

Abstract
Summary: High-throughput omics datasets often contain technical replicates included to account for technical sources of noise in the measurement process. Although summarizing these replicate measurements by using robust averages may help to reduce the influence of noise on downstream data analysis, the information on the variance across the replicate measurements is lost in the averaging process and therefore typically disregarded in subsequent statistical analyses.
We introduce RepExplore, a web-service dedicated to exploit the information captured in the technical replicate variance to provide more reliable and informative differential expression and abundance statistics for omics datasets. The software builds on previously published statistical methods, which have been applied successfully to biomedical omics data but are difficult to use without prior experience in programming or scripting. RepExplore facilitates the analysis by providing a fully automated data processing and interactive ranking tables, whisker plot, heat map and principal component analysis visualizations to interpret omics data and derived statistics.
Availability and implementation: Freely available at http://www.repexplore.tk
Contact: enrico.glaab@uni.lu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Technical noise is a common limitation in many high-throughput biological experiments. Both mass spectrometry devices for proteomics and metabolomics profiling as well as gene and protein microarray platforms can only provide a limited reproducibility (Albrethsen, 2007; Chen et al., 2007). In combination with the biological variance observed across different omics samples under the same condition, the technical variability can significantly aggravate the statistical analysis of the data, increasing the risk for spurious and misinterpreted results.
A common approach to reduce the influence of noise on the statistical analysis of omics data is to use technical replicate measurements, e.g. for mass spectrometry data collecting three technical replicates per biological sample is a typical setting. During data pre-processing the replicate measurements are summarized to average values, by determining the mean, median or a trimmed mean, to reduce the influence of noise in downstream data analysis. However, the variance across replicate measurements often differs significantly between the biological samples and this data on measurement uncertainty is not retained by the summarization and consequently not considered in following statistical analyses.
Approaches to exploit technical variance information to improve robustness and sensitivity in downstream data analyses have been developed in recent years for differential expression analysis (Liu et al., 2006), principal component analysis (PCA; Sanguinetti et al., 2005) and differential pathway analysis (Glaab and Schneider, 2012). To enable users with limited or no programming experience to benefit from these new techniques to propagate variance information to downstream analyses, we have developed RepExplore, a web-service to analyze proteomics and metabolomics data with technical and biological replicates. The software takes advantage of available replicate variance data to derive more robust and informative differential expression and abundance statistics, whisker plot and PCA visualizations for omics data interpretation. All results, including interactive ranking tables, 2D and 3D PCA visualizations, bar charts and heat maps are generated automatically within few minutes for a typical dataset.
2 Workflow and methods
Analyzing omics data with RepExplore requires only the upload of a tab-delimited dataset containing both technical and biological replicates (all parameter settings on the web-interface are optional). The data is processed automatically and the results can be explored interactively in the web-browser.
Input: The only input required for RepExplore is a pre-processed proteomics or metabolomics dataset of log-scale intensity measurements in tab-delimited format with labels for biological and technical replicates (example data can be downloaded or analyzed directly on the main web-interface). Optionally, the user can choose to include further normalization procedures, e.g. to ensure that all samples have the same median value (using a median scaling normalization) or to remove dependencies between the signal variance and average signal intensity (using a variance-stabilizing normalization, see Huber et al., 2002).
Processing: After submitting an analysis task, the data is processed in the background and a temporary status page is loaded, redirecting the user to the results page after a short waiting time (typically up to a few minutes depending on the dataset size; for large datasets with a limit of 100 MB the status page can be bookmarked). During the statistical data processing, information on measurement uncertainty derived from the variance across technical replicates is exploited using the probability of positive log ratio (PPLR) statistic (Liu et al., 2006; Pearson et al., 2009) to score the differential abundance/expression of biomolecules across the biological conditions. This method takes both summarized point estimates and variation across the replicates into account to obtain a more robust ranking of biomolecules [for comparison, results on the mean-summarized replicates are generated additionally by applying the widely used empirical Bayes moderated t-statistic, here referred to as eBayes (Smyth, 2004)]. Similarly, to generate PCA results, the replicate variance data is extracted and used to reduce the influence of noise on the PCA computation (see Sanguinetti et al., 2005).
Output: The main result of a submitted analysis is an interactive, sortable ranking table, listing the PPLR and eBayes significance scores and the fold-changes as effect size measure for each biomolecule and allowing the user to generate whisker plots for all table entries of interest. If the user has chosen to generate a PCA visualization, a 2D plot of the first two principal components is shown, revealing potential grouping patterns among the samples or facilitating the recognition of outlier samples. Additionally, the user can view a navigable 3D PCA visualization (Glaab et al., 2010) of the first three principal components by using a VRML browser plugin or an offline VRML-viewer (see Tutorial section on the web-page). Finally, to investigate the separability of sample sub-groups a web-based, interactive heat map visualization using average linkage hierarchical clustering is provided for the top-ranked biomolecules (Deu-Pons et al., 2014).
Methods and previous validation: In functional genomics datasets the measured signal for a biomolecule on logarithmic scale is commonly assumed to have an approximate normal distribution (Karpievitch et al., 2009; Sabatine et al., 2005; Sjögren et al., 2007) and to depend on the mean expression/abundance level μi and the between-replicate variance λi for biological conditions indexed by i. If the technical replicate variance νij for condition i and replicate j is taken into account additionally and assumed to follow a normal distribution centered at zero, the measured signal yij can be modeled as follows (Liu et al., 2006):
| (1) |
where the parameters μi and λi are to be determined. The PPLR approach estimates these parameters using a variational Expectation-Maximization (EM) algorithm, modeling them as independent and λ as shared across the biological conditions. The parameter estimates are then used to calculate a differential expression/abundance score, reflecting the PPLR between specified conditions in the input data.
In the same spirit, to reduce the influence of technical noise in PCA, a further dedicated approach has been developed to exploit replicate variance information for PCA computation (Sanguinetti et al., 2005). This method is derived from the interpretation of PCA as the maximum likelihood solution of a probabilistic factor analysis model (Tipping and Bishop, 1999) into which the technical variance is integrated as an additional term (for the detailed derivation, see Sanguinetti et al., 2005). Optimal model parameters are again estimated using an iterative EM algorithm.
These statistical methods have previously been validated on benchmark omics datasets, resulting in improved accuracy in identifying differential abundance patterns (Liu et al., 2006) and tighter sample clusterings (Sanguinetti et al., 2005). In the Supplementary Material, we use multiple proteomic and metabolomic datasets to compare the results obtained from the PPLR method with the eBayes approach, a modification of the classical t-statistic using an empirical Bayes method to shrink the estimated sample variances towards a pooled estimate, providing a more stable inference for small numbers of samples (Smyth, 2004). As a final supplemental analysis, we compare the PPLR results obtained for different numbers of technical replicates on simulated data, showing that the ranking statistics improve with increasing numbers of replicates.
3 Results
To illustrate RepExplore’s features and the results obtainable on typical experimental data, we have applied the software to a metabolomics dataset comparing wild-type samples from the plant Arabidopsis thaliana against the mutant mapk phosphatase 1 (mkp1), which is more resistant to bacterial infection (Anderson et al., 2014, see datasets overview in the Supplementary Material).
As shown in the whisker plot in Figure 1a, for the top-ranked metabolite identified using a standard eBayes analysis with mean-summarized intensities (l-valine) the overlap of the value ranges for the technical replicates across the two sample groups covers the complete value range of the wild-type samples (only the summarized intensity values are non-overlapping and would suggest a significant difference in the metabolite abundance between the groups). By contrast, for the top-ranked metabolite according to the PPLR score (l-proline), the value ranges of the technical replicates do not display any overlap across the sample groups and the overall replicate variance is significantly smaller (see Fig. 1b). Thus, the whisker plots reveal that the evidence for the induction of l-proline is more reliable than for l-valine, highlighting the benefit of accounting for replicate variance information within the differential abundance statistic.
Fig. 1.
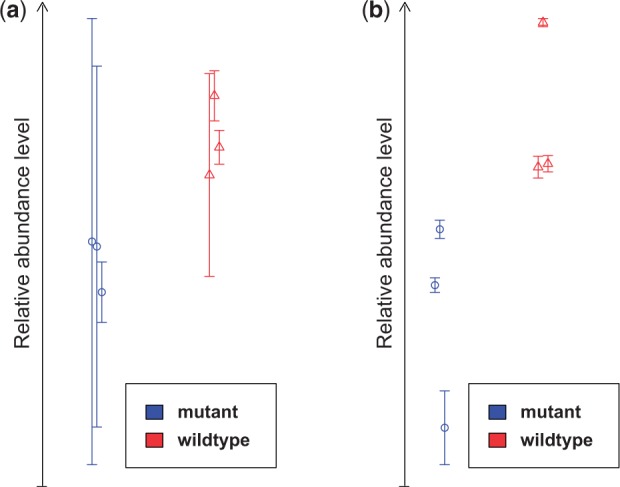
(a) Whisker plot for the top differentially abundant metabolite (l-valine) in the Arabidopsis dataset according to the eBayes approach applied to the mean-summarized replicates; (b) Whisker plot for the top differentially abundant metabolite (l-proline) according to the PPLR score (circle and triangle symbols represent the sample averages of mutant, resp. wild-type samples, vertical lines represent the technical error per biological sample)
Ranking tables of metabolites comparing the PPLR and eBayes statistics, heat map visualizations of the metabolite abundance differences between the knockdown and wild-type samples, and further whisker plots for this and other datasets are provided in the Supplementary Materials. The same metabolomics and proteomics example datasets can also be analyzed in an automated fashion on the RepExplore web-application, which enables an interactive exploration of the results (ranking tables are sortable and support the generation of whisker plots for chosen metabolites; the 3D PCA plots provides zoom, pan and rotate functionality, and meta-information is displayed when clicking on a chosen column/row entry in a heat map or on a data point in the 3D plots).
In summary, RepExplore interlinks the automated application of statistical analyses exploiting technical replicate variance information with web-based features to facilitate data exploration via interactive ranking tables and visualizations of the differential expression/abundance patterns. In addition to the public web-application, an exposed programmatic web-service API can be used to control the software, enabling an efficient analysis of multiple large-scale omics datasets.
4 Implementation
Statistical data processing and analysis methods were all implemented in the R statistical programming language. The web-application providing access to these statistics is written in PHP and runs on an Apache web-server. To guide the user on how to use the software, a detailed tutorial, help windows for specific features and example datasets from different case/control and wild-type/knockout studies are provided on the web page at http://www.repexplore.tk.
Conflict of Interest: none declared.
Supplementary Material
References
- Albrethsen J. (2007) Reproducibility in protein profiling by MALDI-TOF mass spectrometry. Clin. Chem. , 53, 852–858. [DOI] [PubMed] [Google Scholar]
- Anderson J.C., et al. (2014) Decreased abundance of type III secretion system-inducing signals in arabidopsis mkp1 enhances resistance against pseudomonas syringae. Proc. Natl. Acad. Sci. USA , 111,6846–6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.J., et al. (2007) Reproducibility of microarray data: a further analysis of microarray quality control (MAQC) data. BMC Bioinformatics , 8, 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deu-Pons J., et al. (2014) jheatmap: an interactive heatmap viewer for the web. Bioinformatics , 30, 1757–1758. [DOI] [PubMed] [Google Scholar]
- Glaab E., et al. (2010) vrmlgen: an R package for 3D data visualization on the web. J. Stat. Soft. , 36, 1–18. [Google Scholar]
- Glaab E., Schneider R. (2012) PathVar: analysis of gene and protein expression variance in cellular pathways using microarray data. Bioinformatics, 28, 446–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W., et al. (2002) Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics , 18, S96–S104. [DOI] [PubMed] [Google Scholar]
- Karpievitch Y., et al. (2009) A statistical framework for protein quantitation in bottom-up MS-based proteomics. Bioinformatics , 25, 2028–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., et al. (2006) Probe-level measurement error improves accuracy in detecting differential gene expression. Bioinformatics , 22, 2107–2113. [DOI] [PubMed] [Google Scholar]
- Pearson R., et al. (2009) puma: a Bioconductor package for propagating uncertainty in microarray analysis. BMC Bioinformatics , 10, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatine M.S., et al. (2005) Metabolomic identification of novel biomarkers of myocardial ischemia. Circulation , 112, 3868–3875. [DOI] [PubMed] [Google Scholar]
- Sanguinetti G., et al. (2005) Accounting for probe-level noise in principal component analysis of microarray data. Bioinformatics , 21, 3748–3754. [DOI] [PubMed] [Google Scholar]
- Sjögren A., et al. (2007) Weighted analysis of general microarray experiments. BMC Bioinformatics , 8, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G.K. (2004) Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. , 3, article 3. [DOI] [PubMed] [Google Scholar]
- Tipping M.E., Bishop C.M. (1999) Probabilistic principal component analysis. J. R. Stat. Soc. Series B Stat. Methodol. , 61, 611–622. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.