

Abstract
Tumour formation is blocked by two barriers, replicative senescence and crisis1. Senescence is triggered by short telomeres and is bypassed by disruption of tumour suppressive pathways. After senescence bypass, cells undergo crisis, during which almost all of the cells in the population die. Cells that escape crisis harbor unstable genomes and other parameters of transformation. The mechanism of cell death during crisis remained elusive. We show that cells in crisis undergo spontaneous mitotic arrest, resulting in death during mitosis or in the following cell cycle. The phenotype was induced by loss of p53 function, and suppressed by telomerase overexpression. Telomere fusions triggered mitotic arrest in p53-compromised non-crisis cells, indicating such fusions as the underlying cause. Exacerbation of mitotic telomere deprotection by partial TRF2 knockdown2 increased the ratio of cells that died during mitotic arrest and sensitized cancer cells to mitotic poisons. We propose a crisis pathway wherein chromosome fusions induce mitotic arrest, resulting in mitotic telomere deprotection and cell death, thereby eliminating precancerous cells from the population.
Replicative senescence is induced by partially deprotected telomeres, which activate a DNA damage response (DDR) without telomere fusions2. Crisis requires the bypass of senescence through loss of checkpoints and causes massive cell death concomitant with further telomere shortening and spontaneous telomere fusions. However, the mechanism of cell death was not understood. Mitotic arrest is associated with spindle assembly checkpoint (SAC) independent telomere deprotection3, and we therefore set out to test whether prolonged mitosis could play a role.
We monitored mitotic duration using live cell imaging. Mitosis in primary IMR-90 fibroblasts lasted <45 min. However, IMR-90 fibroblasts expressing HPV16 E6 and E7, which inhibit p53 and Rb4, displayed variable mitotic duration upon senescence bypass (Fig. 1a, b). Prolonged mitosis, defined as mitosis of >2 h, became prominent in pre-crisis cells (Extended Data Fig. 1a). Spontaneous mitotic arrest also occurred in pre-crisis cells following expression of E6 or dominant-negative p53dd (Extended Data Fig. 1b), indicating that loss of p53 function was required (Fig. 1c, d and Extended Data Fig. 1c, d).
Fig. 1. Spontaneous mitotic arrest upon bypass of senescence.
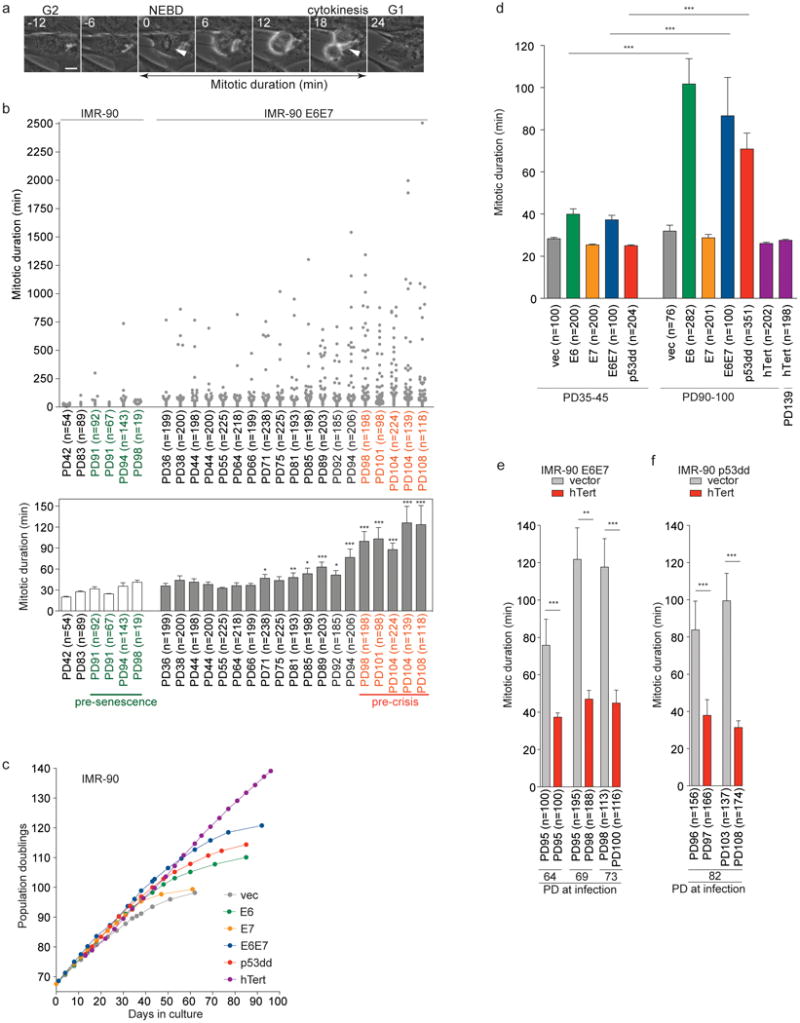
a, Representative movie frames of a normal mitosis. Arrows, nuclear envelope break down and cytokinesis. Scale Bar, 10 µm. b, Mitotic duration of individual indicated cells (upper panel). Mean mitotic duration ± s.e.m. (lower panel) (Man-Whitney test, compared to IMR-90 E6E7 PD36). c, Growth curves of indicated IMR-90 derivatives. d, Mitotic duration of indicated derivatives as in b. Results were reproduced independently at least twice. e, f, Mitotic duration of IMR-90 E6E7 (e) or p53dd (f) cells expressing hTert as in b. Cells were infected at indicated PD. *P<0.05, **P<0.005, ***P<0.0001. Mann-Whitney test.
Overexpressing hTERT5,6 prevented senescence in IMR-90 cells (Fig. 1c, d and Extended Data Fig. 1c, d). Telomere elongation in IMR-90 E6E7 or p53dd cells also suppressed mitotic arrest (Fig. 1e, f and Extended Data Fig. 2a-c), confirming telomere shortening as the cause. Reversine inhibition of MPS17 suppressed mitotic arrest (Extended Data Fig. 1e), indicating dependence on the SAC. Hesperadin, an Aurora B kinase inhibitor required for activation of the SAC upon tensionless kinetochore-microtubule attachment8, suppressed mitotic arrest (Extended Data Fig. 1e), suggesting abnormal kinetochore-microtubule attachment.
To determine if telomere fusion causes mitotic arrest, we used two independent guide RNAs (sgTRF2-1 and -2)9, which efficiently reduced TRF2 expression and induced telomere fusions in young IMR-90 E6E7 cells (Fig. 2a and Extended Data Fig. 2d, e). These guide RNAs also led to mitotic arrest, comparable to IMR-90 E6E7 cells around PD108 (Fig. 2b and Extended Data Fig. 2f). Suppression of both telomere fusion and mitotic arrest by a resistant TRF2 (TRF2RsgRNA) excludes off-target effects (Fig. 2c, d and Extended Data Fig. 3a-c).
Fig. 2. Telomere fusions induce mitotic arrest.
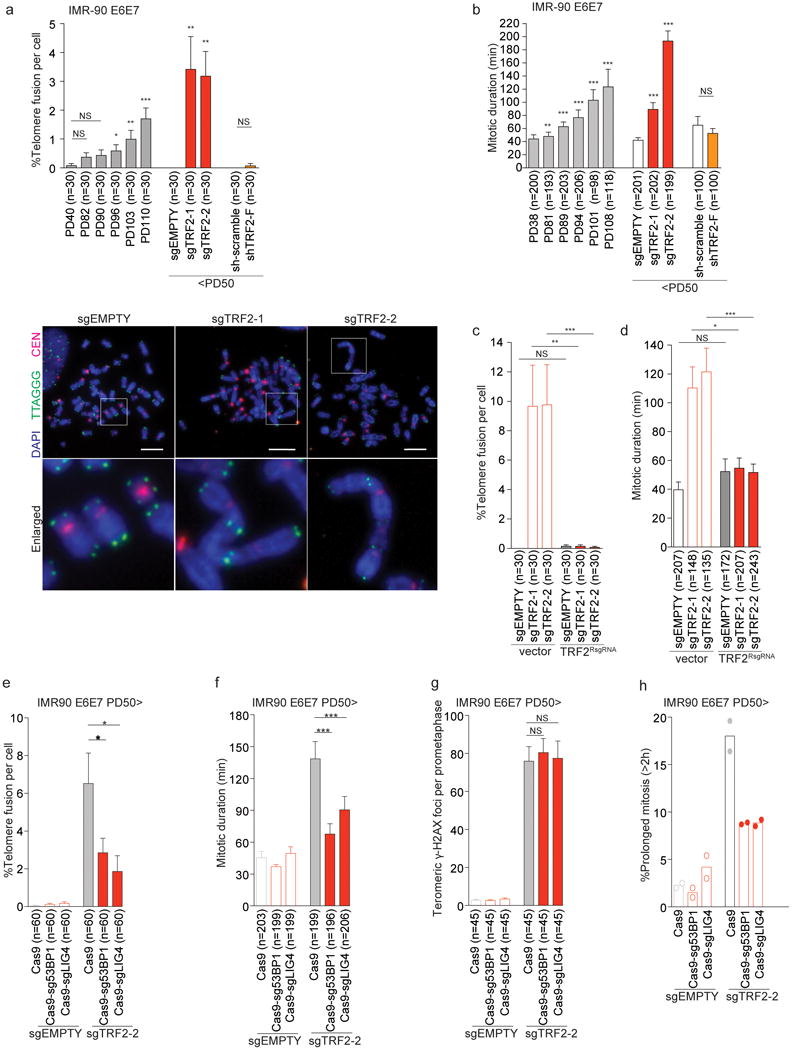
a, Mean percentage of telomeric fusions per cell ± s.e.m. in IMR-90 E6E7 and derivatives 7 days post infection around PD45. Representative metaphases below. Scale Bar, 10 µm. b, Mean mitotic duration ± s.e.m. in the cell lines from a. Mitotic duration data of growing IMR-90 E6E7 cells are the same as Fig. 1b. c, d, Percentage of telomeric fusion (c) and mitotic duration (d) in indicated IMR90 E6E7 cells (mean ± s.e.m.). e, f, g, Percentage of telomeric fusion (e), mitotic duration (f) and mean number of meta-TIF ± s.e.m. (g) in indicated cells (mean ± s.e.m.). h, Scatter plots with bars showing the mean percentage of cells with prolonged mitosis (two independent experiments). *P<0.05, **P<0.005, ***P<0.0001. NS, not significant. Mann-Whitney test.
To address whether telomeric DDR or telomere fusion induces mitotic arrest, we deleted TRF2 in young IMR-90 E6E7 cells lacking 53BP1 or Ligase 4 (Extended Data Fig. 3d, e)10,11. Suppression of 53BP1 or Ligase 4 strongly reduced fusion frequency (Fig. 2e) and prevented mitotic arrest (Fig. 2f, h), but did not reduce the number of deprotected telomeres (Fig. 2g and Extended Data Fig. 3f), thereby separating mitotic delay from DDR. Both telomere fusion and mitotic arrest phenotypes were suppressed by ATM inhibitor12,13,14 (Extended Data Fig. 3g-j), again indicating that telomere fusion underlies mitotic arrest. ATM inhibition did not suppress mitotic arrest induced by Taxol15 (Extended Data Fig. 3k and l), confirming that the inhibitor does not perturb the SAC. Additionally, cells expressing shTRF2-F, which causes telomere deprotection in the absence of fusion2, did not undergo arrest (Fig. 2a, b and Extended Data Fig. 2e, f). These data are consistent with the observation that senescent cells, while harboring a number of unfused deprotected telomeres2,16 do not display mitotic arrest (Fig. 1b and Extended Data Fig. 1a).
Deletion of TRF2 increased anaphase bridge frequency and pericentrin foci (Extended Data Fig. 4a, b) 7 d post infection, indicating multipolar mitosis when cells display telomere fusions and mitotic arrest. Accordingly, sgTRF2-2 cells exhibit unaligned metaphase chromosomes (Extended Data Fig. 4c), suggesting a chromosome congression defect. Tetraploidy did not increase as dramatically as the pericentrin foci (Extended Data Fig. 4d), ruling out tetraploidization and centrosome amplification due to anaphase bridges and cytokinesis failure, as the cause of multipolarity. We conclude that multipolarity contributes to the mitotic arrest phenotype, although it is not clear how telomere fusions drive centrosome abnormality.
Mitotic arrest in young IMR-90 cells induces mitotic telomere deprotection3. To examine whether spontaneous mitotic arrest in pre-crisis cells also induces telomere deprotection we used premature sister separation, either cause or consequence of mitotic arrest17,18, as a marker of prolonged mitosis. Indeed, cells in pre-crisis displayed increased premature chromatid separation (Fig. 3a). Telomeric γ-H2AX foci analysis (meta-TIF)19,20 revealed that metaphase spreads with separated chromatids displayed increased telomere deprotection (Fig. 3b, c and Extended Data Fig. 5a). Accordingly, suppression of TRF2 in young IMR90 E6E7 cells not only caused fusion and prolonged mitosis (Fig. 2a, b), but also increased premature separation (Extended Data Fig 5b).
Fig. 3. Cell fate decision during telomere crisis.

a, Mean percentage ± s.d. of metaphases with separated chromatids or diplochromosomes in IMR-90 E6E7 cells (n=3, >100 metaphases per experiment). For one-way ANOVA separated chromatids, P<0.0001; diplochromosomes, not significant. Right, representative images. b, c, Meta-TIF analysis of pre-crisis IMR-90 E6E7 cells. (b). Scatter blots show mean telomeric γ-H2AX foci ± s.e.m. on attached chromatids (n=67) or separated chromatids (n=8), ***P<0.0001, Mann-Whitney tests. (c). d, The three distinct fates of prolonged mitosis. e, f, Mean mitotic duration of prolonged mitosis associated with the indicated fate (e) and ratio of each fate after indicated duration of mitotic arrest (f) in the pre-crisis IMR-90 E6E7 cells shown in Fig. 1b. **P<0.005, Fisher's exact test. g, Cell cycle phase at cell death in pre-crisis IMR-90 E6E7 cells (left) and fate of the previous mitosis prior to death in interphase or mitosis (<2 h) (right). Scale bar, 10 µm.
Cells exhibiting premature separation were mostly near-diploid, excluding the possibility that they have an increased number of telomeric ends (Extended Data Fig. 5c). We conclude that prolonged mitosis in pre-crisis is associated with a telomere DDR and that mitotic arrest occurs in near-diploid cells. Consistently the ratio of tetraploid cells in pre-crisis cultures did not increase as prominently as the percentage of cells undergoing mitotic arrest (Extended Data Fig. 5d, e). We also hardly observed diplochromosomes, which are a consequence of two rounds of DNA replication without mitosis (endoreduplication) and thereby tetraploid (Fig. 3a).
Next we asked how prolonged mitotic arrest affects cellular fate in pre-crisis. Live cell imaging indicated three potential outcomes: cytokinesis, slippage (mitotic exit without cytokinesis), and cell death (Fig. 3d)21. Mitotic duration correlated with cellular fate, as cells that spent the least amount of time in mitosis underwent cytokinesis, and cells that resided longer in mitosis tended to slip or die (Fig. 3e). The cell death ratio increased significantly from 19% in cells arrested for 2 – 6 h to 50% in cells arrested for 6 – 10 h and to 69% in cells arrested >10 h (Fig. 3f).
However, we noted that cells also died during interphase. Live cell imaging revealed that 32% (86/266) of death occurred after prolonged mitosis (>2 h), 14% (38/266) of cells died during mitosis lasting <2 h and 53% (142/266) of cells died in interphase (Fig. 3g). To address whether cells that died during a short mitosis or interphase were associated with prolonged mitosis in the previous cell cycle, we traced the cells in question to the mitosis before death. Of the cells that succumb in interphase, 46% of the traceable previous mitosis was prolonged (18/39) (Fig. 3g). Of the cells that died during a short mitosis, 29% of the previous mitosis was prolonged (4/14) (Fig. 3g), indicating that cell death during either short mitosis or in interphase follows prolonged mitosis in the previous cycle.
Partial knockdown of TRF2 exacerbates mitotic telomere deprotection in young IMR-90 E6E7 cells2. To understand whether mitotic telomere deprotection is the cause of death in pre-crisis, we tested whether a partial knockdown of TRF2 would enhance cell death upon spontaneous mitotic arrest. IMR-90 E6E7 cells were transduced with shTRF2-F at PD45, PD70 or PD852 (Extended Data Fig. 6a). The resulting partial suppression of TRF2 did not affect cell growth dynamics in the younger populations, but slowed down growth in the population infected at PD85 (Fig. 4a-c). However, in all settings, the cells entered crisis prematurely (Fig. 4a-c). γ-H2AX foci analysis around PD100 revealed that shTRF2-F cells suffer from increased numbers of TIF, which was greatly enhanced on separated chromatids (Fig. 4d and Extended Data Fig. 6b, c). Accordingly, live cell imaging revealed shTRF2-F expression increased the cell death ratio especially after a short period of mitotic arrest (Fig. 4e). In contrast, TRF2 overexpression (Extended Data Fig. 6d), which partially suppresses mitotic telomere deprotection3, reduced cell death after mitotic arrest, suppressed TIF and delayed crisis (Extended Data Fig. 6e, f, g). Neither shTRF2-F nor TRF2 overexpression affected mitotic duration (Extended Data Fig. 6h, i). shTRF2-F also did not increase fusion frequency (Extended Data Fig. 6j), thereby attributing the increased cell death to exaggerated loss of TRF2, not to increased fusion formation. Accordingly, loss of TRF2 in cells lacking 53BP1 significantly increased cell death and shortened the mitotic duration prior to cell death upon colcemid-induced mitotic arrest (Extended Data Fig. 3d and 7a, b). Furthermore, inhibition of Aurora B kinase by hesperadin, which suppresses mitotic telomere deprotection3 (Extended Data Fig. 7c), greatly reduced cell death upon colcemid exposure (Extended Data Fig. 7d, e), supporting a model, where amplified telomere deprotection induced by mitotic arrest triggers cell death.
Fig. 4. Mitotic telomere deprotection dictates cellular fate upon mitotic arrest.
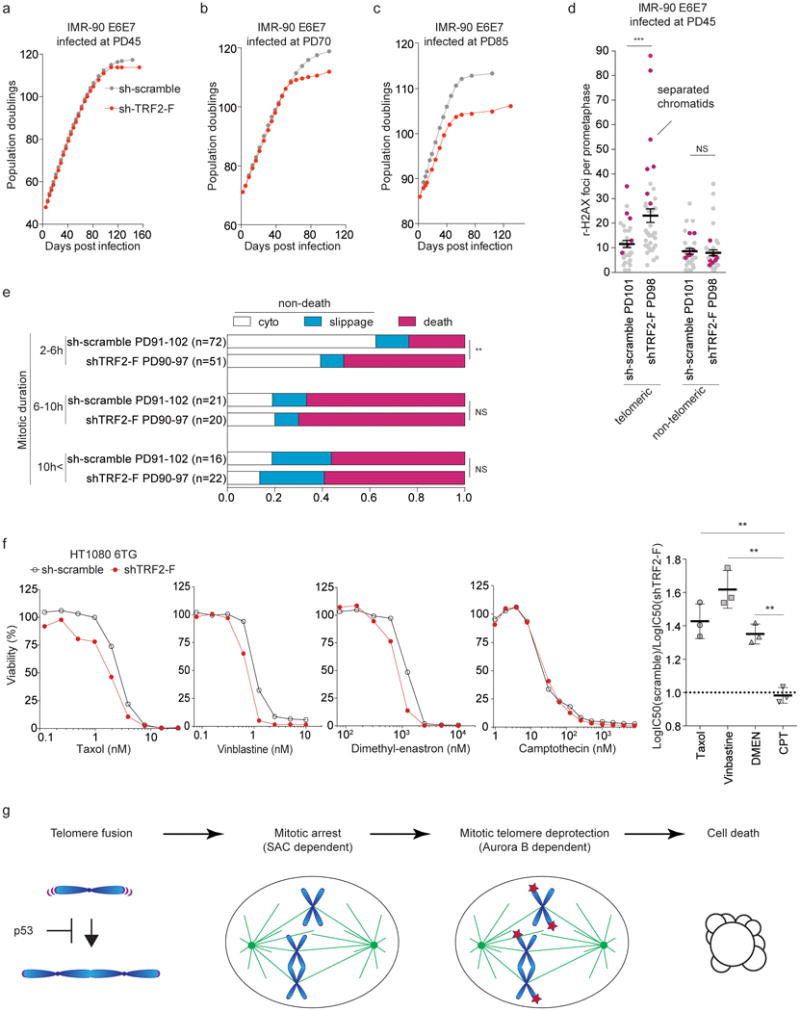
a-c, Growth curves of indicated cells. d, Scatter plots show mean telomeric and non-telomeric γ-H2AX foci ± s.e.m. in individual indicated cells shown in Fig. 4a (n>30). IF-FISH, as in Fig. 3b. Magenta, metaphases with separated chromatids. e, Ratio of indicated fates in pre-crisis IMR-90 E6E7 cells expressing sh-scramble and shTRF2-F infected at PD 85, analyzed as in Fig. 3f (Fisher's exact test). f, Viability assay of HT1080 6TG expressing sh-scramble and shTRF2-F. Right, the ratio between LogIC50 of sh-scramble and shTRF2-F from three independent experiments (two-tailed t-test). g, Model of mitotic cell death pathway during crisis. *P<0.05, **P<0.005, ***P<0.0001. NS, not significant.
This model predicts that shTRF2-F would sensitize cells to drugs that induce mitotic arrest. Therefore HT1080 6TG cells expressing sh-scramble or shTRF2-F were exposed to Taxol, vinblastine, dimethyl-enastron22 and as a control the topoisomerase I inhibitor camptothecin. Expression of shTRF2-F significantly sensitized HT1080 6TG cells to Taxol, vinblastine, and dimethyl-enastron, all of which induce mitotic arrest (Fig. 4f). No such effect was observed upon exposure to camptothecin (Fig. 4f), confirming that TRF2 levels affect cellular fate specifically upon mitotic arrest.
Here we show that chromosome end-to-end fusions during crisis cause spontaneous mitotic arrest, amplifying telomere deprotection, which determines cellular fate. We suggest that telomere deprotection upon spontaneous mitotic arrest is the underlying molecular signal that leads to cell death in crisis (Fig. 4g). While we cannot rule out a role of fusion breakage cycles and the resulting chromosomal abnormalities as cause of death23, several observations argue against it: Only few fusions are observed in crisis cells (Fig. 2a), which is enough to trigger prolonged mitosis (Fig. 2b). Disruption of TRF2 leads to the rapid onset of prolonged mitosis, arguing against long-term effects of fusion breakage cycles (Fig. 2a). Cells succumb to death in the first prolonged mitosis, or in the following cell cycle (Fig. 3f, g), ruling out a long-term process. Increasing damage signals without increasing fusion frequency causes more death (Fig. 4e and Extended Data Fig 7a), arguing for signaling from deprotected telomeres as the cause for death. Since most cell death during pre-crisis is associated with mitotic arrest, we propose that prolonged mitosis is the main mechanism that limits cellular life span upon bypass of senescence. These findings might also offer a clinical opportunity, since exacerbation of mitotic telomere deprotection sensitises cancer cells to mitotic drugs. Mitotic arrest, however, has also been associated with tumourigenesis in checkpoint-compromised cells24. Similarly, bone marrow failure and cancer in individuals with telomeropathies are frequent, which could potentially be explained by mitotic arrest resulting from overly short telomeres25,26. Telomere-driven spontaneous mitotic arrest and the resulting mitotic telomere deprotection in the pre-crisis stage may thus function as a double-edged sword, explaining both cell death and chromosome instability upon bypass of senescence.
Methods
Cell culture and treatment
Human IMR-90 primary lung fibroblasts (ATCC) and their derivatives were grown in Glutamax-DMEM (Gibco) supplemented with 0.1 mM nonessential Amino Acids and 15% Fetal Bovine Serum. HT1080 6TG cells were grown in Glutamax-DMEM supplemented with 0.1 mM Nonessential Amino Acids and 10% Bovine Growth Serum. All cells were grown at 7.5% CO2 and 3% O2. Colcemid (Life Technologies), Taxol (A. G. Scientific), vinblastine (A. G. Scientific), dimethylenastron (A. G. Scientific), hesperadin (Selleck), reversine (Selleck) and camptothecin (Selleck) were used at indicated concentration. ATM inhibitor (KU-55933) (Tocris) was used at 10 μM. FACS analysis was performed as described27. Cells were tested for mycoplasma contamination and found negative.
Live cell imaging
Live-imaging was performed in 8 well µ-slide chambers (iBidi) on a Zeiss inverted fluorescent microscope with a 20× 0.8NA air objective at 37°C and 7.5% CO2 (XLmulti S1 module, Zeiss). Images were captured with an AxioCam MRm (Zeiss) using Axio Vision software (Zeiss) typically every 6 min for at least 48 h. Mitotic duration was defined as movie frames from nuclear envelope break down or a previous frame of cell rounding to a frame of cytokinesis, slippage or mitotic cell death. Prolonged mitosis was defined as a mitosis that continues for more than 2 h. Cells that escaped from a movie screen during prolonged mitosis were included in the mitotic duration analysis but excluded from the cell fate analysis.
For shTRF2-F, image capture was started 5 d after infection. For sgEMPTY, sgTRF2-1 and sgTRF2-2, image capture was started 7 d after infection. Where indicated, population doubling is the one at their seeding, typically 1 d prior to the starting date of image capture. Typically more than 2 independent movies were analyzed to confirm reproducibility except for Fig.1b, where one movie was analyzed per data point. Where indicated, Taxol, colcemid and hesperadin were added to the culture right before starting live imaging.
Vectors and viral Infections
Target sequences of CRISPR/Cas9 are as follows28: sgTRF2-1, 5′-ACTGCATAACCCGCAGCAAT-(PAM)-3′; sgTRF2-2, 5′-TGTCTGTCGCGGATTGAAGA-(PAM)-3′; sg53BP1, 5′-CAGAATCATCCTCTAGAACC-(PAM)-3′; and sgLIG4, 5′-TGGCGTCGAAACATACTGAG-(PAM)-3′. Target sequences of sg53BP1 and sgLIG4 were first cloned into LentiCRISPR vector (Addgene plasmid 49535), followed by recloning of the guide RNA expression cassette (U6 promoter, target sequence and gRNA scaffold) into NheI site of LentiCas9-Blast vector (Addgene plasmid 52962). BsmBI-digested LentiCRISPR vector and LentiGuide-puro (Addgene plasmid 52963) was incubated with T4 PNK (NEB), Klenow fragment (NEB), and then T4 DNA ligase (NEB) to generate LentiCRISPR-sgEMPTY and LentiGuide-sgEMPTY, respectively. Silent mutations that pWZL-TRF2RsgRNA carries are; 5′-G CTC CTC AGA GTG ATG CAA T-3′ for sgTRF2-1, and 5′-TGC CTC AGC AGA ATC GAG GA-3′ for sgTRF2-2.
IMR-90 cells were infected with pLXSN3-HPV16E6, pLXSN3-HPV16E7, pLXSN3-HPV16E6E7, pLXSN3-p53dd, and pBabe-hTert retroviral vectors as described29, and subjected to long term culturing in the presence of 600 μg/ml G418 (Mediatech, Inc.) (for pLXSN3) or 2 μg/ml puromycin (Mediatech, Inc.) (for pBabe). TRF2RsgRNA-expressing IMR-90 E6E7 cells were selected in the presence of 100 ng/ml hygromycin (Mediatech, Inc.) for 6 d and subjected to LentiCRISPR infection.
Lentiviral vectors pseudotyped with VSV glycoprotein were generated by the Salk Institute Gene Transfer, Targeting and Therapeutics (GT3) Core using a modified protocol30. For sh-scramble (Addgene plasmid 1864), shTRF2-F, LentiCRISPR-sgEMPTY, LentiCRISPR-sgTRF2-1, LentiCRISPR-sgTRF2-2, LentiCas9-Blast, LentiCas9-Blast-sg53BP1, LentiCas9-Blast-sgLIG4, LentiGuide-sgEMPTY, and LentiGuide-sgTRF2-2, cells were plated in growth media containing 4 μg/ml polybrene and lentivirus and cultured for two days. Puromycin and Blasticidin were added to the culture at 2 μg/ml and 10 μg/ml, respectively, and infected cells were selected for more than 3 d before analysis. Where indicated, ATMi was added 4 d after infection and medium was refreshed every 1-2 d. For fusion assay, cells were harvested 7 d after infection or at indicated PD.
Antibodies
Primary antibodies: anti-γ-H2AX (613402 clone 2F3, Biolegend); anti-TRF1 (Karlseder lab); anti-TRF2 (Karlseder lab); anti-53BP1 (H-300) (sc-22760, Santa Cruz); anti-LIG4 (ab80514, Abcam); anti-pericentrin (ab4448, Abcam); anti-MPM-2 (05-368MG, Millipore); anti-γ-tubulin (T6557, Sigma-Aldrich); anti-H3S10P (D2C8) (3377, Cell Signaling); anti-GAPDH (A300-641A, Bethyl); anti-p53 (sc-126, Santa Cruz).
Secondary antibodies: HPR-linked anti-mouse or anti-rabbit (NXA931 or NA934V; GE Healthcare); Alexa-488-conjugated anti-rabbit (Invitrogen); Alexa-594-conjugated anti-mouse (Invitrogen).
Western blotting
Western blots were performed as described3.
Immunofluorescence and Telomere-Centromere FISH on metaphase spreads
Cytocentrifugation, IF and telomeric FISH were performed as described previously20. For telomere and centromere double-staining, alexa-488-conjugated telomeric PNA probe (TelC-A488, PNA Bio Inc.) and Cy3-conjugated centromeric PNA probe (CENT-Cy3, PNA Bio Inc.) were used. Percentage of telomere fusion per chromosome end was analyzed as described20. For pericentrin foci analysis, cells in early mitosis (pro-, prometa- and metaphase) were selected according to chromosome shape from MPM-2 positive cells. For chromosome alignment analysis, cells in which most H3S10-P positive chromosomes align between (and do not overlap with) two γ-tubulin foci were selected as metaphase. Cells were analyzed 7 days after infection or at indicated PD.
Telomere blots
Double-stranded telomere analysis was performed as described29.
Viability assay
Premixed WST-1 Cell Proliferation Reagent (Clontech) was used for viability assay according to manufacture's instruction. HT1080 6TG cells infected with either sh-scramble or shTRF2 were seeded in 96-well plates at 6 d post infection and exposed to Taxol, vinblastine, dimethyl-enastron, and camptothecin at 7 d post infection for 48 h. Triplicate wells were analyzed for each drug concentration. The results were reproduced by three independent experiment. LogIC50 value was analysed by log(inhibitor) vs. normalized response – variable slope method using Prism 6 software.
Statistical methods
Each figure legend shows number of samples per experiment and number of experiments that were analysed independently. Two-tailed unpaired t-test and two-tailed Mann-Whitney tests were used to compare two data sets, where Gaussian distribution is assumed and not assumed, respectively. To detect trend among multiple data sets in Fig. 3a, Extended Data Fig. 5a, and Extended Data Fig. 7c, one-way ANOVA was used. Mitotic duration data sets of IMR-90 E6E7 cells in Fig. 1b were analysed by Kruskal-Wallis test to detect trend (P<0.0001), in addition to Mann-Whitney test as described above. For statistical analysis of cellular fate after mitotic arrest, ratio of death versus non-death (cytokinesis and slippage) were analysed by two-tailed Fisher's exact test. For Fig. 3f, data from short (2 – 6 h) and middle mitotic arrest (6 – 10 h) were combined and compared to that of long mitotic arrest (>10 h). The null hypothesis was rejected when P values were less than 0.05. No randomization was performed. All statistical analysis was performed using Prism 6 software.
Extended Data
Extended Data Figure 1.
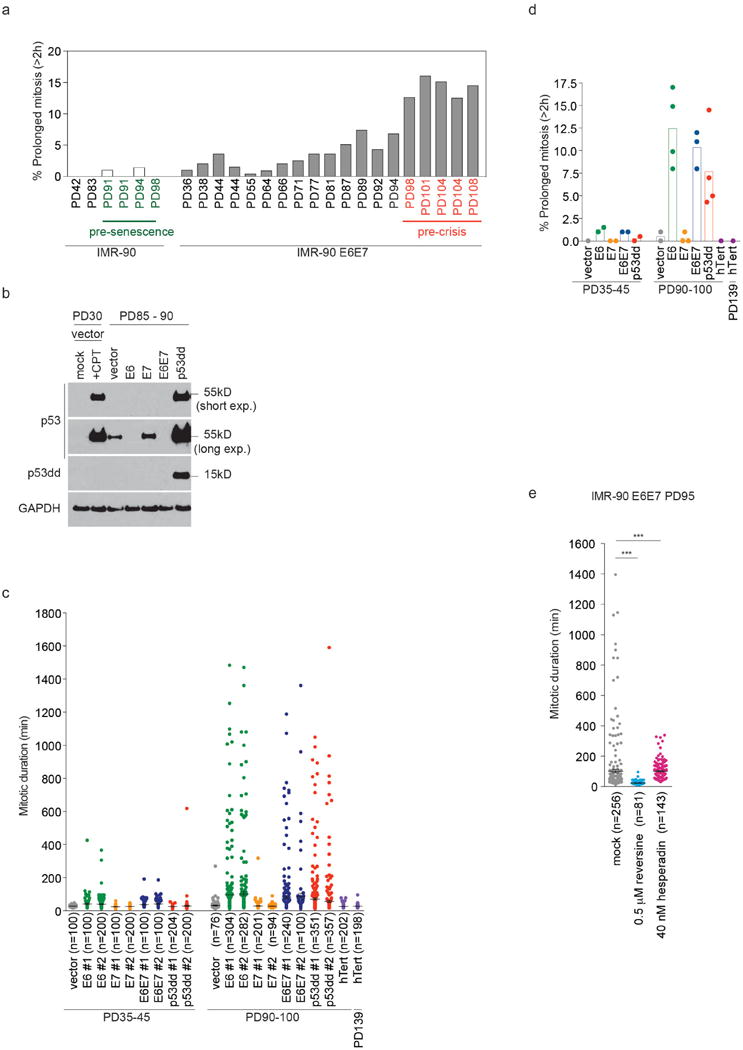
a, Percentage of cells that spend more than 2 h in mitosis (prolonged mitosis), shown in Fig. 1b. b, Effect of indicated oncogenes on p53 expression. c, Scatter plots show mean mitotic duration ± s.e.m. of individual IMR-90 derivative cells analyzed in Fig. 1d. d, Scatter plots with bars show mean percentage of prolonged mitosis analyzed in Fig. 1d (1 – 4 independent experiments). e, Scatter plots show mean mitotic duration of IMR-90 E6E7 PD95 cells exposed to reversine and hesperadin ± s.e.m. (***P<0.0001, Mann-Whitney test). The result was reproduced in two independent experiments.
Extended Data Figure 2.
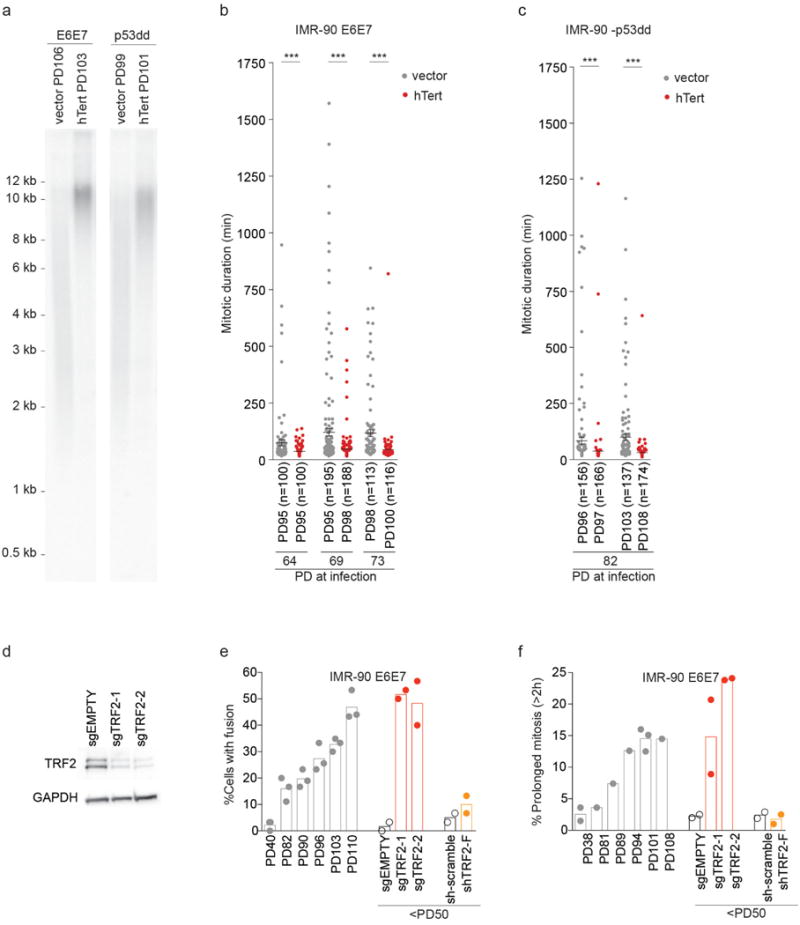
a, Telomere elongation by hTert in IMR-90 E6E7 and p53dd cells shown in Fig. 1e and f. IMR-90 E6E7 and p53dd cells were infected at PD73 and PD82, respectively, and analyzed at the indicated PD. b, c, Scatter plots show mean mitotic duration ± s.e.m. of individual IMR-90 E6E7 (b) and p53dd (c) cells expressing hTert at indicated PD shown in Fig. 1e, f. d, Effect of sgTRF2 on TRF2 expression 7 d after infection. e, f, Scatter plots with bars show mean percentage of cells with telomeric fusion (e) and prolonged mitosis (f) in IMR-90 E6E7 derivatives shown in Fig. 2a, b (1 – 3 independent experiments). ***P<0.0001. Mann-Whitney test.
Extended Data Figure 3.
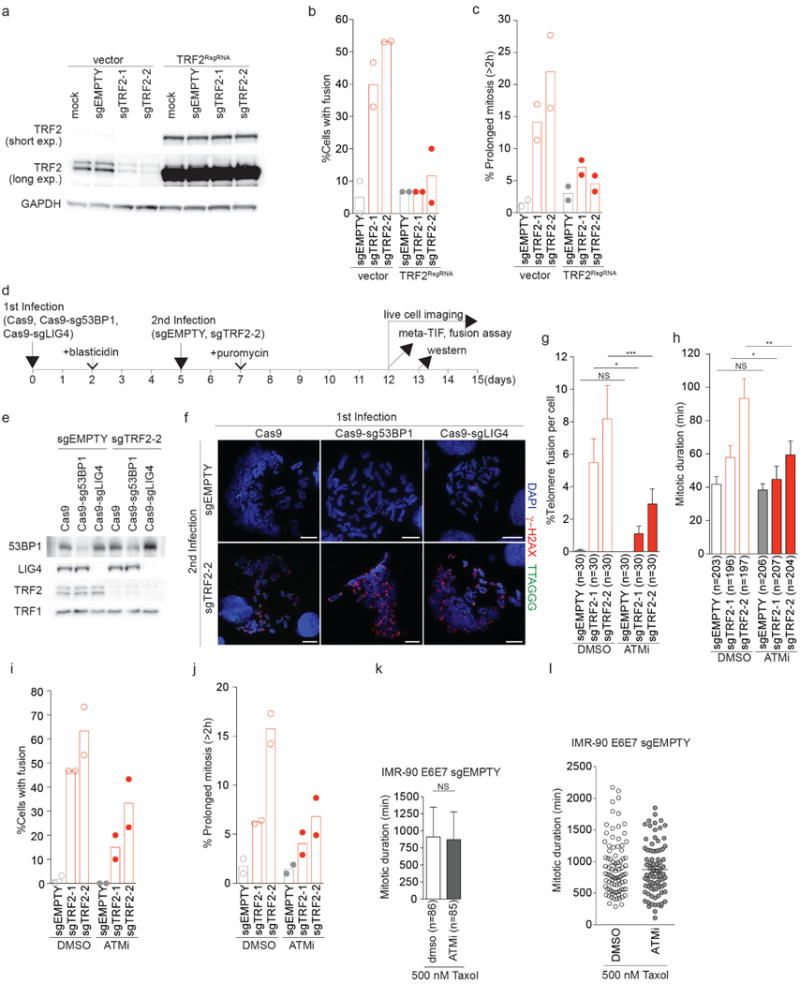
a, Effect of sgTRF2 on TRF2 expression in cells expressing sgTRF2-resistant TRF2 (TRF2RsgRNA) 9 d after infection with CRISPR/Cas9. b, c, Scatter plots with bars show mean percentage of cells with telomeric fusion (b) and prolonged mitosis (c) in IMR-90 E6E7 expressing sgTRF2 in the presence of TRF2RsgRNA shown in Fig. 2c, d (2 independent experiments). d, Schematic of 53BP1 or Ligase 4 suppression experiment in the presence of sgEMPTY or sgTRF2-2. e, Western analysis of IMR90 E6E7 cells expressing Cas9, Cas9-sg53BP1 or Cas9-sgLig4 in the background of sgEMPTY or sgTRF2-2. f, Representative meta-TIF images of cells suppressed for 53BP1 or Ligase 4 in the presence of sgEMPTY or sgTRF2-2 as described in (d). g, h, Percentage of telomeric fusion (g) and mitotic duration (h) in IMR-90 E6E7 expressing sgEMPTY and sgTRF2 in the presence of DMSO or ATM inhibitor (mean ± s.e.m.). i, j, Scatter plots with bars showing mean percentage of cells with telomeric fusion (i) and prolonged mitosis (j) in IMR-90 E6E7 expressing sgTRF2 in the presence of ATM inhibitor shown in g, h (2 independent experiments). k, Mean mitotic duration ± s.e.m. of IMR-90 E6E7 sgEMPTY cells exposed to 500 nM Taxol in the presence of DMSO or ATMi. l, Scatter plots show mean mitotic duration of individual cells shown in (j). NS, not significant; *P<0.05, **P<0.005, ***P<0.0001, Mann-Whitney tests. Results were reproduced in at least two independent experiments.
Extended Data Figure 4.
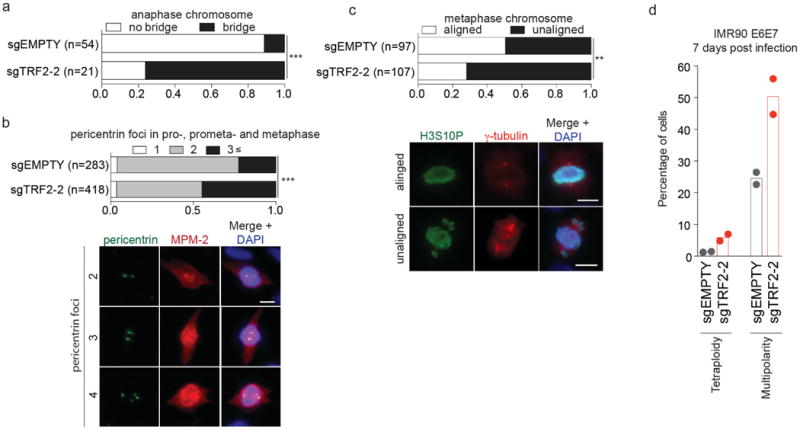
Ratio of anaphase chromosome with or without anaphase bridge (a), pericentrin foci in pro-, prometa- and metaphase (b) and metaphase chromosome with or without unaligned chromosome (c) in IMR-90 E6E7 expressing sgEMPTY and sgTRF2-2 7 days after infection (Fisher's exact test, for pericentrin foci, 1 and 2 foci vs ≥3 foci). Representative images from sgEMPTY cells are shown below (b, c). Results were reproduced in two independent experiments. d, Scatter plots with bars show mean percentage of cells that posses tetraploidy (FACS analysis) and multipolarity (≥3 pericentrin foci as in (j)) in IMR-90 E6E7 cells expressing sgEMPTY and sgTRF2-2 7 d after infection (two independent experiments). **P<0.005, ***P<0.0001. Fisher's exact test. Scale bar, 10 µm.
Extended Data Figure 5.
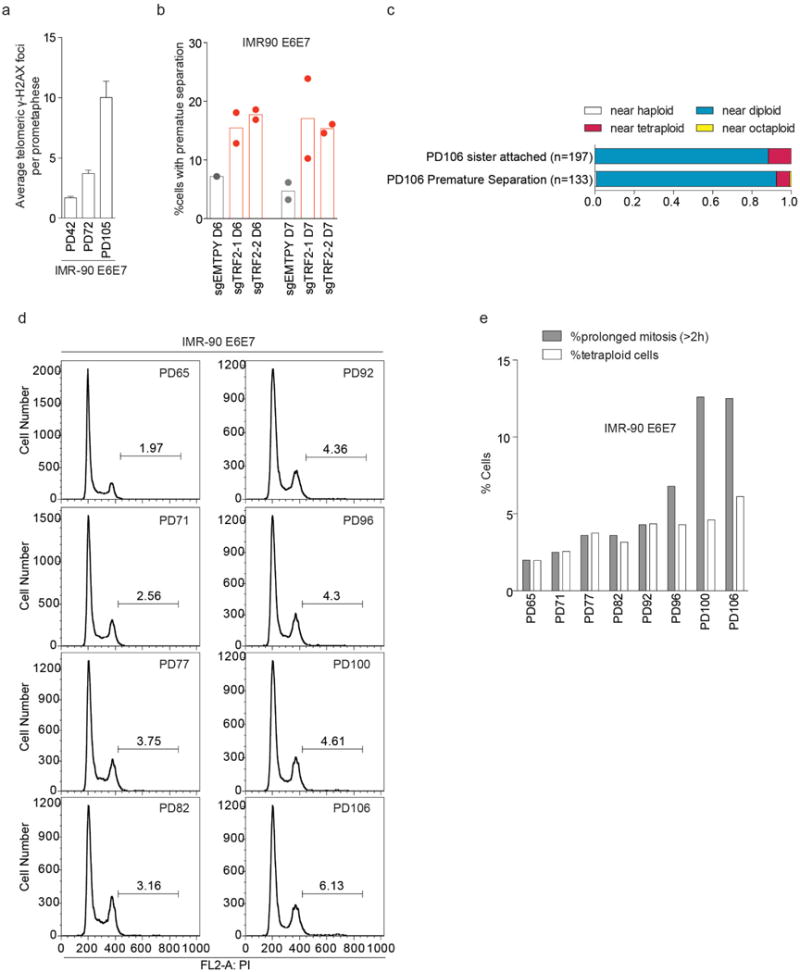
a, Bar charts show mean of three independent experiments of average number of telomeric γ-H2AX foci ± s.e.m. in IMR-90 E6E7 at PD42, 72 and 105 as analyzed in Fig. 4d (n=25 per experiment, P=0.0008, one-way ANOVA). b, Bars show the percentage of young IMR90 E6E7 cells with separated sister chromatids expressing sgEMPTY or sgTRF2-1/2 6 or 7 d post infection with CRISPR/Cas9. Results from two independent experiments except for sgEMPTY D6 (at least 31 metaphases per experiment) are shown. c, Ratio of ploidy of mitotic IMR-90 E6E7 cells at PD106. Cells with attached and separated sister chromatids are plotted separately. d, FACS analysis of cells shown in Fig. 1b. e, Bars show percentage of prolonged mitosis and tetraploid cells (4N<) in IMR-90 E6E7 cells at indicated PD. Percentage of prolonged mitosis are the same data sets as Extended Data Fig. 1a.
Extended Data Figure 6.
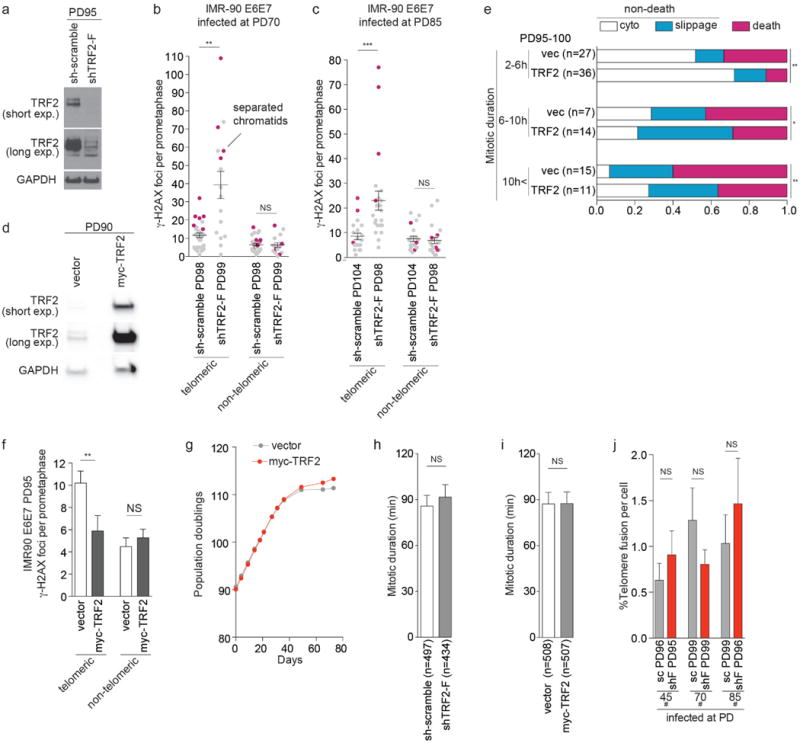
a, Effect of shTRF2-F on TRF2 expression 7 d after infection. b, c, Telomeric and non-telomeric γ-H2AX foci in individual pre-crisis IMR-90 E6E7 cells expressing sh-scramble and shTRF2-F shown in Fig. 4b. (b) and (c) were analyzed as in Fig. 4d (n>16, mean ± s.e.m.). Metaphases with separated chromatids are shown in magenta. d, Effect of myc-TRF2 on TRF2 expression 7 d after infection. e, Ratio of mitotic cell fate in pre-crisis IMR-90 E6E7-expressing TRF2 cells, analyzed as in Fig. 3f (Fisher's exact test, death vs non-death). f, Meta-TIF analysis of pre-crisis IMR-90 E6E7 cells expressing a control vector or myc-TRF2. Bars show mean telomeric and non-telomeric γ-H2AX foci ± s.e.m. (n=25, Mann-Whitney tests). g, Growth curve of IMR-90 E6E7 cells expressing a control vector or myc-TRF2-F infected at PD90. h, i, Mean mitotic duration ± s.e.m. of IMR-90 E6E7 cells expressing sh-scramble and shTRF2-F (h) and myc-TRF2 (i) analyzed in Fig. 4e and Extended Data Fig. 6e, respectively (Mann-Whitney test). j, Mean percentage of telomere fusion ± s.e.m. of pre-crisis IMR-90 E6E7 expressing sh-scramble and shTRF2-F analyzed in Fig. 4a-c (Mann-Whitney test). *P<0.05, **P<0.005, ***P<0.0001. NS, not significant.
Extended Data Figure 7.
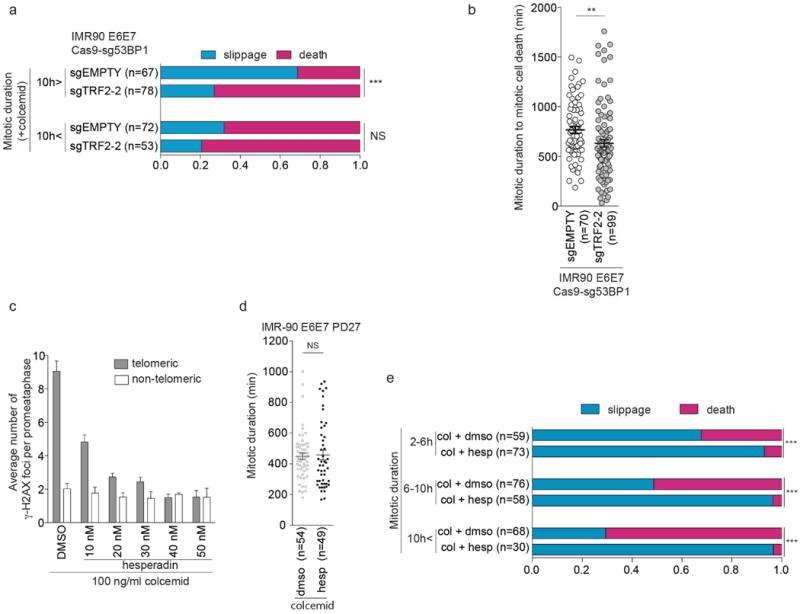
a, Ratio of mitotic slippage and cell death in IMR-90 E6E7 Cas9-sg53BP1 cells expressing sgEMPTY and sgTRF2-2 in the presence of colcemid, analyzed as in Fig. 3f (Fisher's exact test, death vs slippage). b, Scatter plots show mean mitotic duration ± s.e.m. prior to cell death of individual IMR-90 E6E7 Cas9-sg53BP1 cells expressing sgEMPTY and sgTRF2-2 in the presence of colcemid (Mann-Whitney test). c, Bars show mean of three independent experiments of average telomeric and non-telomeric γ-H2AX foci ± s.d. in IMR-90 E6E7 cells at PD45 exposed to colcemid in the presence of DMSO or hesperadin at indicated concentrations for 24 h analyzed as in Fig. 4d (50 metaphase per experiment). For one-way ANOVA telomeric foci, P<0.0001; non-telomeric foci, not significant. d, Scatter plots show mean mitotic duration ± s.e.m. of IMR-90 E6E7 cells at PD27 exposed to 100 ng/ml colcemid in the presence of DMSO or 40 nM hesperadin ± s.e.m. (Mann-Whitney test). e, Ratio of mitotic cell fate in IMR-90 E6E7 cells around PD45 exposed to 100 ng/ml colcemid in the presence of DMSO or 40 ng/ml hesperadin (Fisher's exact test, death versus slippage). **P<0.005, ***P<0.0001. NS, not significant.
Acknowledgments
All data are archived at the Salk Institute. We thank the Salk Institute's J. Fitzpatrick of the Waitt Advanced Biophotonics Center and members of GT3 Core, C. O'Shea, G. Wahl, F. Zhang, and D. Sabatini for support and Karlseder lab members for comments. M.T.H. was supported by the Human Frontier Science Program and the Japan Society for the Promotion of Science Postdoctoral Fellowships for Research Abroad. A.J.C. was supported by an NIH NRSA T32 Fellowship (5T32CA009370). T.R. was supported by the Glenn Center for Research on Aging and CIRM training grant TG2-01158. The Salk Institute Cancer Center Core Grant (P30CA014195), the NIH (R01GM087476, R01CA174942), the Donald and Darlene Shiley Chair, the Highland Street Foundation, the Fritz B. Burns Foundation, the Emerald Foundation and the Glenn Center for Research on Aging support J.K.
Footnotes
Author Contributions: M.T.H. and A.J.C. designed and performed experiments, and wrote the manuscript, T.R. performed experiments, J.K. designed experiments and wrote the manuscript.
Author Information: The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
References
- 1.Wright WE, Pereira-Smith OM, Shay JW. Reversible cellular senescence: implications for immortalization of normal human diploid fibroblasts. Mol Cell Biol. 1989;9:3088–3092. doi: 10.1128/mcb.9.7.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cesare AJ, Hayashi MT, Crabbe L, Karlseder J. The telomere deprotection response is functionally distinct from the genomic DNA damage response. Mol Cell. 2013;51:141–155. doi: 10.1016/j.molcel.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayashi MT, Cesare AJ, Fitzpatrick JA, Lazzerini-Denchi E, Karlseder J. A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat Struct Mol Biol. 2012;19:387–394. doi: 10.1038/nsmb.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le Poole IC, et al. Generation of a human melanocyte cell line by introduction of HPV16 E6 and E7 genes. In Vitro Cell Dev Biol Anim. 1997;33:42–49. doi: 10.1007/s11626-997-0021-6. [DOI] [PubMed] [Google Scholar]
- 5.Bodnar AG, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 6.Meyerson M, et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997;90:785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- 7.Santaguida S, Tighe A, D'Alise AM, Taylor SS, Musacchio A. Dissecting the role of MPS1 in chromosome biorientation and the spindle checkpoint through the small molecule inhibitor reversine. J Cell Biol. 2010;190:73–87. doi: 10.1083/jcb.201001036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hauf S, et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore–microtubule attachment and in maintaining the spindle assembly checkpoint. The Journal of cell biology. 2003;161:281. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimitrova N, Chen YCM, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smogorzewska A, Karlseder J, Holtgreve-Grez H, Jauch A, de Lange T. DNA ligase IV-dependent NHEJ of deprotected mammalian telomeres in G1 and G2. Curr Biol. 2002;12:1635–1644. doi: 10.1016/s0960-9822(02)01179-x. [DOI] [PubMed] [Google Scholar]
- 12.Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1068–1071. doi: 10.1038/nature06065. [DOI] [PubMed] [Google Scholar]
- 13.Guo X, et al. Dysfunctional telomeres activate an ATM-ATR-dependent DNA damage response to suppress tumorigenesis. EMBO J. 2007;26:4709–4719. doi: 10.1038/sj.emboj.7601893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hickson I, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer research. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 15.Schiff PB, Horwitz SB. Taxol stabilizes microtubules in mouse fibroblast cells. Proc Natl Acad Sci U S A. 1980;77:1561–1565. doi: 10.1073/pnas.77.3.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaul Z, Cesare AJ, Huschtscha LI, Neumann AA, Reddel RR. Five dysfunctional telomeres predict onset of senescence in human cells. EMBO Rep. 2012;13:52–59. doi: 10.1038/embor.2011.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoque MT, Ishikawa F. Cohesin defects lead to premature sister chromatid separation, kinetochore dysfunction, and spindle-assembly checkpoint activation. J Biol Chem. 2002;277:42306–42314. doi: 10.1074/jbc.M206836200. [DOI] [PubMed] [Google Scholar]
- 18.Stevens D, Gassmann R, Oegema K, Desai A. Uncoordinated loss of chromatid cohesion is a common outcome of extended metaphase arrest. PLoS One. 2011;6:e22969. doi: 10.1371/journal.pone.0022969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–1556. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 20.Cesare AJ, et al. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat Struct Mol Biol. 2009;16:1244–1251. doi: 10.1038/nsmb.1725. [DOI] [PubMed] [Google Scholar]
- 21.Rieder CL, Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell. 2004;7:637–651. doi: 10.1016/j.devcel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 22.Gartner M, et al. Development and biological evaluation of potent and specific inhibitors of mitotic Kinesin Eg5. Chembiochem. 2005;6:1173–1177. doi: 10.1002/cbic.200500005. [DOI] [PubMed] [Google Scholar]
- 23.Gisselsson D, et al. Telomere-mediated mitotic disturbances in immortalized ovarian epithelial cells reproduce chromosomal losses and breakpoints from ovarian carcinoma. Genes Chromosomes Cancer. 2005;42:22–33. doi: 10.1002/gcc.20094. [DOI] [PubMed] [Google Scholar]
- 24.Dalton WB, Yang VW. Role of prolonged mitotic checkpoint activation in the formation and treatment of cancer. Future Oncol. 2009;5:1363–1370. doi: 10.2217/fon.09.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012;13:693–704. doi: 10.1038/nrg3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Batista LF, Artandi SE. Understanding telomere diseases through analysis of patient-derived iPS cells. Curr Opin Genet Dev. 2013;23:526–533. doi: 10.1016/j.gde.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karlseder J. p53- and ATM-Dependent Apoptosis Induced by Telomeres Lacking TRF2. Science (New York, NY) 1999;283:1321–1325. doi: 10.1126/science.283.5406.1321. [DOI] [PubMed] [Google Scholar]
- 28.Shalem O, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karlseder J. Senescence Induced by Altered Telomere State, Not Telomere Loss. Science (New York, NY) 2002;295:2446–2449. doi: 10.1126/science.1069523. [DOI] [PubMed] [Google Scholar]
- 30.Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat Protoc. 2006;1:241–245. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]