


Abstract
Mutations in human mitochondrial DNA (mtDNA) can cause mitochondrial disease and have been associated with neurodegenerative disorders, cancer, diabetes and aging. Yet our progress toward delineating the precise contributions of mtDNA mutations to these conditions is impeded by the limited availability of faithful transmitochondrial animal models. Here, we report a method for the isolation of mutations in mouse mtDNA and its implementation for the generation of a collection of over 150 cell lines suitable for the production of transmitochondrial mice. This method is based on the limited mutagenesis of mtDNA by proofreading-deficient DNA-polymerase γ followed by segregation of the resulting highly heteroplasmic mtDNA population by means of intracellular cloning. Among generated cell lines, we identify nine which carry mutations affecting the same amino acid or nucleotide positions as in human disease, including a mutation in the ND4 gene responsible for 70% of Leber Hereditary Optic Neuropathies (LHON). Similar to their human counterparts, cybrids carrying the homoplasmic mouse LHON mutation demonstrated reduced respiration, reduced ATP content and elevated production of mitochondrial reactive oxygen species (ROS). The generated resource of mouse mtDNA mutants will be useful both in modeling human mitochondrial disease and in understanding the mechanisms of ROS production mediated by mutations in mtDNA.
INTRODUCTION
A mammalian genome consists of two components: nuclear DNA and mitochondrial DNA. Nuclear DNA (nDNA) is organized into chromosomes of which two sets are present per cell: one from each parent. In contrast, mitochondrial DNA (mtDNA) inheritance in animals is almost exclusively maternal, and this DNA species is highly redundant, with hundreds to thousands of copies present in a typical cell. Human mtDNA is a 16 569 bp circular molecule crucial for proper mitochondrial function and cellular ATP production. It encodes 13 protein components of the mitochondrial oxidative phosphorylation (OXPHOS) system. These polypeptides are encoded using an alternate genetic code distinct from that used to encode nuclear genes, so they require a separate translational apparatus, some components of which (22 tRNAs and 2 rRNAs) also are encoded in mtDNA (1). In many (but not all, (2)) cell types, the bulk of ATP is produced by OXPHOS in mitochondria. Since mtDNA encodes components for four of the five mitochondrial respiratory complexes, it is not surprising that mutations in mtDNA have been associated with various human pathologies, such as mitochondrial disease (3–5), diabetes (6–8), cancer (9,10), neurodegenerative disorders (11), and others.
Mitochondrial diseases are a diverse group of genetic disorders, which can be caused by mutations in either nuclear genes or mtDNA (12) and have prevalence of at least 1 in 5000 (13). A recent release of the MITOMAP (www.mitomap.org) lists 584 mtDNA mutations associated with pathological conditions in humans, and epidemiological studies indicate that at least one in 200 healthy humans may harbor a pathogenic mtDNA mutation that may cause disease in the offspring of female carriers (14). While anecdotal reports describe positive prognoses for select patients treated with various vitamins, cofactors or reagents (15,16), there is no reliable treatment or cure for these often fatal disorders (17).
There are several features unique to mitochondrial disorders caused by pathogenic mtDNA mutations. The mtDNA mutations present in many patients with mitochondrial disease exist in a heteroplasmic state, a condition in which a mixture of mutant and wild type (WT) mtDNA is present in a cell, usually in unequal proportions. Typically, there is a threshold for the content of mutated mtDNA genomes below which patients are asymptomatic. Heteroplasmy is associated with variability in mutation content between different tissues of a patient and even between different cells of the same tissue (18–21). Heteroplasmy is a dynamic phenomenon, meaning that the content of mutant mtDNA can change in the tissue over the time, most notably during aging (22). In mitochondrial disease, the same phenotype can be induced by distinct mutations in mtDNA (e.g. MELAS is caused by mutations at positions 583, 3302, 3303, 3243, 3250, 3256, 3260, 3271, 4332, 8316, 12147, 12299, 13513, etc.) (23). Conversely, the same mtDNA mutation can have clinically different presentations (e.g. patients with the A3243G mutation can present with either classical MELAS, chronic progressive external ophthalmoplegia (CPEO) or with diabetes and deafness) (24–27). Also, mtDNA mutations often demonstrate incomplete penetrance, indicating modulation of the disease phenotype by nuclear and/or environmental factors (28). Studies investigating the molecular basis of these and other unique facets of mitochondrial disease are complicated in an outbred cohort of human patients, which provides limited tissue availability. Therefore, faithful animal models can provide unique insights into the molecular pathogenesis and the mechanisms of inheritance of mitochondrial disorders.
Animal models of human disease are instrumental in developing and testing new therapeutic modalities. While the development of mouse models of human mitochondrial diseases resulting from mutations in nDNA is relatively straightforward, the development of models for diseases caused by mtDNA mutations has proven technically difficult. Several groups have shown that mtDNA mutations can be transferred from cultured cells into mice, thereby creating transmitochondrial mice (29–31). Thus, if a collection of mouse cell lines carrying mtDNA mutations homologous to those found in human diseases were available, mouse models for these diseases could be generated. However, neither such cell lines, nor methods enabling the generation of a sufficiently diverse library of mouse mtDNA mutations that could be screened for homologs of human pathogenic mutations, have been reported yet. Previously, mutations in mouse mtDNA were isolated by selecting for resistance to various inhibitors of mitochondrial metabolism (32,33). Also, several mutations were induced by chemical mutagens followed by segregation using EtBr-induced mtDNA depletion (34). However, these approaches are limited by the availability of appropriate inhibitors and by each mutagen's narrow spectrum of mutations, and therefore do not allow for the generation of an exhaustive collection of mtDNA mutations (35).
Here, we describe an application of regulated expression of the proofreading-deficient mouse DNA-polymerase γ (mPolG exo-) for limited mutagenesis of mouse mtDNA, and report successful generation of a number of clones containing homoplasmic single non-synonymous mutations, suitable for transfer into animals, using this approach.
MATERIALS AND METHODS
Cells and culture conditions
3T3#52 are NIH3T3 cells transduced with retrovirus rv2641 (36) encoding rtTA Advanced. UNG cells are WT C57Bl/6j mouse embryonic fibroblasts immortalized with SV40 large T-antigen. The UNG/2641 cell line is a Tet-On derivative of UNG. 3T3#52, UNG/2641 and their derivatives were propagated in complete Dulbecco's modified Eagle medium (DMEM) supplemented with 10% (v/v) fetal calf serum (FCS), 50 μg/ml uridine, 1 mM pyruvate and 50 μg/ml gentamycin at 37°C in a humidified atmosphere containing 5% (v/v) CO2.
Plasmids and viral constructs are described in Supplementary Methods.
Determination of mtDNA copy number
Precise determination of mtDNA copy number was performed using duplex TaqMan qPCR essentially as described previously (37) using EcoRI-digested total cellular DNA. To generate standard curves, a separate linearized calibrator plasmid containing cloned nuclear and mitochondrial targets was used.
mtDNA depletion/intracellular cloning of mutant mtDNA molecules
Intracellular cloning of mtDNA molecules was achieved by, first, depleting mtDNA to the level of less than one molecule per cell with ethidium bromide (EtBr). A range of 2–10 ug/ml EtBr and 3–5 ug/ml EtBr was determined to be effective for depleting mtDNA in UNG/2641 and 3T3#52 cells, respectively. mtDNA copy number was monitored during depletion by qPCR and EtBr treatment halted when ≤1 mtDNA per cell was achieved. Approximately 1000–25 000 cells were then seeded on 150-mm dishes and cultured for 5 days. The cells were then cultured in selection media (complete DMEM with 10% (v/v) dialyzed FCS but lacking uridine and pyruvate) in order to select against rho zero cells and until colonies appeared.” Colonies were picked into 24-well plates, expanded, total DNA was extracted and used to determine complete mtDNA sequence in each clone.
Production of viral supernatants and transduction of target cells
Production of viral supernatants and transduction of target cells has been described previously (36). Antibiotics were used for selection at the following concentrations: puromycin, 2 μg/ml; G418, 1 mg/ml or hygromycin, 400 μg/ml.
Cellular respiration
Respiration in attached cells was measured using an XF-24 extracellular flux analyzer (Seahorse Biosciences, Billerica, MA, USA) as described previously (37).
Mitochondrial membrane potential
Membrane potential was measured with tetramethylrhodamine methyl ester TMRM (50 nM) as described previously (38). Membrane potential is expressed as percent of CCCP-sensitive fluorescence compared to control (e.g. WT) cells.
Cytoplasmic hybrids (cybrids)
Cybrids were produced using established techniques (39), with minor modifications. Briefly, donor cells were enucleated by centrifugation at 8000g for 20 min in 35-mm dishes in DMEM containing 10 μg/ml cytochalasin B at 37°C. After centrifugation, cells were washed three times with complete DMEM, and overlaid with 1 × 106 of 3T3 or UNG ρo cells, which were produced by extended treatment with 10 μg/ml EtBr. After 3–8 h attachment, fusion was induced by addition of 50% (w/w) PEG1500, 10% DMSO (v/w)—in DMEM for 1 min at room temperature. After fusion, cells were allowed to grow in complete DMEM for 24 h, after which they were trypsinized, and dilutions were plated in selective DMEM medium without uridine or pyruvate (see above).
Mitochondrial ROS
ROS were measured with MitoSOX Red (Invitrogen, Carlsbad, CA, USA). Cells were loaded with 5 μM MitoSOX in DMEM for 30 min at 37°C in an atmosphere of 5% (v/v) CO2. After treatment, cells were washed, trypsinized and immediately subjected to flow cytometry on a BD FACS Aria with excitation at 561 nm and an emission bandpass filter 582/15. The amount of ROS produced was expressed as percent fluorescence relative to control (e.g. WT) values.
mtDNA mutation loads
Mutation loads were determined by PCR-cloning-sequencing approach as described earlier for human cells (38). Three different ∼1 kb regions of mtDNA were amplified with the following primers using high-fidelity Phusion PCR mix (Fisher Scientific, Pittsburgh, PA, USA): 1f (AAA GCA TCT GGC CTA CAC CCA GAA) plus 1r (ACC CTC GTT TAG CCG TTC ATG CTA), 2f (AAA GCC CAC TTC GCC ATC ATA TTC) plus 2r (TAC TGT TGC TTG ATT TAG TCG GCC) and 3f (AGC CCA TGT TGA AGC TCC AAT TGC) plus 3r (TGT GGT GGT GTA CAG TGG GAA GTT). PCR fragments were cloned into EcoRV-digested pBluescriptII plasmid, and inserts sequenced with T3 and T7 primers using Big Dye v3.0 chemistry. The resulting mutation loads were expressed as average number of mutations per mitochondrial genome.
mtDNA sequencing
To derive a complete sequence, the mtDNA was first amplified as two overlapping fragments 8653 and 9453 bp using primers ShortF + ShortR and LongF and LongR (Supplementary Table S1). The PCR fragments were then treated with ExoSap-IT (Affymetrix, Santa Clara, CA, USA) and sequenced using BigDye® Terminator v3.1 Cycle Sequencing Kit (Life Technologies) and a set of 24 primers spanning mouse mitochondrial genome (Supplementary Table S1). Reaction products were subjected to capillary runs at the Functional Biosciences (Madison, WI, USA), and sequences were aligned to the NIH3T3 mitochondrial genome (GenBank AY999076) using SeqMan Pro software (DNA Star, Madison, WI). The mitochondrial genomes of 3T3#52 and UNG/2641 cell lines differ in the length of the polyA tract in the MTTR gene (10 residues in the 3T3#52 and 9 residues in the UNG/2641).
Cellular ATP content
ATP was measured using ADP/ATP Glo kit (Promega, Madison, WI, USA) according to manufacturer's recommendations.
Statistical analyses
Pairwise comparisons were made using unpaired two-tailed T-test assuming unequal variance. Multiple comparisons were performed using one-way ANOVA with post-hoc Tukey test.
RESULTS AND DISCUSSION
Mutagenesis workflow
Since the spectrum of DNA alterations induced by a given chemical mutagen is limited, there exists need for a method of mutagenesis capable of producing more diverse mutations in mtDNA. Much of the natural DNA sequence diversity (including naturally occurring mtDNA mutations) is generated by replication errors. Previous studies demonstrated that expression of the PolG exo- results in accumulation of mtDNA mutations (40–43). However, these mutations were of limited utility since constitutive expression of the defective DNA polymerase resulted in a highly heteroplasmic population of mtDNA molecules containing multiple (up to 10) mutations. Both the high level of genetic heterogeneity in generated mtDNA populations and the presence of multiple mutations in each mtDNA molecule would confound interpretation of the effects of individual mutations. Therefore, regulated expression of a proofreading-deficient mPolG exo- (36) was used to limit mutagenesis to 1–2 mutations per mtDNA molecule.
Conceptually, our workflow for the production of clones containing single, homoplasmic non-synonymous mtDNA mutations consists of four basic steps (Figure 1):
Engineering a cell line with inducible expression of mPolG exo-. Clonal isolation of doubly transduced cells and verification of inducibility by western blotting.
Brief induction of mPolG exo- expression with doxycycline, or slow accumulation of mtDNA mutations via leaky expression, followed by confirmation of the mtDNA mutation load (1–2 mutations per mtDNA molecule).
Depletion of mtDNA to ≤1 mtDNA molecule per cell by inhibiting mtDNA replication with EtBr. Depending on the extent and uniformity of the depletion, some cells may lose all mtDNA (ρ0 phenotype).
Release of the replication block to allow repopulation of cells with mtDNA accompanied by cloning of the resulting cells. A uniform depletion will result predominantly in three types of clones: homoplasmic WT clones, homoplasmic clones with mutations in their mtDNA, and ρ0 clones (Figure 1), which can be analyzed by sequencing.
Figure 1.
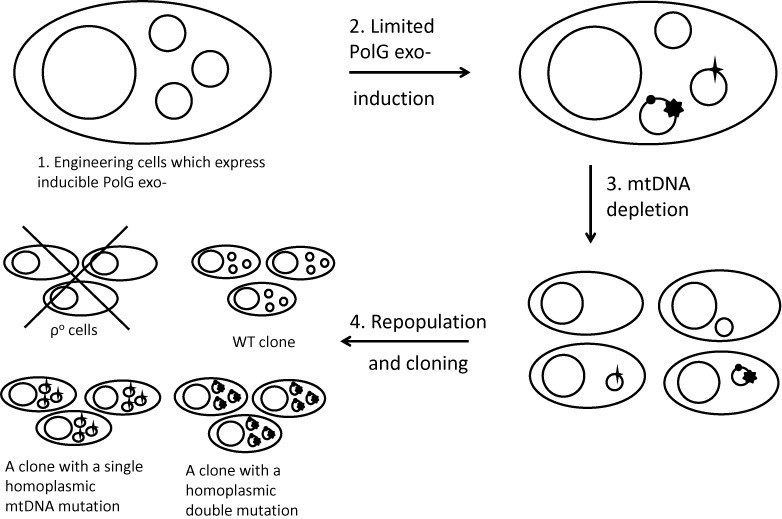
mtDNA mutagenesis workflow. The process consists of four principal steps: (1) Engineering cells for inducible expression of PolG exo-; (2) Limited mtDNA mutagenesis; (3) mtDNA depletion and (4) mtDNA repopulation and cloning. Larger circles, nuclei; smaller circles, mtDNA molecules. mtDNA mutations are designated by filled shapes (star, circle, heptagon) over the circle designating an mtDNA molecule. A cross over ρ0 cells indicates that they do not survive in selective media.
Iterations of the system
The first generation of the system for mouse mtDNA mutagenesis relied solely on doxycycline- regulated expression of the myc-tagged PolGexo− (Supplementary Figure S1A). This system was created by transducing 3T3#52 cells with a lentivirus encoding a doxycycline-inducible PolGexo− transgene. A large proportion of resulting clones demonstrated high inducibility of PolGexo− (Supplementary Figure S1D), and induction of PolGexo− was uniform (Supplementary Figure S1E)
Although the first generation of the system was used successfully to generate a number of clones containing a single, homoplasmic, non-synonymous mtDNA mutations, long-term culturing of these clones could potentially lead to the slow accumulation of mtDNA mutations due to the ‘leaky’ expression (low-level expression without inducer) of the mPolGexo− transgene. Therefore, a second generation of the system was developed (Supplementary Figure S1B). In the second generation system, PolG exo- is flanked by LoxP sites allowing the expression of the mPolGexo− transgene to be terminated by Cre recombinase delivered either by stable transfection with a plasmid, or by transduction with a retrovirus.
While the second generation system proved to be a reliable means for mouse mtDNA mutagenesis, incomplete excision mediated by Cre recombinase (44,45) proved to be a limitation. Therefore, a third generation system was developed (Supplementary Figure S1C). Similar to the second generation system, the third generation system relies on termination of PolG exo- expression by Cre-mediated excision. However, in the third generation system a promoterless hygromycin resistance gene was inserted immediately downstream of the second LoxP site thereby placing the hygromycin resistance gene under control of the Tet-On promoter upon excision of the PolG exo-. Therefore, only those cells that were successfully excised would be resistant to hygromycin upon induction with doxycycline (Supplementary Figure S1C).
Implementation and characterization of the system
The system was employed for the mutagenesis of UNG/2641 and 3T3#52 cells. These two cell lines possess complementary properties: 3T3#52 cells demonstrate tight control over PolG exo- with low levels of leaky expression, which makes them superior candidates for the controlled accumulation of mutations. However, mtDNA depletion in these cells proceeds more slowly, with target copy number ∼1 copy per cell achieved in around 18 days (Supplementary Figure S2A). On the other hand, UNG/2641 cells have relatively high leaky expression of the PolG exo- and accumulate mutations in mtDNA even without induction, yet they are more resistant to EtBr and deplete of mtDNA more easily, although with higher concentrations of this reagent (Supplementary Figure S2B). These properties make UNG/2641 cells a superior host for intracellular cloning of the mutant mtDNA molecules. Both parent cell lines provided a range of mtDNA mutations. Using the methodology described above, we isolated 101 clones containing single non-synonymous homoplasmic mtDNA mutations (Supplementary Table S2) and 56 clones containing single non-synonymous heteroplasmic mtDNA mutations (Supplementary Table S3).
The potential to mutagenize any base in mtDNA is the biggest advantage of using the PolG exo- based mutagenesis over previously used approaches. Wanrooij et al. examined distribution of PolG exo- induced mtDNA mutations in a ∼1000 bp region of the light strand origin of replication, and concluded that, although the distribution of mutation frequencies was not perfectly uniform, every base could be mutated with the exception of those which are critical for mtDNA replication (46). This observation is not likely to be a major limitation of the proposed method, since the same regulatory bases are not likely to be mutated in human disease because of replicative disadvantage and negative selection against mtDNA molecules containing such mutations. The distribution of the 812 base substitutions identified in this study (including those found in clones containing multiple mutations and therefore not reflected in Supplementary Tables S2–S4) was fairly uniform throughout the mtDNA genome with the exception of the regulatory region (D-loop), in which few mutations were detected (Figure 2A).
Figure 2.
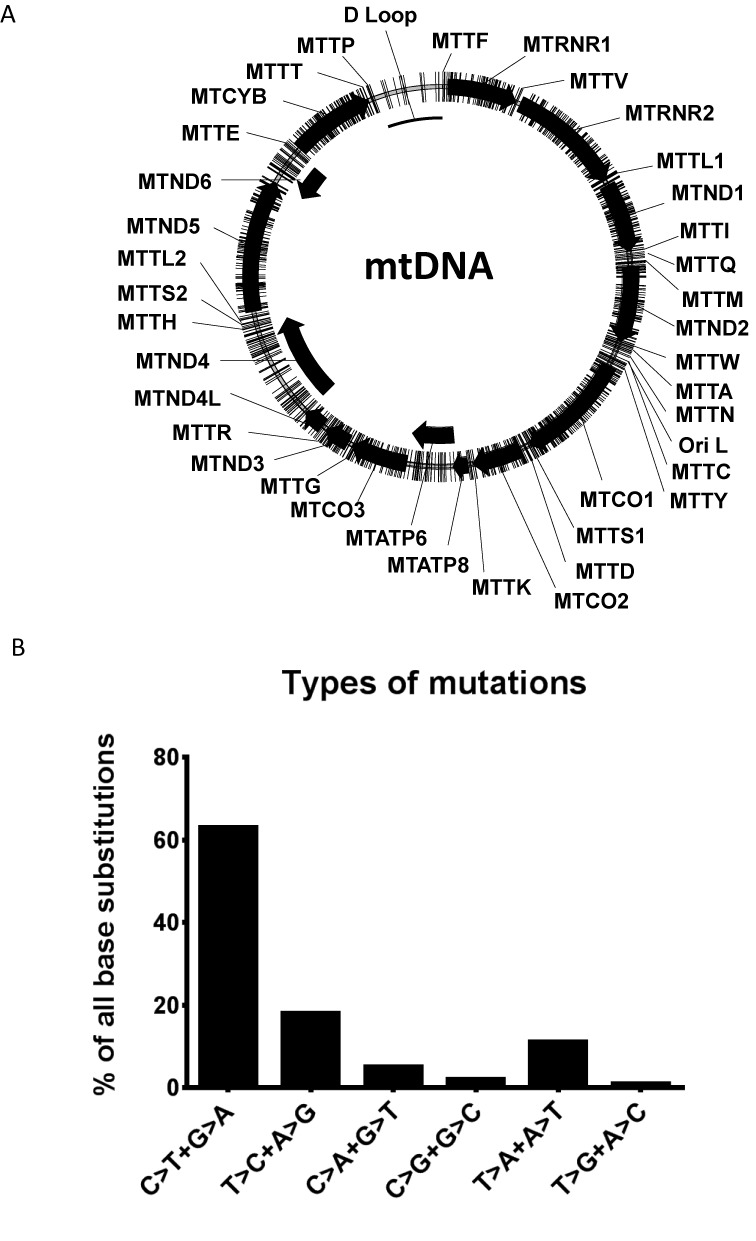
Characteristics of mtDNA mutagenesis with PolG exo-. (A) Mapping of all detected mutations of mouse mtDNA. Mutations are designated as lines crossing the contour of the circular mtDNA molecule. The polypeptide-encoding genes are designated as black arrows. (B) Frequencies of various types of mutations induced by PolG exo- in mouse mtDNA. Since it is impossible to determine on which DNA strand a mutation occurred, complementary mutations are grouped (e.g. C>T transition on the Heavy strand is equivalent to G>A transition on the Light strand. Therefore C>T and G>A transitions are grouped together).
Of the 812 unique point mutations, 619 (76.2%) were found in polypeptide-encoding genes. This is consistent with expectations based on the fraction of mtDNA encoding polypeptides (70%). Of the 619 point mutations in protein-encoding genes, 202 were found in the first position of the codon, 210 in the second and 207 in the third. This observation suggests that mutations induced by PolG exo- are well-tolerated in cultured cells. Indeed, one would expect a bias toward the third position of the codon (synonymous mutations) if a sizable fraction of the mutations had detrimental effect on cell viability. Overall, 69 mutations (8%) were identified in genes encoding tRNA, which is consistent with the fraction of mtDNA occupied by tRNA genes (9%). The fraction of mutations in rRNA genes (13%) is also consistent with the fraction of mtDNA occupied by these genes (15.6%). Finally, the mutation spectrum (Figure 2B) was generally consistent with the spectrum of spontaneous mtDNA mutations, except for higher frequency of A>T+T>A transversions, which is likely due to the proofreading defect in mPolGexo- (47).
Assuming perfectly random distribution of mutations, uniform depletion and repletion and uniform growth rates of all mutants, the method is expected to yield up to 36% of clones with single mutations. The actual yield of such clones varied in different libraries between 8% and 25%, suggesting non-uniformity of mutagenesis and potential for further improvements to the method.
Homoplasmic versus heteroplasmic mutations
Both homo- and heteroplasmic mtDNA mutations have been reported to cause human disease. Analysis of all pathogenic mtDNA mutations listed in the recent (11 April 2014) release of the MITOMAP database reveals that approximately half of all known pathogenic mtDNA mutations cause disease in the homoplasmic state, and other half cause disease in the heteroplasmic state. About 10% of all reported mutations can cause disease in either state. Therefore, while cells carrying a particular mtDNA mutation in the homoplasmic state may be better suited for the characterization of induced biochemical defects, cell lines carrying mouse mtDNA mutations in either the homo- or heteroplasmic state are suitable for the generation of mouse models of human disease.
Utility of the same mutation in both the homo- and heteroplasmic states indicates that (1) interconversion of heteroplasmic mutations into homoplasmic and vice versa can be useful, and (2) isolation of cell lines with altered (increased or decreased) levels of heteroplasmy may be useful for modeling threshold effects, heteroplasmy inheritance and tissue variability of mutation loads. Partial mtDNA depletion has been previously reported as an effective means for inducing transitions from the hetero- to homoplasmic state and to isolate clones with altered levels of heteroplasmy in cultured cells (48). Accordingly, we used intracellular cloning of mtDNA molecules to convert a number of heteroplasmic mutations (e.g. G3864A, G11663T, A11687G, A12107T, C13871T, C14650G and others) into the homoplasmic state. In the process, clones with altered levels of heteroplasmy were isolated as well (Figure 3).
Figure 3.
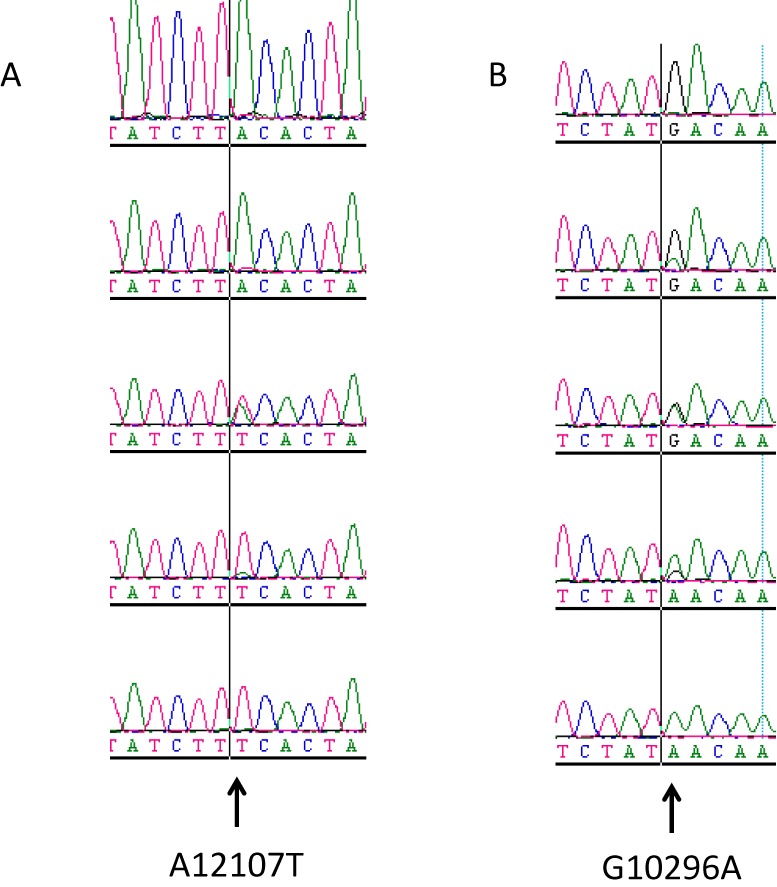
Examples of mtDNA segregation during intracellular cloning. Heteroplasmic clones containing mutations A12107T (A) or G10296A (B) were subjected to intracellular cloning as described in the ‘Materials and Methods’ section. Chromatogram traces of representative clones spanning a range of mutation loads between 0 and 100% are presented for each mutation.
Our attempts to segregate mutations in clones containing two or more heteroplasmic mutations by repeating intracellular cloning of the mtDNA led to the observation that cells contained only two different mtDNA species in the vast majority of heteroplasmic cell lines isolated. We also observed that cell lines containing up to three heteroplasmic mtDNA mutations could be made homoplasmic in most cases for at least one of the mutations present by repeating intracellular cloning. Therefore, cell lines containing two or three non-synonymous heteroplasmic mtDNA mutations (Supplementary Table S4) are useful for isolating cell lines with a single homoplasmic non-synonymous mutation.
The converse effect, i.e. dilution of the homoplasmic mutant mtDNA with the WT counterpart in order to obtain heteroplasmic cells, can be achieved by incomplete inactivation of the mitochondria with Rhodamine 6G in embryonic stem (ES) cells followed by fusion with cytoplasts containing homoplasmic mutant mtDNA (29). Alternatively, this can be achieved by electrofusion of single-cell embryos containing WT mtDNA with cytoplasts containing homoplasmic mutant mtDNA as described previously (49,50).
Validation of the resource
For initial validation of the generated collection of mtDNA mutants, baseline respiration rates were examined in 15 clones containing homoplasmic missense mutations with a Seahorse XF-24 extracellular flux analyzer. As expected of an unbiased collection generated without enrichment for functional mutants, clones exhibited a range of respiratory activities with 60% of the mutants retaining WT rates of oxygen consumption, and the remaining 40% demonstrating various degrees of impairment of respiratory chain activity in vivo (Figure 4A).
Figure 4.

Validation of the resource. (A) respiratory profiling of the 14 missense mutants. Note the predominance of near-WT activities and a range of respiratory activities in the remaining mutants. (B) Respiratory profiling of nonsense mutants. (B) Respiration by tRNA mutants. (C) ROS production by tRNA mutants. (D) Membrane potential in tRNA mutants. (E) Respiration of homoplasmic nonsense mutants. (F) ROS production by nonsense mutants. Dashed horizontal line, WT (100%). (G and H) Increased ROS production in nonsense mutants depends on mtDNA. mtDNA depletion by mUNG1 (G) is accompanied by reduced ROS production (H). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant. (A–D) One-way ANOVA with post-hoc Tukey test. (G and H) Two-tailed unpaired Student's t-test assuming unequal variances.
We further examined respiration rates and reactive oxygen species (ROS) production in several cell lines harboring homoplasmic mutations in tRNA genes. Except for the clone carrying C7746T mutation in the MTTK, respiratory rates for the homoplasmic tRNA mutants tested did not differ significantly from the WT (Figure 4B). Interestingly, the C7746T mutation in mouse mtDNA affects the base homologous to the one immediately adjacent to A8344 in the T-loop of the human MTTK. The A8344G mutation is found in over 80% of the patients with Myoclonic Epilepsy and Ragged Red Muscle Fibers (MERRF) (51). A transmitochondrial mouse model for MTTK has been reported recently, which reproduced many salient features of the disease, yet no ragged red fibers were reported in affected animals (39). Therefore, C7746T mutation may be used to further refine a mouse model for MERRF.
Elevated ROS production is often considered a hallmark of mitochondrial dysfunction, even though in some experimental systems mitochondrial dysfunction caused by mtDNA mutations is not associated with elevated ROS (52). Therefore, we examined levels of mitochondrial ROS in tRNA mutants. Even though most tRNA mutants had normal levels of respiration, ∼2/3 of them showed reduced ROS production as compared to WT (Figure 4C). The reduction in ROS production correlated well with reduced membrane potential in the same set of mutants (Figure 4D). This may indicate a mild defect in mitochondrial translation, which is compensated for by more efficient electron transfer, and therefore reduced ‘escape’ of electrons to mediate one-electron oxygen reduction and formation of ROS.
Finally, respiration and ROS production in a subset of homoplasmic nonsense mutants were examined (Figure 4E and F). A few of the nonsense mutants retained residual respiration, which was most pronounced in the MTND1 W185* and MTND6 G68* mutants. While the respiratory flux trace of the MTND1 W185* mutant was typical of other nonsense mutants in that it lacked a response to an uncoupler (Supplementary Figure S3A), the MTND6 G68* mutant retained 44% of WT respiratory activity and responded to mitochondrial respiratory chain inhibitors in a predictable way, despite the presence of an inactivating nonsense mutation (Supplementary Figure S3B and C). This is consistent with previous studies reporting nuclear suppression of nonsense mutations in MTND6 and other mtDNA-encoded subunits (53,54). Therefore, our collection of nonsense mutants can be useful in studying nuclear suppression of mtDNA mutations. Interestingly, a significant fraction of our nonsense mutants demonstrated increased ROS production (Figure 4F). To test whether this increased ROS production was dependent on mtDNA, two mutants with favorable growth characteristics, MTND4, R432fs and MTND5, W196*, were transduced with either empty retroviral vector, or a retrovirus encoding mitochondrially targeted human uracil-N-glycosylase carrying a Y147A mutation (mUNG1). Previously, we have demonstrated that this mutant enzyme induces rapid depletion of mtDNA (38). Indeed, transduction with mUNG1 was accompanied by the loss of mtDNA (Figure 4G) and reduced ROS production (Figure 4H). Therefore, in these mutants, increased ROS production is mediated, at least in part, by mtDNA.
Mouse equivalents of pathogenic human mutations
A primary objective of this study was to produce mouse cell lines harboring mtDNA mutations that could then be used to generate mouse models of human mitochondrial disorders. Therefore, we compared the mtDNA mutations generated in this study (Supplementary Tables S2 and S3) to those reported in MITOMAP in order to identify mtDNA mutations that affect the same position in the polypeptide chain or the same base in the RNA as reported in human disease. This search produced nine candidates (55–66), or about 5% of all single non-synonymous mutants (Table 1). Thus, a method based on limited mutagenesis of mouse mtDNA by PolG exo- can be used for the isolation of mutations suitable for modeling human mitochondrial disease.
Table 1. Mutations at positions equivalent to those affected in human disease.
| Locus | Disease | Mouse mutation | Human allele | Reference |
|---|---|---|---|---|
| MTND1 | LHON/MELASa overlap | G2820A (E24K) | G3376A (E24K) | (55) |
| MTND1 | MELAS/LS/ LDYTa | G3142A (G131E) | G3697A (G131C) | (56–65) |
| MTTW | Hypertension factor | G4977A | G5540A | (60) |
| MTTC | Myopathy/DEAFa | A5214T | G5783A | (61) |
| MTCO1 | Prostate cancer | G6582A (V419M) | A7158G (I419V) | (62) |
| MTCO2 | HCMa susceptibility | T7125C (V38A) | G7697A (V38I) | (63) |
| MTATP6 | Ataxia syndromes | T8435C (L170S) | T9035C (L170P) | (64) |
| MTCO3 | EXITa | T9189C (S195P) | T9789C (S195P) | (65) |
| MTND4 | LHONa | G11186A (R340H) | G11778A (R340H) | (66) |
aLHON, Leber Hereditary Optic Neuropathy; MELAS, Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-like episodes; DEAF, Maternally inherited DEAFness or aminoglycoside-induced DEAFness; HCM, Hypertrophic CardioMyopathy; EXIT, Exercise Intolerance; LS, Leigh syndrome; LDYT, Leber's hereditary optic neuropathy and dystonia.
One of the more interesting mtDNA mutations generated in this study is the G11186A mutation, resulting in an amino acid substitution R340H in the MTND4 gene. This mutation is equivalent to G11778A in the human gene for MTND4, which results in the same amino acid substitution. In the human population, G11778A is the most common Leber Hereditary Optic Neuropathy ((LHON) mutation, accounting for ∼70% of all cases worldwide (67,68). The G11186A mutation was initially isolated in a heteroplasmic state along with a second mutation, C15581T, in the D-loop region which was homoplasmic. However, through intracellular cloning of mtDNA we were able to segregate the two mtDNA species present in this clone, thus producing two homoplasmic clones: one containing a D-loop mutation C15581T, and another containing G11186A (mouse LHON, or mLHON) in addition to C15581T. The isolation of two mtDNA variants, C15581 and C15581 + G11186A, from the same heteroplasmic clone illustrates the progressive nature of mutagenesis in our system. In this case, the G11186A mutation arose apparently after the C15581T mutation had occurred. The C15581 nucleotide in mouse mtDNA is equivalent to A16180 in the human D-loop, a reported (MITOMAP) polymorphic site not associated with disease. To evaluate effects of G11186A and C15581T mutations on mitochondrial function, we compared respiratory profiles in WT cells, cells carrying a D-loop mutation only, and cells carrying both LHON and D-loop mutations. As expected, respiratory rates of WT and D-loop mutant were similar and significantly higher than that of the LHON + D-loop mutant (Figure 5A). After subtraction of non-mitochondrial respiration, respiratory rates of the WT and D-loop mutant were nearly identical, while respiration in the double mutant was only 24% of WT values (Figure 5B).
Figure 5.
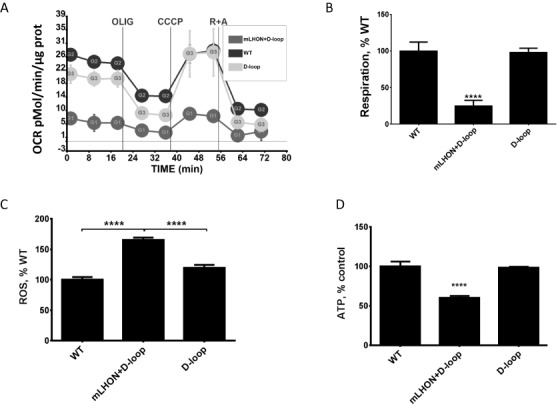
Properties of cybrids containing mouse D-loop and LHON + D-loop mutations. (A and B) Respiration is suppressed in the LHON + D-loop double mutant, but not in the D-loop mutant. (C) ROS production is elevated in the LHON+D-loop double mutant, but not in the D-loop mutant. (D) ATP content is reduced in the LHON + D-loop double mutant, but not in the D-loop mutant. ****P < 0.0001, one-way ANOVA with post-hoc Tukey test.
Elevated mitochondrial ROS production as a consequence of respiratory chain defects is believed to play a pivotal role in the pathophysiology of LHON (69). Cybrids containing mtDNA from LHON patients demonstrate increased ROS production (70), and antioxidants appear to be protective against neuronal defects induced by LHON mutation (71). Therefore, we compared mitochondrial ROS production in cybrids containing WT mtDNA to cybrids containing the D-loop mutation alone or D-loop + mLHON mutations. Like in human cybrids, ROS production was increased in cybrids containing mLHON mutation (Figure 5C). Finally, reduced ATP content was reported in immortalized lymphoblasts from LHON patients (72). Similarly, reduced ATP levels were observed in cybrids containing mLHON (Figure 5D). Overall, these results suggest that the properties of cybrids containing human and mouse LHON mutations are similar, and that mouse G11186A mutation is a suitable candidate for the generation of a mouse model of LHON.
CONCLUSIONS
The method for the generation of random mutations in mouse mtDNA described here enables the production of a large number of homo- and heteroplasmic clones with single and multiple non-synonymous mutations. Using this method, we were able to generate a collection of cell lines with mutations in mtDNA that far exceeds the combined number of cell lines generated so far using alternative methods. The mutant cell lines generated in this study should prove useful for generating mouse models of human disease, and represent a valuable resource for basic studies aimed at unraveling molecular mechanisms of respiratory chain (dys)function, in particular of ROS production as a consequence of impairment of the respiratory chain. They may also be useful in studies aimed at identifying nuclear suppressors of mtDNA mutations. Considering that proofreading-deficient human DNA-polymerase γ efficiently introduces mutations into human mtDNA (40), we suggest that, when appropriately modified, the method described here can be used for the isolation of mtDNA mutations in human cell lines as well as in cell lines from other species.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors wish to acknowledge Viktoriya Pastukh for technical assistance. Olga Alexeeva is acknowledged for writing a script for the correspondence of nucleotide and amino acid substitutions in human and mouse genomes and for critical reading of the manuscript.
FUNDING
National Institutes of Health [HL66299 and OD010944]; University of South Alabama College of Medicine Dean's office. Funding for open access charge: University of South Alabama College of Medicine Dean's office.
Conflict of interest statement. None declared.
REFERENCES
- 1.Herrmann J.M., Longen S., Weckbecker D., Depuydt M. Biogenesis of mitochondrial proteins. Adv. Exp. Med. Biol. 2012;748:41–64. doi: 10.1007/978-1-4614-3573-0_3. [DOI] [PubMed] [Google Scholar]
- 2.Noll T., Wissemann P., Mertens S., Krutzfeldt A., Spahr R., Piper H.M. Hypoxia tolerance of coronary endothelial cells. Adv. Exp. Med. Biol. 1990;277:467–476. doi: 10.1007/978-1-4684-8181-5_52. [DOI] [PubMed] [Google Scholar]
- 3.Ylikallio E., Suomalainen A. Mechanisms of mitochondrial diseases. Ann. Med. 2012;44:41–59. doi: 10.3109/07853890.2011.598547. [DOI] [PubMed] [Google Scholar]
- 4.Schapira A.H. Mitochondrial diseases. Lancet. 2012;379:1825–1834. doi: 10.1016/S0140-6736(11)61305-6. [DOI] [PubMed] [Google Scholar]
- 5.Zheng J., Ji Y., Guan M.X. Mitochondrial tRNA mutations associated with deafness. Mitochondrion. 2012;12:406–413. doi: 10.1016/j.mito.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Maassen J.A., Janssen G.M., ‘t Hart L.M. Molecular mechanisms of mitochondrial diabetes (MIDD) Ann. Med. 2005;37:213–221. doi: 10.1080/07853890510007188. [DOI] [PubMed] [Google Scholar]
- 7.Bannwarth S., Abbassi M., Valero R., Fragaki K., Dubois N., Vialettes B., Paquis-Flucklinger V. A novel unstable mutation in mitochondrial DNA responsible for maternally inherited diabetes and deafness. Diabetes Care. 2011;34:2591–2593. doi: 10.2337/dc11-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Supale S., Li N., Brun T., Maechler P. Mitochondrial dysfunction in pancreatic beta cells. Trends Endocrinol. Metab. 2012;23:477–487. doi: 10.1016/j.tem.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 9.Wallace D.C. Mitochondria and cancer. Nat. Rev. Cancer. 2012;12:685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu M. Somatic mitochondrial DNA mutations in human cancers. Adv. Clin. Chem. 2012;57:99–138. doi: 10.1016/b978-0-12-394384-2.00004-8. [DOI] [PubMed] [Google Scholar]
- 11.Milone M. Mitochondria, diabetes, and Alzheimer's disease. Diabetes. 2012;61:991–992. doi: 10.2337/db12-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Russell O., Turnbull D. Mitochondrial DNA disease-molecular insights and potential routes to a cure. Exp. Cell Res. 2014;325:38–43. doi: 10.1016/j.yexcr.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaefer A.M., Taylor R.W., Turnbull D.M., Chinnery P.F. The epidemiology of mitochondrial disorders-past, present and future. Biochim. Biophys. 2004;1659:115–120. doi: 10.1016/j.bbabio.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Elliott H.R., Samuels D.C., Eden J.A., Relton C.L., Chinnery P.F. Pathogenic mitochondrial DNA mutations are common in the general population. Am. J. Hum. Genet. 2008;83:254–260. doi: 10.1016/j.ajhg.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mahoney D.J., Parise G., Tarnopolsky M.A. Nutritional and exercise-based therapies in the treatment of mitochondrial disease. Curr. Opin. Clin. Nutr. Metab. Care. 2002;5:619–629. doi: 10.1097/00075197-200211000-00004. [DOI] [PubMed] [Google Scholar]
- 16.Marriage B., Clandinin M.T., Glerum D.M. Nutritional cofactor treatment in mitochondrial disorders. J. Am. Diet. Assoc. 2003;103:1029–1038. doi: 10.1016/s0002-8223(03)00476-0. [DOI] [PubMed] [Google Scholar]
- 17.Smith P.M., Ross G.F., Taylor R.W., Turnbull D.M., Lightowlers R.N. Strategies for treating disorders of the mitochondrial genome. Biochim. Biophys. Acta. 2004;1659:232–239. doi: 10.1016/j.bbabio.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 18.Lertrit P., Noer A.S., Byrne E., Marzuki S. Tissue segregation of a heteroplasmic mtDNA mutation in MERRF (myoclonic epilepsy with ragged red fibers) encephalomyopathy. Hum. Genet. 1992;90:251–254. doi: 10.1007/BF00220072. [DOI] [PubMed] [Google Scholar]
- 19.Matthews P.M., Hopkin J., Brown R.M., Stephenson J.B., Hilton-Jones D., Brown G.K. Comparison of the relative levels of the 3243 (A–>G) mtDNA mutation in heteroplasmic adult and fetal tissues. J. Med. Genet. 1994;31:41–44. doi: 10.1136/jmg.31.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poulton J., Marchington D.R. Segregation of mitochondrial DNA (mtDNA) in human oocytes and in animal models of mtDNA disease: clinical implications. Reproduction. 2002;123:751–755. doi: 10.1530/rep.0.1230751. [DOI] [PubMed] [Google Scholar]
- 21.Ozawa M., Nonaka I., Goto Y. Single muscle fiber analysis in patients with 3243 mutation in mitochondrial DNA: comparison with the phenotype and the proportion of mutant genome. J. Neurol. Sci. 1998;159:170–175. doi: 10.1016/s0022-510x(98)00152-x. [DOI] [PubMed] [Google Scholar]
- 22.Greaves L.C., Nooteboom M., Elson J.L., Tuppen H.A., Taylor G.A., Commane D.M., Arasaradnam R.P., Khrapko K., Taylor R.W., Kirkwood T.B., et al. Clonal expansion of early to mid-life mitochondrial DNA point mutations drives mitochondrial dysfunction during human ageing. PLoS Genet. 2014;10:e1004620. doi: 10.1371/journal.pgen.1004620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thajeb P., Dai D., Chiang M.F., Shyu W.C. Genotype-phenotype correlation of maternally inherited disorders due to mutations in mitochondrial DNA. Taiwan J. Obstet. Gynecol. 2006;45:201–207. doi: 10.1016/S1028-4559(09)60225-4. [DOI] [PubMed] [Google Scholar]
- 24.Smith P.R., Dronsfield M.J., Mijovic C.H., Hattersley A.T., Yeung V.T., Cockram C., Chan J.C., Barnett A.H., Bain S.C. The mitochondrial tRNA[Leu(UUR)] A to G 3243 mutation is associated with insulin-dependent and non-insulin-dependent diabetes in a Chinese population. Diabet. Med. 1997;14:1026–1031. doi: 10.1002/(SICI)1096-9136(199712)14:12<1026::AID-DIA514>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki Y., Taniyama M., Muramatsu T., Atsumi Y., Hosokawa K., Asahina T., Shimada A., Murata C., Matsuoka K. Diabetes mellitus associated with 3243 mitochondrial tRNA(Leu(UUR)) mutation: clinical features and coenzyme Q10 treatment. Mol. Aspects Med. 1997;18(Suppl.):S181–S188. doi: 10.1016/s0098-2997(97)00041-1. [DOI] [PubMed] [Google Scholar]
- 26.Hsu C.C., Chuang Y.H., Tsai J.L., Jong H.J., Shen Y.Y., Huang H.L., Chen H.L., Lee H.C., Pang C.Y., Wei Y.H., et al. CPEO and carnitine deficiency overlapping in MELAS syndrome. Acta Neurol. Scand. 1995;92:252–255. doi: 10.1111/j.1600-0404.1995.tb01697.x. [DOI] [PubMed] [Google Scholar]
- 27.Ciafaloni E., Ricci E., Shanske S., Moraes C.T., Silvestri G., Hirano M., Simonetti S., Angelini C., Donati M.A., Garcia C., et al. MELAS: clinical features, biochemistry, and molecular genetics. Ann. Neurol. 1992;31:391–398. doi: 10.1002/ana.410310408. [DOI] [PubMed] [Google Scholar]
- 28.Giordano C., Iommarini L., Giordano L., Maresca A., Pisano A., Valentino M.L., Caporali L., Liguori R., Deceglie S., Roberti M., et al. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber's hereditary optic neuropathy. Brain. 2014;137:335–353. doi: 10.1093/brain/awt343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sligh J.E., Levy S.E., Waymire K.G., Allard P., Dillehay D.L., Nusinowitz S., Heckenlively J.R., MacGregor G.R., Wallace D.C. Maternal germ-line transmission of mutant mtDNAs from embryonic stem cell-derived chimeric mice. Proc. Natl. Acad. Sci. U.S.A. 2000;97:14461–14466. doi: 10.1073/pnas.250491597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Inoue K., Nakada K., Ogura A., Isobe K., Goto Y., Nonaka I., Hayashi J.I. Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat. Genet. 2000;26:176–181. doi: 10.1038/82826. [DOI] [PubMed] [Google Scholar]
- 31.McKenzie M., Trounce I.A., Cassar C.A., Pinkert C.A. Production of homoplasmic xenomitochondrial mice. Proc. Natl. Acad. Sci. U.S.A. 2004;101:1685–1690. doi: 10.1073/pnas.0303184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bai Y., Attardi G. The mtDNA-encoded ND6 subunit of mitochondrial NADH dehydrogenase is essential for the assembly of the membrane arm and the respiratory function of the enzyme. EMBO J. 1998;17:4848–4858. doi: 10.1093/emboj/17.16.4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bai Y., Shakeley R.M., Attardi G. Tight control of respiration by NADH dehydrogenase ND5 subunit gene expression in mouse mitochondria. Mol. Cell. Biol. 2000;20:805–815. doi: 10.1128/mcb.20.3.805-815.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Acin-Perez R., Bayona-Bafaluy M.P., Fernandez-Silva P., Moreno-Loshuertos R., Perez-Martos A., Bruno C., Moraes C.T., Enriquez J.A. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol. Cell. 2004;13:805–815. doi: 10.1016/s1097-2765(04)00124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bayona-Bafaluy M.P., Movilla N., Perez-Martos A., Fernandez-Silva P., Enriquez J.A. Functional genetic analysis of the mammalian mitochondrial DNA encoded peptides: a mutagenesis approach. Methods Mol. Biol. 2008;457:379–390. doi: 10.1007/978-1-59745-261-8_28. [DOI] [PubMed] [Google Scholar]
- 36.Alexeyev M.F., Fayzulin R., Shokolenko I.N., Pastukh V. A retro-lentiviral system for doxycycline-inducible gene expression and gene knockdown in cells with limited proliferative capacity. Mol. Biol. Rep. 2010;37:1987–1991. doi: 10.1007/s11033-009-9647-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shokolenko I.N., Fayzulin R.Z., Katyal S., McKinnon P.J., Wilson G.L., Alexeyev M.F. Mitochondrial DNA ligase is dispensable for the viability of cultured cells but essential for mtDNA maintenance. J. Biol. Chem. 2013;288:26594–26605. doi: 10.1074/jbc.M113.472977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shokolenko I.N., Wilson G.L., Alexeyev M.F. Persistent damage induces mitochondrial DNA degradation. DNA Repair. 2013;12:488–499. doi: 10.1016/j.dnarep.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimizu A., Mito T., Hayashi C., Ogasawara E., Koba R., Negishi I., Takenaga K., Nakada K., Hayashi J. Transmitochondrial mice as models for primary prevention of diseases caused by mutation in the tRNA(Lys) gene. Proc. Natl. Acad. Sci. U.S.A. 2014;111:3104–3109. doi: 10.1073/pnas.1318109111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spelbrink J.N., Toivonen J.M., Hakkaart G.A., Kurkela J.M., Cooper H.M., Lehtinen S.K., Lecrenier N., Back J.W., Speijer D., Foury F., et al. In vivo functional analysis of the human mitochondrial DNA polymerase POLG expressed in cultured human cells. J. Biol. Chem. 2000;275:24818–24828. doi: 10.1074/jbc.M000559200. [DOI] [PubMed] [Google Scholar]
- 41.Trifunovic A., Wredenberg A., Falkenberg M., Spelbrink J.N., Rovio A.T., Bruder C.E., Bohlooly Y.M., Gidlof S., Oldfors A., Wibom R., et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 42.Kujoth G.C., Hiona A., Pugh T.D., Someya S., Panzer K., Wohlgemuth S.E., Hofer T., Seo A.Y., Sullivan R., Jobling W.A., et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 43.Zhang D., Mott J.L., Chang S.W., Denniger G., Feng Z., Zassenhaus H.P. Construction of transgenic mice with tissue-specific acceleration of mitochondrial DNA mutagenesis. Genomics. 2000;69:151–161. doi: 10.1006/geno.2000.6333. [DOI] [PubMed] [Google Scholar]
- 44.Forlino A., Porter F.D., Lee E.J., Westphal H., Marini J.C. Use of the Cre/lox recombination system to develop a non-lethal knock-in murine model for osteogenesis imperfecta with an alpha1(I) G349C substitution. Variability in phenotype in BrtlIV mice. J. Biol. Chem. 1999;274:37923–37931. doi: 10.1074/jbc.274.53.37923. [DOI] [PubMed] [Google Scholar]
- 45.Weis B., Schmidt J., Lyko F., Linhart H.G. Analysis of conditional gene deletion using probe based Real-Time PCR. BMC Biotechnol. 2010;10:75. doi: 10.1186/1472-6750-10-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wanrooij S., Miralles Fuste J., Stewart J.B., Wanrooij P.H., Samuelsson T., Larsson N.G., Gustafsson C.M., Falkenberg M. In vivo mutagenesis reveals that OriL is essential for mitochondrial DNA replication. EMBO Rep. 2012;13:1130–1137. doi: 10.1038/embor.2012.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vermulst M., Wanagat J., Kujoth G.C., Bielas J.H., Rabinovitch P.S., Prolla T.A., Loeb L.A. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 2008;40:392–394. doi: 10.1038/ng.95. [DOI] [PubMed] [Google Scholar]
- 48.D'Aurelio M., Pallotti F., Barrientos A., Gajewski C.D., Kwong J.Q., Bruno C., Beal M.F., Manfredi G. In vivo regulation of oxidative phosphorylation in cells harboring a stop-codon mutation in mitochondrial DNA-encoded cytochrome c oxidase subunit I. J. Biol. Chem. 2001;276:46925–46932. doi: 10.1074/jbc.M106429200. [DOI] [PubMed] [Google Scholar]
- 49.Jenuth J.P., Peterson A.C., Fu K., Shoubridge E.A. Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat. Genet. 1996;14:146–151. doi: 10.1038/ng1096-146. [DOI] [PubMed] [Google Scholar]
- 50.Jenuth J.P., Peterson A.C., Shoubridge E.A. Tissue-specific selection for different mtDNA genotypes in heteroplasmic mice. Nat. Genet. 1997;16:93–95. doi: 10.1038/ng0597-93. [DOI] [PubMed] [Google Scholar]
- 51.Shoffner J.M., Lott M.T., Lezza A.M., Seibel P., Ballinger S.W., Wallace D.C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell. 1990;61:931–937. doi: 10.1016/0092-8674(90)90059-n. [DOI] [PubMed] [Google Scholar]
- 52.Trifunovic A., Hansson A., Wredenberg A., Rovio A.T., Dufour E., Khvorostov I., Spelbrink J.N., Wibom R., Jacobs H.T., Larsson N.G. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. U.S.A. 2005;102:17993–17998. doi: 10.1073/pnas.0508886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deng J.H., Li Y., Park J.S., Wu J., Hu P., Lechleiter J., Bai Y. Nuclear suppression of mitochondrial defects in cells without the ND6 subunit. Mol. Cell. Biol. 2006;26:1077–1086. doi: 10.1128/MCB.26.3.1077-1086.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Srivastava S., Barrett J.N., Moraes C.T. PGC-1alpha/beta upregulation is associated with improved oxidative phosphorylation in cells harboring nonsense mtDNA mutations. Hum. Mol. Genet. 2007;16:993–1005. doi: 10.1093/hmg/ddm045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blakely E.L., de Silva R., King A., Schwarzer V., Harrower T., Dawidek G., Turnbull D.M., Taylor R.W. LHON/MELAS overlap syndrome associated with a mitochondrial MTND1 gene mutation. Eur. J. Hum. Genet. 2005;13:623–627. doi: 10.1038/sj.ejhg.5201363. [DOI] [PubMed] [Google Scholar]
- 56.Spruijt L., Smeets H.J., Hendrickx A., Bettink-Remeijer M.W., Maat-Kievit A., Schoonderwoerd K.C., Sluiter W., de Coo I.F., Hintzen R.Q. A MELAS-associated ND1 mutation causing leber hereditary optic neuropathy and spastic dystonia. Arch. Neurol. 2007;64:890–893. doi: 10.1001/archneur.64.6.890. [DOI] [PubMed] [Google Scholar]
- 57.Valente L., Piga D., Lamantea E., Carrara F., Uziel G., Cudia P., Zani A., Farina L., Morandi L., Mora M., et al. Identification of novel mutations in five patients with mitochondrial encephalomyopathy. Biochim. Biophys. Acta. 2009;1787:491–501. doi: 10.1016/j.bbabio.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 58.Morava E., Hamel B., Hol F., Rodenburg R., Smeitink J. Mitochondrial dysfunction in Stuve-Wiedemann syndrome in a patient carrying an ND1 gene mutation. Am. J. Med. Genet. A. 2006;140:2248–2250. doi: 10.1002/ajmg.a.31452. [DOI] [PubMed] [Google Scholar]
- 59.Kirby D.M., McFarland R., Ohtake A., Dunning C., Ryan M.T., Wilson C., Ketteridge D., Turnbull D.M., Thorburn D.R., Taylor R.W. Mutations of the mitochondrial ND1 gene as a cause of MELAS. J. Med. Genet. 2004;41:784–789. doi: 10.1136/jmg.2004.020537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Silvestri G., Mongini T., Odoardi F., Modoni A., deRosa G., Doriguzzi C., Palmucci L., Tonali P., Servidei S. A new mtDNA mutation associated with a progressive encephalopathy and cytochrome c oxidase deficiency. Neurology. 2000;54:1693–1696. doi: 10.1212/wnl.54.8.1693. [DOI] [PubMed] [Google Scholar]
- 61.Feigenbaum A., Bai R.K., Doherty E.S., Kwon H., Tan D., Sloane A., Cutz E., Robinson B.H., Wong L.J. Novel mitochondrial DNA mutations associated with myopathy, cardiomyopathy, renal failure, and deafness. Am. J. Med. Genet. A. 2006;140:2216–2222. doi: 10.1002/ajmg.a.31436. [DOI] [PubMed] [Google Scholar]
- 62.Petros J.A., Baumann A.K., Ruiz-Pesini E., Amin M.B., Sun C.Q., Hall J., Lim S., Issa M.M., Flanders W.D., Hosseini S.H., et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 2005;102:719–724. doi: 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wei Y.L., Yu C.A., Yang P., Li A.L., Wen J.Y., Zhao S.M., Liu H.X., Ke Y.N., Campbell W., Zhang Y.G., et al. Novel mitochondrial DNA mutations associated with Chinese familial hypertrophic cardiomyopathy. Clin. Exp. Pharmacol. Physiol. 2009;36:933–939. doi: 10.1111/j.1440-1681.2009.05183.x. [DOI] [PubMed] [Google Scholar]
- 64.Sikorska M., Sandhu J.K., Simon D.K., Pathiraja V., Sodja C., Li Y., Ribecco-Lutkiewicz M., Lanthier P., Borowy-Borowski H., Upton A., et al. Identification of ataxia-associated mtDNA mutations (m.4052T>C and m.9035T>C) and evaluation of their pathogenicity in transmitochondrial cybrids. Muscle Nerve. 2009;40:381–394. doi: 10.1002/mus.21355. [DOI] [PubMed] [Google Scholar]
- 65.Horvath R., Schoser B.G., Muller-Hocker J., Volpel M., Jaksch M., Lochmuller H. Mutations in mtDNA-encoded cytochrome c oxidase subunit genes causing isolated myopathy or severe encephalomyopathy. Neuromuscul. Disord. 2005;15:851–857. doi: 10.1016/j.nmd.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 66.Wallace D.C., Singh G., Lott M.T., Hodge J.A., Schurr T.G., Lezza A.M., Elsas L.J., Nikoskelainen E.K. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. 1988;242:1427–1430. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- 67.Cwerman-Thibault H., Augustin S., Ellouze S., Sahel J.A., Corral-Debrinski M. Gene therapy for mitochondrial diseases: Leber Hereditary Optic Neuropathy as the first candidate for a clinical trial. Comptes Rendus Biol. 2014;337:193–206. doi: 10.1016/j.crvi.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 68.Yu-Wai-Man P., Votruba M., Moore A.T., Chinnery P.F. Treatment strategies for inherited optic neuropathies: past, present and future. Eye. 2014;28:521–537. doi: 10.1038/eye.2014.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yen M.Y., Wang A.G., Wei Y.H. Leber's hereditary optic neuropathy: a multifactorial disease. Progr. Retinal Eye Res. 2006;25:381–396. doi: 10.1016/j.preteyeres.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 70.Floreani M., Napoli E., Martinuzzi A., Pantano G., De Riva V., Trevisan R., Bisetto E., Valente L., Carelli V., Dabbeni-Sala F. Antioxidant defences in cybrids harboring mtDNA mutations associated with Leber's hereditary optic neuropathy. FEBS J. 2005;272:1124–1135. doi: 10.1111/j.1742-4658.2004.04542.x. [DOI] [PubMed] [Google Scholar]
- 71.Sala G., Trombin F., Beretta S., Tremolizzo L., Presutto P., Montopoli M., Fantin M., Martinuzzi A., Carelli V., Ferrarese C. Antioxidants partially restore glutamate transport defect in leber hereditary optic neuropathy cybrids. J. Neurosci. Res. 2008;86:3331–3337. doi: 10.1002/jnr.21773. [DOI] [PubMed] [Google Scholar]
- 72.Qian Y., Zhou X., Liang M., Qu J., Guan M.X. The altered activity of complex III may contribute to the high penetrance of Leber's hereditary optic neuropathy in a Chinese family carrying the ND4 G11778A mutation. Mitochondrion. 2011;11:871–877. doi: 10.1016/j.mito.2011.06.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.