPK kinase signaling cascade, triggers phosphorylation and release of RPL13A from the 60S ribosomal subunit (Mukhopadhyay et al., 2008). RPL13A subsequently associates with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Jia et al., 2012), glutamyl-prolyl tRNA synthetase (EPRS), and NS1-associated protein 1 (NSAP1) (Sampath et al., 2004) to form the active GAIT complex, which binds to a defined element within 3′ untranslated region (3′ UTR) of target mRNAs consequently leading to translational inhibition (Mazumder et al., 2003; Mukhopadhyay et al., 2009). Interestingly, DAPK1 and ZIPK, the kinases responsible for RPL13A phosphorylation, are also targets of the GAIT complex, thus forming feedback regulatory loop (Mukhopadhyay et al., 2008).
As mentioned above, inflammatory response is a double-edged sword that kills both pathogens and host cells. Identification of RPL13A as a negative regulator of inflammatory proteins suggests that this RP could be a repressor of inflammatory signaling. It is noteworthy mentioning that ZIPK-mediated phosphorylation and release of RPL13A from the 60S ribosomal subunit often occur between 12 and 16 h after IFN-γ treatment (Mukhopadhyay et al., 2009). This belated formation of the GAIT complex allows elimination of pathogens and removal of damaged cells upon IFN-γ stimulation at the early stage of inflammation, but prevents excessive accumulation of inflammatory proteins, as thus contributing to inflammation resolution, a regenerative process, in which the inflamed tissues are completely restored back to normal tissues. Interestingly, the anti-inflammation role of RPL13A is well in accordance with the aforementioned tumor suppressive function of RPs, as inflammatory response plays an essential role at different stages of tumorigenesis, and prolonged expression of inflammatory genes promotes tumor progression. In this regard, RPL13A is a bodyguard to not only protect host tissues from inflammatory injury, but also prevent cancerous growth of the inflamed cells.
Another example of RP involvement in immune signaling is RPS3 that selectively modulates NF-κB target gene expression. NF-κB is a family of transcription factors that were originally identified to regulate genes crucial for immune response, but later on shown to also regulate genes implicated in cell survival or proliferation. The NF-κB family consists of five members, RelA (p65), RelB, c-Rel, NF-κB1 (p50), and NF-κB2 (p52), forming homo- or heterodimers (Ghosh and Karin, 2002). All family members share a conserved N-terminal Rel domain, while only RelA, RelB, and c-Rel possess a transactivation domain in their C-termini (Plaksin et al., 1993; Guan et al., 2005). Interestingly, RPS3 was identified as a non-Rel component of the NF-κB complex by directly binding to the RelA subunit, and required for enhanced DNA binding ability and selectively regulating the expression of transcriptional target genes of this family (Wan et al., 2007). Thus far, two major signals have been found to initiate the RPS3 regulation of expression of NF-κB target genes. First, TNF-α-stimulated expression of cystathionine γ-lyase (CSE) mediates sulfhydration of RelA, and this modification facilitates the interaction of Rel-A with RPS3 (Sen et al., 2012). The other pathway is through IκB kinase β (Ikkβ)-dependent phosphorylation of RPS3 at serine-309, resulting in nuclear translocation of RPS3 (Wan et al., 2011). Given that Ikkβ also activates NF-κB by mediating ubiquitination and proteasome degradation of the master inhibitors of NF-κB (IκBs), the Ikkβ–RPS3 cascade provides an alternative mechanism that selectively activates NF-κB in a RPS3-dependent manner (Wan et al., 2011). Due to the essential role of RPS3 in eliciting immune signaling, this RP becomes a target of bacterial virulence protein. It has been found that the virulence protein NleH1 of the foodborne pathogen Escherichia coli strain O157:H7 specifically binds to and suppresses RPS3-mediated NF-κB activation by inhibiting phosphorylation and nuclear accumulation of RPS3, as thus impairing host innate immune responses (Gao et al., 2009; Wan et al., 2011). Given the fact that RPS3 also activates the p53 tumor suppressive pathway as mentioned above, this RP is regarded as one of the most fascinating RPs with pivotal multifunctions (Gao and Hardwidge, 2011).
RPs are not always the gatekeeper of our immune system, but also act as Trojan Horses in the body, because some RPs, such as RPS6 (Huang et al., 2012), RPS19 (Haque and Mir, 2010; Cheng et al., 2011), RPS25 (Landry et al., 2009), and RPL22 (Wood et al., 2001), have been found to facilitate translation initiation of viral transcripts. Interestingly, depletion of each of these RPs selectively represses viral transcript translation, but not general translation, indicating that a preferential engagement of these RPs in bolstering viral translation.
Together, these lines of evidence indicate that some RPs play diverse roles in host immune response by either boosting immune signaling or facilitating pathogen production under different circumstances (Table 1).
Tissue-specific roles of ribosomal proteins in development and diseases
In additional to implementing a number of extraribosomal functions aforementioned, RPs are also implicated in the development of multiple human diseases, named ribosomopathies (Narla and Ebert, 2010; Ellis, 2014; Wang et al., 2015). Although the ribosome machinery is ubiquitously produced in all cells and tissues, its impairment caused by RS and the detrimental consequence due to this impairment may not happen equally to all cells, tissues, or organs. Indeed, a number of studies have shown that RS triggered by different RP mutations is actually involved in developmental defects of specific tissues or organs. The first example is the RPS19 mutation, which causes RPS19 haploinsufficiency. In 1999, mutation of RPS19 was found to impair ribosome biogenesis and to be highly associated with a congenital bone marrow failure syndrome, Diamond–Blackfan anemia (DBA), and a selective decrease or absence of erythroid precursors (Draptchinskaia et al., 1999). Later on, more mutations in other RP-encoding genes, including RPS7 (Gazda et al., 2008), RPS10 (Doherty et al., 2010), RPS15 (Gazda et al., 2008), RPS17 (Cmejla et al., 2007), RPS24 (Gazda et al., 2006; Campagnoli et al., 2008), RPS26 (Doherty et al., 2010), RPS27A (Gazda et al., 2008), RPS28 (Gripp et al., 2014), RPS29 (Mirabello et al., 2014), RPL5 (Gazda et al., 2008; Cmejla et al., 2009), RPL11 (Gazda et al., 2008; Cmejla et al., 2009), RPL15 (Landowski et al., 2013), RPL35A (Farrar et al., 2008), and RPL36 (Gazda et al., 2008), were identified in DBA patients. Moreover, one allele deletion of RPS14 was found to be pathogenic to an acquired DBA-like disease, called 5q-syndrome (Ebert et al., 2008). Mutation/deregulation of these RP genes has been shown to selectively activate p53 through RS in hematopoietic progenitor cells, leading to p53-dependent cell cycle arrest of these cells (Dutt et al., 2011). When TP53, the p53-encoding gene, was further deleted in the DBA or 5q syndrome animal models, the impaired erythroid linage was remarkably restored (Barlow et al., 2010; Jaako et al., 2011). Although it was clear that abnormal activation of p53 is causative to the manifestations of these two types of ribosomopathies (Narla and Ebert, 2010), the mechanism underlying the specific p53 activation in the hematopoietic stem cells still remains elusive.
In addition to the function of those RPs in the hematopoietic progenitor cell development, many RPs have also been found to be required for the formation of other tissues or organs. For example, among the DBA patients, only those with mutated RPL5 exhibit cleft lip and/or palate, which indicates that RPL5, but not other DBA-related RP genes, is engaged in this regional craniofacial development (Gazda et al., 2008). The most recently discovered ribosomopathy, isolated congenital asplenia (ICA), with the absence of a spleen at birth has been found to be associated with haploinsufficiency of RPSA (Bolze et al., 2013). Because lacking a spleen, ICA patients are prone to suffering from life-threatening bacterial infections, but they have no other observable developmental defects. RPL10 mutation is associated with the pathogenesis of autism probably by affecting brain development (Klauck et al., 2006). Moreover, mutation in RPL21 could be pathogenic to hereditary hypotrichosis simplex (Zhou et al., 2011). All these findings suggest that some RPs are selectively required for the development of specific tissues or organs.
Indeed, animal studies further confirm this notion in laboratories. Aside from the animal models with phenotypes of the aforementioned ribosomopathies, increasing evidence has shown that more RP genes are important for controlling specific developmental processes. Three development-defective mouse mutants, Tail short (Ts), Tail-short Shionogi (Tss), and Rabo torcido (Rbt), display similar severe developmental defects, including an abnormally short and kinky tail, a midline facial cleft and/or cleft palate, exencephaly, various eye abnormalities, and pronounced axial skeletal patterning defects. All the three mutants were mapped to the chromosome 11, where Rpl38 was identified responsible for the development-defective phenotypes in the three mouse lines (Kondrashov et al., 2011). Remarkably, re-expression of functional Rpl38 in Rpl38-deficient mice restored most abnormal phenotypes. Mechanistically, Rpl38 regulates a subset of homeobox (Hox) mRNA expression by mediating the formation of 80S–Hox mRNA complex. The Hox gene family is a group of evolutionally conserved genes controlling the anterior–posterior patterning during embryogenesis. Interestingly, this specialized translational control of Hox genes is not observed in other RP gene-haploinsufficient mouse models, whereas Rpl38-mutated mice do not display any defective hematopoietic progenitor cells typically shown in DBA or 5q-mouse models (Barlow et al., 2010; Jaako et al., 2011). However, some manifestations, such as the facial defects, do exhibit in both Rpl38-haploinsufficient and DBA models/patients, implying that some RPs might also play overlapping roles during development.
Another interesting example is Rpl22 deletion. This deletion selectively impairs αβ-lineage T cell development (Anderson et al., 2007), though Rpl22 is not essential for translation or cell survival, because unlike other reported RP genes (Matsson et al., 2004), deletion of Rpl22 in mice does not cause a lethal phenotype (Anderson et al., 2007). Homozygous deletion of Rpl22 triggered restricted p53 activation in the αβ-lineage progenitor cells, retarding T cell development, but further depleting p53 completely rescued this specific defect (Anderson et al., 2007). This cell lineage-specific phenotype is strikingly similar to the defect of erythroid development in DBA patients, as both of the defects, though of different cells, could be completely rescued by deleting TP53 (Narla and Ebert, 2010). It is highly possible that the progenitor cells need a large number of ribosomes for their rapid proliferation, and are thereby much more sensitive to RS-induced p53 activation, which is also consistent with an intriguing study showing that mouse embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) undergo p53-dependent apoptosis in response to RS (Morgado-Palacin et al., 2012). However, this mechanism is not universally true for other RP deficiencies, as downregulation of several other RP genes, such as rpl3, rpl6, and rpl23a, in zebrafish impairs the expansion of pancreatic progenitor cells by a p53-independent mechanism (Provost et al., 2012, 2013), which might be a species-specific phenotype.
Then, it remains puzzling how deficiencies of different RPs cause specific phenotypes in different cell types and tissues? Several mechanisms have been proposed to expound this tissue-specific phenomenon. The simplest and most direct explanation could be the dosage requirement of each RP for maintenance of normal physiological events, such as ribosome biogenesis, p53 modulation, and other cellular processes, in different cell types. Another possible mechanism is that ribosomal components of the ribosome may vary in different cell types. It was speculated that the ribosome is a monolithic machine that requires all the RPs and rRNAs for ribosome assembly. Yet, recent studies, creating viable Rpl22−/− or Rpl29−/− homozygous mouse lines, challenged the classical notion (Anderson et al., 2007; Kirn-Safran et al., 2007). It is possible that these two RPs are only needed for specific cell types and may not be present in other cell types. Alternatively, though constitution of the ribosome is universal to all cells, the ribosome-associated RPs may confer specific affinity for mRNA translation. For example, RPL38 controls a subset of Hox mRNA expression by mediating formation of the ribosome–Hox mRNA complex, and as such, plays a role in the anterior–posterior patterning during embryogenesis. An additional example is provided by the finding that haploinsufficiency of DBA-associated RP genes selectively attenuates the internal ribosome entry site (IRES)-mediated translation of Bag1 and Csde1, both of which are required for erythroid differentiation, consequently leading to impaired hematopoiesis (Horos et al., 2012; Garcon et al., 2013). In this scenario, the differentiation status or potential of cells may also account for the tissue-specified functions of RPs. Lastly, the cell-specific functions of RPs might be due to their interplays with some cell type-specific proteins. This idea has been proved, at least in the case of RPS15, in neurodegenerative Parkinson's disease (PD). The pathogenic leucine-rich repeat kinase 2 (LRRK2) mutations are the most common genetic etiology of PD. The G2019S mutation in LRRK2 increases the kinase activity, but the phosphosubstrate that links LRRK2 kinase activity to neurodegeneration had been elusive until RPS15 was identified as a bona fide substrate of LRRK2 (Martin et al., 2014). Phospho-deficiency or partial knockdown of RPS15 prevented pathogenic and LRRK2-stimulated elevation of global protein synthesis and dopamine neuron degeneration in Drosophila and human cells.
Hence, in addition to their essential roles in ribosome biogenesis and protein translation, a large number of RPs can act as individual regulatory proteins to execute numerous extraribosomal functions during the development of specific tissues (Table 1). However, the enigma is why cells need so many house-keeping RPs to perform diverse functions.
Prospects: from basic to clinic
Although perturbation of ribosome biogenesis during embryogenesis causes severe developmental defects, resulting in various human genetic diseases, restricted RS in cancerous cells has profound clinical applications. First, a number of ribosome-free RPs induced by RS can enhance the activities of tumor suppressors. For example, p53 activity is highly boosted by ribosome-free RPs upon RS, as reduction of RPs, such as RPL5, RPL11, and others, dramatically impairs RS-induced p53-dependent cell cycle arrest and apoptosis in multiple tumor cells (Zhang and Lu, 2009; Zhou et al., 2012). Second, the ribosome-free RPs also inactivate oncogenic proteins, such as c-Myc (Dai et al., 2007; Challagundla et al., 2011; Liao et al., 2013; Zhou et al., 2013b). Additionally, rapidly and actively growing and proliferating tumor cells need more ribosome machineries compared with normal somatic cells, as evidenced by the fact that c-Myc-promoted tumor growth is dramatically abolished when the enhanced ribosome biogenesis in tumor is reduced to normal (Barna et al., 2008). This suggests that tumor cells are more sensitive to RS than normal somatic cells, and hence, targeting ribosome biogenesis of the tumor cells could be a reasonable strategy for the development of anti-cancer therapy. Anti-ribosome biogenesis drugs might be less toxic to normal and differentiated cells, as usually they would not cause DNA damage and thus are little genotoxic to normal cells, unlike traditional chemotherapeutic anti-cancer drugs, such as cisplatin and doxorubicin that are genotoxic, though they could induce RS as well. Apparently, selectively targeting ribosome biogenesis may be a more efficient and non-genotoxic strategy for developing new cancer therapeutics. Indeed, several anti-cancer chemotherapeutic agents that selectively target RNA Pol I and thereby induce RS have been identified. Transcription of human GC-rich rDNA is accompanied by the formation of a G-quadruplex DNA/nucleolin complex in non-template strands, which prevents renaturation of template DNA, facilitating RNA Pol I-mediated rDNA transcription. A small molecule CX-3543 has been identified as the first G-quadruplex-interacting agent that competes with nucleolin for binding to the G-quadruplex structures, consequently leading to specific inhibition of RNA Pol I-dependent transcription (Drygin et al., 2009). CX-5461, another specific RNA Pol I inhibitor identified by the same group, prevents the recruitment of SL1 to the RNA Pol I-responsive promoter and represses rDNA transcription initiation (Drygin et al., 2011). Interestingly, this inhibitor displays a remarkable anti-cancer effect as tested in an animal tumor model system by specifically targeting RNA Pol I-specific transcription (Bywater et al., 2012). It can elicit apoptosis in Eμ-Myc lymphoma cells by reducing RNA Pol I-driven transcription rate. More remarkably, CX-5461 administration selectively kills Eμ-Myc lymphoma cells, but not normal B cells, in vivo in a p53-dependent manner. This study again demonstrates that tumor cells are highly responsive to RS, and presents a great example of targeting ribosome biogenesis as a potential anti-cancer therapy.
Besides RNA Pol I, ribosome biogenesis also needs hundreds of other proteins, including RPs, RNA Pol III, and other non-ribosomal nucleolar factors. Theoretically, any of the nucleolar proteins could be a potential target for the development of anti-cancer treatment, as targeting each of them might cause RS. Systematical characterization of the functions of these proteins important for ribosome biogenesis in the nucleoli of cancer cells would effectively facilitate this development in the near future. Therefore, we anticipate that more anti-cancer drugs, and even more anti-immune drugs or gene therapy for genetic diseases, will be developed against ribosomal biogenesis in the coming years or decades.
Funding
H.L. was supported by NIH-NCI grants CA095441 and CA172468, and the Reynolds and Ryan Families chair fund.
Conflict of interest: none declared.
Acknowledgements
We thank the members in the Lu laboratory for active discussion.
References
- Allison A.C., Eugui E.M. (2005). Mechanisms of action of mycophenolate mofetil in preventing acute and chronic allograft rejection. Transplantation 80, S181–S190. [DOI] [PubMed] [Google Scholar]
- Anderson S.J., Lauritsen J.P., Hartman M.G., et al. (2007). Ablation of ribosomal protein L22 selectively impairs αβ T cell development by activation of a p53-dependent checkpoint. Immunity 26, 759–772. [DOI] [PubMed] [Google Scholar]
- Artero-Castro A., Castellvi J., Garcia A., et al. (2011). Expression of the ribosomal proteins Rplp0, Rplp1, and Rplp2 in gynecologic tumors. Hum. Pathol. 42, 194–203. [DOI] [PubMed] [Google Scholar]
- Austin K.M., Leary R.J., Shimamura A. (2005). The Shwachman-Diamond SBDS protein localizes to the nucleolus. Blood 106, 1253–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai D., Zhang J., Xiao W., et al. (2014). Regulation of the HDM2-p53 pathway by ribosomal protein L6 in response to ribosomal stress. Nucleic Acids Res. 42, 1799–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkic M., Crnomarkovic S., Grabusic K., et al. (2009). The p53 tumor suppressor causes congenital malformations in Rpl24-deficient mice and promotes their survival. Mol. Cell. Biol. 29, 2489–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow J.L., Drynan L.F., Hewett D.R., et al. (2010). A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q- syndrome. Nat. Med. 16, 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barna M., Pusic A., Zollo O., et al. (2008). Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 456, 971–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer A.R., Bann D.V., Rice B., et al. (2013). Nucleolar trafficking of the mouse mammary tumor virus gag protein induced by interaction with ribosomal protein L9. J. Virol. 87, 1069–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolze A., Mahlaoui N., Byun M., et al. (2013). Ribosomal protein SA haploinsufficiency in humans with isolated congenital asplenia. Science 340, 976–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boria I., Quarello P., Avondo F., et al. (2008). A new database for ribosomal protein genes which are mutated in Diamond-Blackfan anemia. Hum. Mutat. 29, E263–E270. [DOI] [PubMed] [Google Scholar]
- Boulon S., Westman B.J., Hutten S., et al. (2010). The nucleolus under stress. Mol. Cell 40, 216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger K., Muhl B., Harasim T., et al. (2010). Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J. Biol. Chem. 285, 12416–12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bywater M.J., Poortinga G., Sanij E., et al. (2012). Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 22, 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns C.A., White R.J. (1998). p53 is a general repressor of RNA polymerase III transcription. EMBO J. 17, 3112–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagnoli M.F., Ramenghi U., Armiraglio M., et al. (2008). RPS19 mutations in patients with Diamond-Blackfan anemia. Hum. Mutat. 29, 911–920. [DOI] [PubMed] [Google Scholar]
- Cardenas M.E., Cutler N.S., Lorenz M.C., et al. (1999). The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev. 13, 3271–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanaugh A.H., Hempel W.M., Taylor L.J., et al. (1995). Activity of RNA polymerase I transcription factor UBF blocked by Rb gene product. Nature 374, 177–180. [DOI] [PubMed] [Google Scholar]
- Challagundla K.B., Sun X.X., Zhang X., et al. (2011). Ribosomal protein L11 recruits miR-24/miRISC to repress c-Myc expression in response to ribosomal stress. Mol. Cell. Biol. 31, 4007–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., Dittmer D.P. (2011). Ribosomal protein S6 interacts with the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Virol. 85, 9495–9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D., Zhang Z., Li M., et al. (2007). Ribosomal protein S7 as a novel modulator of p53-MDM2 interaction: binding to MDM2, stabilization of p53 protein, and activation of p53 function. Oncogene 26, 5029–5037. [DOI] [PubMed] [Google Scholar]
- Chen B., Zhang W., Gao J., et al. (2014). Downregulation of ribosomal protein S6 inhibits the growth of non-small cell lung cancer by inducing cell cycle arrest, rather than apoptosis. Cancer Lett. 354, 378–389. [DOI] [PubMed] [Google Scholar]
- Cheng E., Haque A., Rimmer M.A., et al. (2011). Characterization of the Interaction between hantavirus nucleocapsid protein (N) and ribosomal protein S19 (RPS19). J. Biol. Chem. 286, 11814–11824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarmatori S., Scott P.H., Sutcliffe J.E., et al. (2001). Overlapping functions of the pRb family in the regulation of rRNA synthesis. Mol. Cell. Biol. 21, 5806–5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claypool J.A., French S.L., Johzuka K., et al. (2004). Tor pathway regulates Rrn3p-dependent recruitment of yeast RNA polymerase I to the promoter but does not participate in alteration of the number of active genes. Mol. Biol. Cell 15, 946–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cmejla R., Cmejlova J., Handrkova H., et al. (2007). Ribosomal protein S17 gene (RPS17) is mutated in Diamond-Blackfan anemia. Hum. Mutat. 28, 1178–1182. [DOI] [PubMed] [Google Scholar]
- Cmejla R., Cmejlova J., Handrkova H., et al. (2009). Identification of mutations in the ribosomal protein L5 (RPL5) and ribosomal protein L11 (RPL11) genes in Czech patients with Diamond-Blackfan anemia. Hum. Mutat. 30, 321–327. [DOI] [PubMed] [Google Scholar]
- Cordon-Cardo C., Reuter V.E. (1997). Alterations of tumor suppressor genes in bladder cancer. Semin. Diagn. Pathol. 14, 123–132. [PubMed] [Google Scholar]
- Cui D., Li L., Lou H., et al. (2014). The ribosomal protein S26 regulates p53 activity in response to DNA damage. Oncogene 33, 2225–2235. [DOI] [PubMed] [Google Scholar]
- Daftuar L., Zhu Y., Jacq X., et al. (2013). Ribosomal proteins RPL37, RPS15 and RPS20 regulate the Mdm2-p53-MdmX network. PLoS One 8, e68667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai M.S., Lu H. (2004). Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J. Biol. Chem. 279, 44475–44482. [DOI] [PubMed] [Google Scholar]
- Dai M.S., Lu H. (2008). Crosstalk between c-Myc and ribosome in ribosomal biogenesis and cancer. J. Cell. Biochem. 105, 670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai M.S., Zeng S.X., Jin Y., et al. (2004). Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol. Cell. Biol. 24, 7654–7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai M.S., Arnold H., Sun X.X., et al. (2007). Inhibition of c-Myc activity by ribosomal protein L11. EMBO J. 26, 3332–3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai M.S., Sun X.X., Lu H. (2008). Aberrant expression of nucleostemin activates p53 and induces cell cycle arrest via inhibition of MDM2. Mol. Cell. Biol. 28, 4365–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y., Pierson S.E., Dudney W.C., et al. (2010). Extraribosomal function of metallopanstimulin-1: reducing paxillin in head and neck squamous cell carcinoma and inhibiting tumor growth. Int. J. Cancer 126, 611–619. [DOI] [PubMed] [Google Scholar]
- Dai Y., Pierson S., Dudney C., et al. (2011). Ribosomal protein metallopanstimulin-1 impairs multiple myeloma CAG cells growth and inhibits fibroblast growth factor receptor 3. Clin. Lymphoma Myeloma Leuk. 11, 490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Keersmaecker K., Atak Z.K., Li N., et al. (2013). Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat. Genet. 45, 186–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Las Heras-Rubio A., Perucho L., Paciucci R., et al. (2014). Ribosomal proteins as novel players in tumorigenesis. Cancer Metastasis Rev. 33, 115–141. [DOI] [PubMed] [Google Scholar]
- Doherty L., Sheen M.R., Vlachos A., et al. (2010). Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond-Blackfan anemia. Am. J. Hum. Genet. 86, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donati G., Peddigari S., Mercer C.A., et al. (2013). 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep. 4, 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draptchinskaia N., Gustavsson P., Andersson B., et al. (1999). The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet. 21, 169–175. [DOI] [PubMed] [Google Scholar]
- Drygin D., Siddiqui-Jain A., O'Brien S., et al. (2009). Anticancer activity of CX-3543: a direct inhibitor of rRNA biogenesis. Cancer Res. 69, 7653–7661. [DOI] [PubMed] [Google Scholar]
- Drygin D., Lin A., Bliesath J., et al. (2011). Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 71, 1418–1430. [DOI] [PubMed] [Google Scholar]
- Duan J., Ba Q., Wang Z., et al. (2011). Knockdown of ribosomal protein S7 causes developmental abnormalities via p53 dependent and independent pathways in zebrafish. Int. J. Biochem. Cell Biol. 43, 1218–1227. [DOI] [PubMed] [Google Scholar]
- Duncan K.A., Jimenez P., Carruth L.L. (2009). The selective estrogen receptor-α coactivator, RPL7, and sexual differentiation of the songbird brain. Psychoneuroendocrinology 34(Suppl 1), S30–S38. [DOI] [PubMed] [Google Scholar]
- Dutt S., Narla A., Lin K., et al. (2011). Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 117, 2567–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert B.L., Pretz J., Bosco J., et al. (2008). Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 451, 335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis S.R. (2014). Nucleolar stress in Diamond Blackfan anemia pathophysiology. Biochim. Biophys. Acta 1842, 765–768. [DOI] [PubMed] [Google Scholar]
- Farrar J.E., Nater M., Caywood E., et al. (2008). Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond-Blackfan anemia. Blood 112, 1582–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatica A., Tollervey D. (2002). Making ribosomes. Curr. Opin. Cell Biol. 14, 313–318. [DOI] [PubMed] [Google Scholar]
- Filip A.M., Klug J., Cayli S., et al. (2009). Ribosomal protein S19 interacts with macrophage migration inhibitory factor and attenuates its pro-inflammatory function. J. Biol. Chem. 284, 7977–7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fregoso O.I., Das S., Akerman M., et al. (2013). Splicing-factor oncoprotein SRSF1 stabilizes p53 via RPL5 and induces cellular senescence. Mol. Cell 50, 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli S., Di Cara A., Neb-Gulati A., et al. (2009). Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat. Cell Biol. 11, 501–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli S., Ivanenkov V.V., Teng T., et al. (2012). Suprainduction of p53 by disruption of 40S and 60S ribosome biogenesis leads to the activation of a novel G2/M checkpoint. Genes Dev. 26, 1028–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathi K.A., Austin K.M., Lee C.S., et al. (2007). The human Shwachman-Diamond syndrome protein, SBDS, associates with ribosomal RNA. Blood 110, 1458–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X., Hardwidge P.R. (2011). Ribosomal protein s3: a multifunctional target of attaching/effacing bacterial pathogens. Front. Microbiol. 2, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X., Wan F., Mateo K., et al. (2009). Bacterial effector binding to ribosomal protein s3 subverts NF-κB function. PLoS Pathog. 5, e1000708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M., Li X., Dong W., et al. (2013). Ribosomal protein S7 regulates arsenite-induced GADD45α expression by attenuating MDM2-mediated GADD45α ubiquitination and degradation. Nucleic Acids Res. 41, 5210–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcon L., Ge J., Manjunath S.H., et al. (2013). Ribosomal and hematopoietic defects in induced pluripotent stem cells derived from Diamond Blackfan anemia patients. Blood 122, 912–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazda H.T., Grabowska A., Merida-Long L.B., et al. (2006). Ribosomal protein S24 gene is mutated in Diamond-Blackfan anemia. Am. J. Hum. Genet. 79, 1110–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazda H.T., Sheen M.R., Vlachos A., et al. (2008). Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am. J. Hum. Genet. 83, 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S., Karin M. (2002). Missing pieces in the NF-κB puzzle. Cell 109(Suppl), S81–S96. [DOI] [PubMed] [Google Scholar]
- Ghoshal K., Jacob S.T. (1994). Specific inhibition of pre-ribosomal RNA processing in extracts from the lymphosarcoma cells treated with 5-fluorouracil. Cancer Res. 54, 632–636. [PubMed] [Google Scholar]
- Green L., Houck-Loomis B., Yueh A., et al. (2012). Large ribosomal protein 4 increases efficiency of viral recoding sequences. J. Virol. 86, 8949–8958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripp K.W., Curry C., Olney A.H., et al. (2014). Diamond-Blackfan anemia with mandibulofacial dysostosis is heterogeneous, including the novel DBA genes TSR2 and RPS28. Am. J. Med. Genet. A 164A, 2240–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan H., Hou S., Ricciardi R.P. (2005). DNA binding of repressor nuclear factor-κB p50/p50 depends on phosphorylation of Ser337 by the protein kinase A catalytic subunit. J. Biol. Chem. 280, 9957–9962. [DOI] [PubMed] [Google Scholar]
- Guerra-Rebollo M., Mateo F., Franke K., et al. (2012). Nucleolar exit of RNF8 and BRCA1 in response to DNA damage. Exp. Cell Res. 318, 2365–2376. [DOI] [PubMed] [Google Scholar]
- Guo X., Shi Y., Gou Y., et al. (2011). Human ribosomal protein S13 promotes gastric cancer growth through down-regulating p27(Kip1). J. Cell. Mol. Med. 15, 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan K.M., Hannan R.D., Smith S.D., et al. (2000). Rb and p130 regulate RNA polymerase I transcription: Rb disrupts the interaction between UBF and SL-1. Oncogene 19, 4988–4999. [DOI] [PubMed] [Google Scholar]
- Hannan K.M., Brandenburger Y., Jenkins A., et al. (2003). mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol. Cell. Biol. 23, 8862–8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque A., Mir M.A. (2010). Interaction of hantavirus nucleocapsid protein with ribosomal protein S19. J. Virol. 84, 12450–12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz M.I., Landry D.M., Willis A.E., et al. (2013). Ribosomal protein S25 dependency reveals a common mechanism for diverse internal ribosome entry sites and ribosome shunting. Mol. Cell. Biol. 33, 1016–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe S., Bierhoff H., Cado I., et al. (2009). AMP-activated protein kinase adapts rRNA synthesis to cellular energy supply. Proc. Natl Acad. Sci. USA 106, 17781–17786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn H.F., Vousden K.H. (2008). Cooperation between the ribosomal proteins L5 and L11 in the p53 pathway. Oncogene 27, 5774–5784. [DOI] [PubMed] [Google Scholar]
- Horos R., Ijspeert H., Pospisilova D., et al. (2012). Ribosomal deficiencies in Diamond-Blackfan anemia impair translation of transcripts essential for differentiation of murine and human erythroblasts. Blood 119, 262–272. [DOI] [PubMed] [Google Scholar]
- Huang J.Y., Su W.C., Jeng K.S., et al. (2012). Attenuation of 40S ribosomal subunit abundance differentially affects host and HCV translation and suppresses HCV replication. PLoS Pathog. 8, e1002766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iapalucci-Espinoza S., Franze-Fernandez M.T. (1979). Effect of protein synthesis inhibitors and low concentrations of actinomycin D on ribosomal RNA synthesis. FEBS Lett. 107, 281–284. [DOI] [PubMed] [Google Scholar]
- Iizumi Y., Oishi M., Taniguchi T., et al. (2013). The flavonoid apigenin downregulates CDK1 by directly targeting ribosomal protein S9. PLoS One 8, e73219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaako P., Flygare J., Olsson K., et al. (2011). Mice with ribosomal protein S19 deficiency develop bone marrow failure and symptoms like patients with Diamond-Blackfan anemia. Blood 118, 6087–6096. [DOI] [PubMed] [Google Scholar]
- Jang C.Y., Kim H.D., Kim J. (2012). Ribosomal protein S3 interacts with TRADD to induce apoptosis through caspase dependent JNK activation. Biochem. Biophys. Res. Commun. 421, 474–478. [DOI] [PubMed] [Google Scholar]
- Jia J., Arif A., Willard B., et al. (2012). Protection of extraribosomal RPL13a by GAPDH and dysregulation by S-nitrosylation. Mol. Cell 47, 656–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin A., Itahana K., O'Keefe K., et al. (2004). Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol. Cell. Biol. 24, 7669–7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan P., Carmo-Fonseca M. (1998). Cisplatin inhibits synthesis of ribosomal RNA in vivo. Nucleic Acids Res. 26, 2831–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen P., Nishikawa J.L., Breitkreutz B.J., et al. (2002). Systematic identification of pathways that couple cell growth and division in yeast. Science 297, 395–400. [DOI] [PubMed] [Google Scholar]
- Jorgensen P., Rupes I., Sharom J.R., et al. (2004). A dynamic transcriptional network communicates growth potential to ribosome synthesis and critical cell size. Genes Dev. 18, 2491–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C., McLellan M.D., Vandin F., et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalaileh A., Dreazen A., Khatib A., et al. (2013). Phosphorylation of ribosomal protein S6 attenuates DNA damage and tumor suppression during development of pancreatic cancer. Cancer Res. 73, 1811–1820. [DOI] [PubMed] [Google Scholar]
- Kirn-Safran C.B., Oristian D.S., Focht R.J., et al. (2007). Global growth deficiencies in mice lacking the ribosomal protein HIP/RPL29. Dev. Dyn. 236, 447–460. [DOI] [PubMed] [Google Scholar]
- Klauck S.M., Felder B., Kolb-Kokocinski A., et al. (2006). Mutations in the ribosomal protein gene RPL10 suggest a novel modulating disease mechanism for autism. Mol. Psychiatry 11, 1073–1084. [DOI] [PubMed] [Google Scholar]
- Kondoh N., Shuda M., Tanaka K., et al. (2001). Enhanced expression of S8, L12, L23a, L27 and L30 ribosomal protein mRNAs in human hepatocellular carcinoma. Anticancer Res. 21, 2429–2433. [PubMed] [Google Scholar]
- Kondrashov N., Pusic A., Stumpf C.R., et al. (2011). Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 145, 383–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft C., Deplazes A., Sohrmann M., et al. (2008). Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat. Cell Biol. 10, 602–610. [DOI] [PubMed] [Google Scholar]
- Kruhlak M., Crouch E.E., Orlov M., et al. (2007). The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature 447, 730–734. [DOI] [PubMed] [Google Scholar]
- Kuroda K., Takenoyama M., Baba T., et al. (2010). Identification of ribosomal protein L19 as a novel tumor antigen recognized by autologous cytotoxic T lymphocytes in lung adenocarcinoma. Cancer Sci. 101, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam Y.W., Lamond A.I., Mann M., et al. (2007). Analysis of nucleolar protein dynamics reveals the nuclear degradation of ribosomal proteins. Curr. Biol. 17, 749–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambertsson A. (1998). The minute genes in Drosophila and their molecular functions. Adv. Genet. 38, 69–134. [DOI] [PubMed] [Google Scholar]
- Landowski M., O'Donohue M.F., Buros C., et al. (2013). Novel deletion of RPL15 identified by array-comparative genomic hybridization in Diamond-Blackfan anemia. Hum. Genet. 132, 1265–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry D.M., Hertz M.I., Thompson S.R. (2009). RPS25 is essential for translation initiation by the Dicistroviridae and hepatitis C viral IRESs. Genes Dev. 23, 2753–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.B., Kwon I.S., Park J., et al. (2010). Ribosomal protein S3, a new substrate of Akt, serves as a signal mediator between neuronal apoptosis and DNA repair. J. Biol. Chem. 285, 29457–29468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.S., Burdeinick-Kerr R., Whelan S.P. (2013). A ribosome-specialized translation initiation pathway is required for cap-dependent translation of vesicular stomatitis virus mRNAs. Proc. Natl Acad. Sci. USA 110, 324–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao J.M., Zhou X., Gatignol A., et al. (2013). Ribosomal proteins L5 and L11 co-operatively inactivate c-Myc via RNA-induced silencing complex. Oncogene 33, 4916–4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K.H., Kim K.H., Choi S.I., et al. (2011). RPS3a over-expressed in HBV-associated hepatocellular carcinoma enhances the HBx-induced NF-κB signaling via its novel chaperoning function. PLoS One 6, e22258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom M.S., Jin A., Deisenroth C., et al. (2007). Cancer-associated mutations in the MDM2 zinc finger domain disrupt ribosomal protein interaction and attenuate MDM2-induced p53 degradation. Mol. Cell. Biol. 27, 1056–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llanos S., Serrano M. (2010). Depletion of ribosomal protein L37 occurs in response to DNA damage and activates p53 through the L11/MDM2 pathway. Cell Cycle 9, 4005–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohrum M.A., Ludwig R.L., Kubbutat M.H., et al. (2003). Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 3, 577–587. [DOI] [PubMed] [Google Scholar]
- Lopez-Diaz F.J., Gascard P., Balakrishnan S.K., et al. (2013). Coordinate transcriptional and translational repression of p53 by TGF-β1 impairs the stress response. Mol. Cell 50, 552–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv J., Huang X.R., Klug J., et al. (2013). Ribosomal protein S19 is a novel therapeutic agent in inflammatory kidney disease. Clin. Sci. (Lond) 124, 627–637. [DOI] [PubMed] [Google Scholar]
- Macias E., Jin A., Deisenroth C., et al. (2010). An ARF-independent c-MYC-activated tumor suppression pathway mediated by ribosomal protein-Mdm2 Interaction. Cancer Cell 18, 231–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal V., Elenbaas B., Piette J., et al. (1994). The ribosomal L5 protein is associated with mdm-2 and mdm-2-p53 complexes. Mol. Cell. Biol. 14, 7414–7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion R.M., Regev A., Segal E., et al. (2004). Sfp1 is a stress- and nutrient-sensitive regulator of ribosomal protein gene expression. Proc. Natl Acad. Sci. USA 101, 14315–14322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D.E., Soulard A., Hall M.N. (2004). TOR regulates ribosomal protein gene expression via PKA and the Forkhead transcription factor FHL1. Cell 119, 969–979. [DOI] [PubMed] [Google Scholar]
- Martin I., Kim J.W., Lee B.D., et al. (2014). Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson's disease. Cell 157, 472–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsson H., Davey E.J., Draptchinskaia N., et al. (2004). Targeted disruption of the ribosomal protein S19 gene is lethal prior to implantation. Mol. Cell. Biol. 24, 4032–4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer C., Grummt I. (2006). Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene 25, 6384–6391. [DOI] [PubMed] [Google Scholar]
- Mayer C., Zhao J., Yuan X., et al. (2004). mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev. 18, 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder B., Sampath P., Seshadri V., et al. (2003). Regulated release of L13a from the 60S ribosomal subunit as a mechanism of transcript-specific translational control. Cell 115, 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan K.A., Li J.Z., Park C.Y., et al. (2008). Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat. Genet. 40, 963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menne T.F., Goyenechea B., Sanchez-Puig N., et al. (2007). The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat. Genet. 39, 486–495. [DOI] [PubMed] [Google Scholar]
- Meyuhas O. (2000). Synthesis of the translational apparatus is regulated at the translational level. Eur. J. Biochem. 267, 6321–6330. [DOI] [PubMed] [Google Scholar]
- Miller S.A., Brown A.J., Farach-Carson M.C., et al. (2003). HIP/RPL29 down-regulation accompanies terminal chondrocyte differentiation. Differentiation 71, 322–336. [DOI] [PubMed] [Google Scholar]
- Mirabello L., Macari E.R., Jessop L., et al. (2014). Whole-exome sequencing and functional studies identify RPS29 as a novel gene mutated in multicase Diamond-Blackfan anemia families. Blood 124, 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgado-Palacin L., Llanos S., Serrano M. (2012). Ribosomal stress induces L11- and p53-dependent apoptosis in mouse pluripotent stem cells. Cell Cycle 11, 503–510. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay R., Ray P.S., Arif A., et al. (2008). DAPK-ZIPK-L13a axis constitutes a negative-feedback module regulating inflammatory gene expression. Mol. Cell 32, 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay R., Jia J., Arif A., et al. (2009). The GAIT system: a gatekeeper of inflammatory gene expression. Trends Biochem. Sci. 34, 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naora H., Takai I., Adachi M. (1998). Altered cellular responses by varying expression of a ribosomal protein gene: sequential coordination of enhancement and suppression of ribosomal protein S3a gene expression induces apoptosis. J. Cell Biol. 141, 741–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narla A., Ebert B.L. (2010). Ribosomopathies: human disorders of ribosome dysfunction. Blood 115, 3196–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama T., Yamamoto H., Uchiumi T., et al. (2007). Eukaryotic ribosomal protein RPS25 interacts with the conserved loop region in a dicistroviral intergenic internal ribosome entry site. Nucleic Acids Res. 35, 1514–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofir-Rosenfeld Y., Boggs K., Michael D., et al. (2008). Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol. Cell 32, 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares E., Landry D.M., Caceres C.J., et al. (2014). The 5′ untranslated region of the human T-cell lymphotropic virus type 1 mRNA enables cap-independent translation initiation. J. Virol. 88, 5936–5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oristian D.S., Sloofman L.G., Zhou X., et al. (2009). Ribosomal protein L29/HIP deficiency delays osteogenesis and increases fragility of adult bone in mice. J. Orthop. Res. 27, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry R.P., Kelley D.E. (1970). Inhibition of RNA synthesis by actinomycin D: characteristic dose-response of different RNA species. J. Cell. Physiol. 76, 127–139. [DOI] [PubMed] [Google Scholar]
- Perucho L., Artero-Castro A., Guerrero S., et al. (2014). RPLP1, a crucial ribosomal protein for embryonic development of the nervous system. PLoS One 9, e99956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaksin D., Baeuerle P.A., Eisenbach L. (1993). KBF1 (p50 NF-κB homodimer) acts as a repressor of H-2Kb gene expression in metastatic tumor cells. J. Exp. Med. 177, 1651–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provost E., Wehner K.A., Zhong X., et al. (2012). Ribosomal biogenesis genes play an essential and p53-independent role in zebrafish pancreas development. Development 139, 3232–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provost E., Weier C.A., Leach S.D. (2013). Multiple ribosomal proteins are expressed at high levels in developing zebrafish endoderm and are required for normal exocrine pancreas development. Zebrafish 10, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S., Lee S.Y., Gutierrez A., et al. (2012). Inactivation of ribosomal protein L22 promotes transformation by induction of the stemness factor, Lin28B. Blood 120, 3764–3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray P.S., Fox P.L. (2007). A post-transcriptional pathway represses monocyte VEGF-A expression and angiogenic activity. EMBO J. 26, 3360–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanova L., Grand A., Zhang L., et al. (2009). Critical role of nucleostemin in pre-rRNA processing. J. Biol. Chem. 284, 4968–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubbi C.P., Milner J. (2003). Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 22, 6068–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudra D., Zhao Y., Warner J.R. (2005). Central role of Ifh1p-Fhl1p interaction in the synthesis of yeast ribosomal proteins. EMBO J. 24, 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggero D., Pandolfi P.P. (2003). Does the ribosome translate cancer? Nat. Rev. Cancer 3, 179–192. [DOI] [PubMed] [Google Scholar]
- Saeboe-Larssen S., Lyamouri M., Merriam J., et al. (1998). Ribosomal protein insufficiency and the minute syndrome in Drosophila: a dose-response relationship. Genetics 148, 1215–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath P., Mazumder B., Seshadri V., et al. (2004). Noncanonical function of glutamyl-prolyl-tRNA synthetase: gene-specific silencing of translation. Cell 119, 195–208. [DOI] [PubMed] [Google Scholar]
- Sasaki M., Kawahara K., Nishio M., et al. (2011). Regulation of the MDM2-P53 pathway and tumor growth by PICT1 via nucleolar RPL11. Nat. Med. 17, 944–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schawalder S.B., Kabani M., Howald I., et al. (2004). Growth-regulated recruitment of the essential yeast ribosomal protein gene activator Ifh1. Nature 432, 1058–1061. [DOI] [PubMed] [Google Scholar]
- Sen N., Paul B.D., Gadalla M.M., et al. (2012). Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell 45, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Zhai H., Wang X., et al. (2004). Ribosomal proteins S13 and L23 promote multidrug resistance in gastric cancer cells by suppressing drug-induced apoptosis. Exp. Cell Res. 296, 337–346. [DOI] [PubMed] [Google Scholar]
- Shi W., Zhang X., Jiang X., et al. (2011). Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 333, 1630–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuda M., Kondoh N., Tanaka K., et al. (2000). Enhanced expression of translation factor mRNAs in hepatocellular carcinoma. Anticancer Res. 20, 2489–2494. [PubMed] [Google Scholar]
- Silvera D., Formenti S.C., Schneider R.J. (2010). Translational control in cancer. Nat. Rev. Cancer 10, 254–266. [DOI] [PubMed] [Google Scholar]
- Smolock E.M., Korshunov V.A., Glazko G., et al. (2012). Ribosomal protein L17, RpL17, is an inhibitor of vascular smooth muscle growth and carotid intima formation. Circulation 126, 2418–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.X., Dai M.S., Lu H. (2007). 5-fluorouracil activation of p53 involves an MDM2-ribosomal protein interaction. J. Biol. Chem. 282, 8052–8059. [DOI] [PubMed] [Google Scholar]
- Sun X.X., Dai M.S., Lu H. (2008). Mycophenolic acid activation of p53 requires ribosomal proteins L5 and L11. J. Biol. Chem. 283, 12387–12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.X., Wang Y.G., Xirodimas D.P., et al. (2010). Perturbation of 60 S ribosomal biogenesis results in ribosomal protein L5- and L11-dependent p53 activation. J. Biol. Chem. 285, 25812–25821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.X., DeVine T., Challagundla K.B., et al. (2011). Interplay between ribosomal protein S27a and MDM2 protein in p53 activation in response to ribosomal stress. J. Biol. Chem. 286, 22730–22741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe J.E., Brown T.R., Allison S.J., et al. (2000). Retinoblastoma protein disrupts interactions required for RNA polymerase III transcription. Mol. Cell. Biol. 20, 9192–9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi M., Absalon M.J., McLure K.G., et al. (2005). Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 123, 49–63. [DOI] [PubMed] [Google Scholar]
- Taylor A.M., Humphries J.M., White R.M., et al. (2012). Hematopoietic defects in rps29 mutant zebrafish depend upon p53 activation. Exp. Hematol. 40, 228–237 e225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzian T., Dumble M., Arbab F., et al. (2011). Rpl27a mutation in the sooty foot ataxia mouse phenocopies high p53 mouse models. J. Pathol. 224, 540–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai R.Y., McKay R.D. (2005). A multistep, GTP-driven mechanism controlling the dynamic cycling of nucleostemin. J. Cell Biol. 168, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschochner H., Hurt E. (2003). Pre-ribosomes on the road from the nucleolus to the cytoplasm. Trends Cell Biol. 13, 255–263. [DOI] [PubMed] [Google Scholar]
- Tsofack S.P., Meunier L., Sanchez L., et al. (2013). Low expression of the X-linked ribosomal protein S4 in human serous epithelial ovarian cancer is associated with a poor prognosis. BMC Cancer 13, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Riggelen J., Yetil A., Felsher D.W. (2010). MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 10, 301–309. [DOI] [PubMed] [Google Scholar]
- Volarevic S., Stewart M.J., Ledermann B., et al. (2000). Proliferation, but not growth, blocked by conditional deletion of 40S ribosomal protein S6. Science 288, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Vyas K., Chaudhuri S., Leaman D.W., et al. (2009). Genome-wide polysome profiling reveals an inflammation-responsive posttranscriptional operon in γ interferon-activated monocytes. Mol. Cell. Biol. 29, 458–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan F., Anderson D.E., Barnitz R.A., et al. (2007). Ribosomal protein S3: a KH domain subunit in NF-κB complexes that mediates selective gene regulation. Cell 131, 927–939. [DOI] [PubMed] [Google Scholar]
- Wan F., Weaver A., Gao X., et al. (2011). IKKβ phosphorylation regulates RPS3 nuclear translocation and NF-κB function during infection with Escherichia coli strain O157:H7. Nat. Immunol. 12, 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A., Xu S., Zhang X., et al. (2011). Ribosomal protein RPL41 induces rapid degradation of ATF4, a transcription factor critical for tumour cell survival in stress. J. Pathol. 225, 285–292. [DOI] [PubMed] [Google Scholar]
- Wang Z., Hou J., Lu L., et al. (2013). Small ribosomal protein subunit S7 suppresses ovarian tumorigenesis through regulation of the PI3K/AKT and MAPK pathways. PLoS One 8, e79117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Luo J., Nian Q., et al. (2014). Ribosomal protein S14 silencing inhibits growth of acute myeloid leukemia transformed from myelodysplastic syndromes via activating p53. Hematology 19, 225–231. [DOI] [PubMed] [Google Scholar]
- Wang W., Nag S., Zhang X., et al. (2015). Ribosomal proteins and human diseases: pathogenesis, molecular mechanisms, and therapeutic implications. Med. Res. Rev. 35, 225–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanzel M., Russ A.C., Kleine-Kohlbrecher D., et al. (2008). A ribosomal protein L23-nucleophosmin circuit coordinates Mizl function with cell growth. Nat. Cell Biol. 10, 1051–1061. [DOI] [PubMed] [Google Scholar]
- Warner J.R., McIntosh K.B. (2009). How common are extraribosomal functions of ribosomal proteins? Mol. Cell 34, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins-Chow D.E., Cooke J., Pidsley R., et al. (2013). Mutation of the diamond-blackfan anemia gene Rps7 in mouse results in morphological and neuroanatomical phenotypes. PLoS Genet. 9, e1003094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R.J., Trouche D., Martin K., et al. (1996). Repression of RNA polymerase III transcription by the retinoblastoma protein. Nature 382, 88–90. [DOI] [PubMed] [Google Scholar]
- Wood J., Frederickson R.M., Fields S., et al. (2001). Hepatitis C virus 3′X region interacts with human ribosomal proteins. J. Virol. 75, 1348–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X., Zhao Y., He H., et al. (2011). Ribosomal protein S27-like and S27 interplay with p53-MDM2 axis as a target, a substrate and a regulator. Oncogene 30, 1798–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X., Zhao Y., Tang F., et al. (2014). Ribosomal protein S27-like is a physiological regulator of p53 that suppresses genomic instability and tumorigenesis. Elife 3, e02236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadavilli S., Mayo L.D., Higgins M., et al. (2009). Ribosomal protein S3: A multi-functional protein that interacts with both p53 and MDM2 through its KH domain. DNA Repair 8, 1215–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.Y., Qu Y., Zhang Q., et al. (2012). Knockdown of metallopanstimulin-1 inhibits NF-κB signaling at different levels: the role of apoptosis induction of gastric cancer cells. Int. J. Cancer 130, 2761–2770. [DOI] [PubMed] [Google Scholar]
- Yang H.J., Youn H., Seong K.M., et al. (2013a). Phosphorylation of ribosomal protein S3 and antiapoptotic TRAF2 protein mediates radioresistance in non-small cell lung cancer cells. J. Biol. Chem. 288, 2965–2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.Y., Jiang H., Qu Y., et al. (2013b). Metallopanstimulin-1 regulates invasion and migration of gastric cancer cells partially through integrin β4. Carcinogenesis 34, 2851–2860. [DOI] [PubMed] [Google Scholar]
- Zaragoza D., Ghavidel A., Heitman J., et al. (1998). Rapamycin induces the G0 program of transcriptional repression in yeast by interfering with the TOR signaling pathway. Mol. Cell. Biol. 18, 4463–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatsepina O.V., Voronkova L.N., Sakharov V.N., et al. (1989). Ultrastructural changes in nucleoli and fibrillar centers under the effect of local ultraviolet microbeam irradiation of interphase culture cells. Exp. Cell Res. 181, 94–104. [DOI] [PubMed] [Google Scholar]
- Zhai W., Comai L. (2000). Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol. Cell. Biol. 20, 5930–5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Lu H. (2009). Signaling to p53: ribosomal proteins find their way. Cancer Cell 16, 369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Wolf G.W., Bhat K., et al. (2003). Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol. Cell. Biol. 23, 8902–8912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S., Shi M., Hui C.C., et al. (2006). Loss of the mouse ortholog of the Shwachman-Diamond syndrome gene (Sbds) results in early embryonic lethality. Mol. Cell. Biol. 26, 6656–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Wang J., Yuan Y., et al. (2010). Negative regulation of HDM2 to attenuate p53 degradation by ribosomal protein L26. Nucleic Acids Res. 38, 6544–6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.X., Liu Z.X., Sun Y.P., et al. (2013a). Rig-I regulates NF-κB activity through binding to Nf-κb1 3′-UTR mRNA. Proc. Natl Acad. Sci. USA 110, 6459–6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Wang W., Wang H., et al. (2013b). Identification of ribosomal protein S25 (RPS25)-MDM2-p53 regulatory feedback loop. Oncogene 32, 2782–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C., Zang D., Jin Y., et al. (2011). Mutation in ribosomal protein L21 underlies hereditary hypotrichosis simplex. Hum. Mutat. 32, 710–714. [DOI] [PubMed] [Google Scholar]
- Zhou X., Liao J.M., Liao W.J., et al. (2012). Scission of the p53-MDM2 Loop by Ribosomal Proteins. Genes Cancer 3, 298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Hao Q., Liao J., et al. (2013a). Ribosomal protein S14 unties the MDM2-p53 loop upon ribosomal stress. Oncogene 32, 388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Hao Q., Liao J.M., et al. (2013b). Ribosomal protein S14 negatively regulates c-Myc activity. J. Biol. Chem. 288, 21793–21801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Hao Q., Zhang Q., et al. (2014). Ribosomal proteins L11 and L5 activate TAp73 by overcoming MDM2 inhibition. Cell Death Differ. 22, 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y., Poyurovsky M.V., Li Y., et al. (2009). Ribosomal protein S7 is both a regulator and a substrate of MDM2. Mol. Cell 35, 316–326. [DOI] [PMC free article] [PubMed] [Google Scholar]