


Abstract
LiaR is a ‘master regulator’ of the cell envelope stress response in enterococci and many other Gram-positive organisms. Mutations to liaR can lead to antibiotic resistance to a variety of antibiotics including the cyclic lipopeptide daptomycin. LiaR is phosphorylated in response to membrane stress to regulate downstream target operons. Using DNA footprinting of the regions upstream of the liaXYZ and liaFSR operons we show that LiaR binds an extended stretch of DNA that extends beyond the proposed canonical consensus sequence suggesting a more complex level of regulatory control of target operons. We go on to determine the biochemical and structural basis for increased resistance to daptomycin by the adaptive mutation to LiaR (D191N) first identified from the pathogen Enterococcus faecalis S613. LiaRD191N increases oligomerization of LiaR to form a constitutively activated tetramer that has high affinity for DNA even in the absence of phosphorylation leading to increased resistance. Crystal structures of the LiaR DNA binding domain complexed to the putative consensus sequence as well as an adjoining secondary sequence show that upon binding, LiaR induces DNA bending that is consistent with increased recruitment of RNA polymerase to the transcription start site and upregulation of target operons.
INTRODUCTION
Vancomycin-resistant enterococci are important contributors to the rise of multi-drug resistant Hospital-Associated Infections (HAIs) and in 2013 were categorized as a ‘serious’ threat by the Centers for Disease Control and Prevention (CDC). HAIs are common complications of hospitalization and it is postulated that these infections cause over 2 million cases, and at least 23 000 deaths per year in the United States (http://www.cdc.gov/drugresistance/threat-report-2013/). Perhaps, surprisingly, the majority of HAIs are caused by a handful of organisms. The organisms most strongly associated with US hospital infections and to which new antibiotics are urgently needed are frequently referred to as the no ‘ESKAPE’ pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Enterobacter spp.) (1,2). Among the no ‘ESKAPE’ organisms, vancomycin-resistant enterococci (VRE) are of particular concern due to lack of reliable bactericidal therapeutic options. Enterococci (both E. faecalis and E. faecium) have become increasingly problematic in hospitals around the world with more than 80% of hospital-associated E. faecium isolates resistant to ampicillin and vancomycin (3). As a consequence, VRE present a substantial clinical threat and FDA-approved therapies, such as quinupristin/dalfopristin and linezolid, have important limitations due to resistance, toxicities and bacteriostatic effects against the most recalcitrant organisms. In response to the increased frequency of clinical resistance, the lipopetide daptomycin is often used ‘off-label’ as an antibiotic of last resort for enterococci (3).
Using clinical-strain pairs of DAP-resistant and susceptible E. faecalis recovered from a patient before and after therapy, we showed that mutations in liaFSR, encoding a three-component regulatory system, were responsible for the resistance phenotype (4). Using quantitative experimental evolution, we subsequently demonstrated that the most common evolutionary trajectories leading to daptomycin resistance were indeed through changes in the LiaFSR pathway (5). The LiaFSR system is composed of LiaS, a membrane-bound protein histidine kinase; LiaR, a cytosolic response regulator and LiaF, a membrane-bound protein that reduces or attenuates LiaS activity (Figure 1) (6–9).
Figure 1.
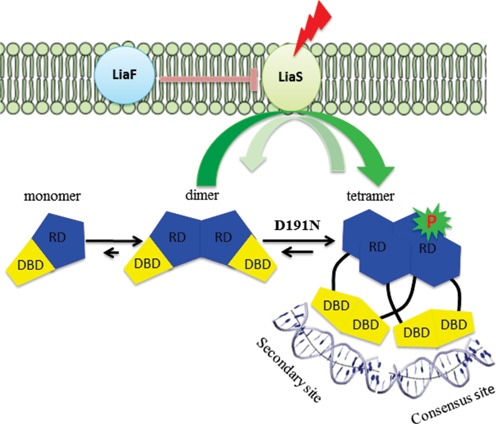
LiaFSR signaling pathway. In the absence of cell membrane-acting antibiotics, the three-component regulatory system, LiaFSR, is turned ‘OFF’ by the negative interaction of LiaF with LiaS. LiaS responds to membrane stress by phosphorylation of LiaR leading to downstream changes in the transcription of several operons to affect membrane homeostasis. E. faecalis LiaR is present largely as a dimer at physiologically relevant concentrations. Phosphorylation, or mutation to a constitutively active analog like LiaRD191N, induces changes in the conformation of the receiver domain leading to release of DNA binding domain and promoting a self-dimerization event to form an active tetramer able to bind extended DNA sequences. Abbreviations: RD, receiver domain; DBD, DNA binding domain.
Mutations to LiaF, LiaR and LiaS have already been identified associated with DAP-resistance in clinical isolates of enterococci and are the most common adaptive changes in response to daptomycin (4,5,10–12). Moreover, we recently showed that deletion of liaR in a DAP-resistant strain of E. faecalis reversed the DAP resistance phenotype and markedly increased susceptibility to other cell-membrane acting antibiotics and antimicrobial peptides with different mechanism of action. Most importantly, the lack of liaR was associated with hypersusceptibility to DAP in a laboratory strain of E. faecalis, supporting the idea of LiaR as a ‘master regulator’ of the enterococcal cell membrane stress response (13).
Using in silico analysis, we identified a potential liaR binding site upstream from a gene cluster encoding three proteins of unknown function (designated liaXYZ, formerly yvlB, pspC, yvlD). Moreover, we previously showed that an adaptive mutation in LiaX (liaX1–289) (5) confers increased resistance to daptomycin, suggesting that this cluster is regulated by LiaR. Thus, the identification of potential LiaR regulatory sequences upstream of the liaFSR and liaXYZ operons along with our earlier observation that mutation of liaX was sufficient to increase resistance to daptomycin formed the basis for the inclusion of the liaXYZ operon in this study. Based on earlier studies of B. subtilis liaRS, we reasoned that adaptive changes to LiaR would likely activate the liaFSR regulon more strongly or even produce a constitutive ‘on’ response that remodels the membrane to reduce antibiotic susceptibility (6).
To elucidate the physicochemical basis for E. faecalis LiaR-mediated changes in daptomycin susceptibility, we used a combination of biophysical and structural approaches to map and quantitate the binding of LiaR and several LiaR variants to the upstream regulatory sequences of both the liaFSR and liaXYZ operons. Our studies reveal that activation of LiaR hinges on a dimer to tetramer transition (Figure 1) that allows LiaR to recognize complex upstream regulatory regions that extend beyond the predicted consensus sequence. Strikingly, the adaptive mutation LiaRD191N shifts LiaR into the activated tetramer even in the absence of phosphorylation leading to a constitutively ‘on’ state. Crystal structures of the LiaR DNA binding domain bound to both the consensus sequence and an adjoining secondary site suggest that LiaR binding significantly bends DNA as part of its potential recruitment of RNA polymerase. As a ‘master’ regulator of the cell envelope stress response, LiaR, as well as other components of the LiaFSR system, may prove to be excellent targets for the development of new strategies and drugs that alter membrane adaptation and, perhaps, would prevent the evolution of resistance in enterococci or other organisms thereby extending the efficacy of current drugs. It is also clear that an increasing number of well-studied transcriptional regulators such as ArcA, BvgA, ComA and UhpA (14–17) may also bind to more complex and nuanced DNA regulatory structures. Presumably such sophisticated regulation reflects robust tuning through evolutionary selection of regulators and their accompanying networks to allow rapid adaptation to changing environments (18).
MATERIALS AND METHODS
Expression and purification of E. faecalis LiaR and its variants
The LiaR gene and truncated LiaR gene (residues 140–206 of LiaR from E. faecalis S613, LiaR(DBD)) were amplified by PCR from genomic DNA S613 and cloned into pETDuet vector. Site-directed mutagenesis of LiaR was performed using the Stratagene QuikChange™ Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). Recombinant proteins were expressed in Escherichia coli BL21 (DE3) at 30°C in EnPressoTM or EnPresso B culture medium (Biosilta, Oulu, Finland) and purified with a HisTrap affinity column (GE Healthcare) followed by the anion-exchange chromatography and gel filtration for the final step (Supplemental Information).
DNA footprint analysis by automated capillary electrophoresis (DFACE)
The DFACE procedure was performed as described previously (Supplemental Information) (19,20).
Determination of DNA binding affinity by Microscale Thermophoresis
The affinity of LiaR and LiaR variants for duplex DNAs were measured by MicroScale Thermophoresis (MST) (21). MST experiments were performed on a Monolith NT.115 system (Nanotemper Technologies) using 25% LED and 100% IR-laser power. Measurements were also carried out on 25% LED power and 90% IR-Laser power for comparison. The resulting Kd values based on average from six independent MST measurements. Data analyses were performed using Nanotemper Analysis software, v.1.5.41.
Analytical ultracentrifugation
Sedimentation equilibrium analytical ultracentrifugation (SEQ) experiments were performed at 20°C using a Beckman XL-I instrument with an AnTi60 rotor. Data analysis was performed using Sedphat 10.40 (22,23). For each protein, the SEQ profiles were globally fitted to a either a dimer–tetramer (LiaR, LiaRD50E and LiaRD191N) or a monomer–dimer (LiaR(DBD), LiaR(DBD)D191N) equilibrium model.
Structure determination of LiaR(DBD) and LiaR(DBD)D191N
Crystals of DBD LiaR suitable for data collection were obtained in 0.1 M Tris pH 8.6, 0.2 M LiSO4, 20% (w/v) PEG 3.350, 0.05% Tween-20, 10% glycerol, 10 mM praseodymium (III) acetate hydrate. The diffraction data sets were collected at Argonne National Laboratory's Advanced Photon Source (ANL APS) beamline 21-ID-F on a MarMosaic 225 CCD detector. The structure of LiaR(DBD) protein was solved by molecular replacement (24) using the DNA binding domains of beryllofluoride-activated VraR from Staphylococcus aureus (PDB code: 4IF4) as an initial search model. The solution from molecular replacement suggested two molecules in the asymmetric unit and a Matthews coefficient of 3.03 (59.5% solvent content) (25). The initial model was submitted to phenix.autobuild and phenix.refine for automatic building and structure refinement (26). Phenix.Xtriage (26) analysis was performed for initial data characterization. Data analysis suggested that pseudo-merohedral twining by the twin law, −h,l,k, was possible; however, no significant pseudotranslation was detected by Patterson analysis. Structural refinement was carried out in Phenix: phenix.refine (26) with the twin specific target function. The initial model was improved by iterative rebuilding using (2Fo − Fc) a map made with Coot (27). The applicable pseudo-merohedral twin law, −h,l,k, was applied from the beginning and throughout refinement in Phenix.refine and the twin fraction reached a final value of 0.49. Water molecules were added using phenix.refine program and by manual inspection of 2Fo − Fc electron density maps. The structure of the LiaRDBD(D191N) protein was solved by molecular replacement (24) using the LiaRDBD structure as an initial search model. Structural refinement was carried out in Phenix: phenix.refine with the twin specific target function (−h,l,k, twin fraction is 0.49) (Supplemental Information). Ramachandran plots and root mean square deviations from ideality for bond angles and lengths for LiaRDBD and LiaR(DBD)D191N were determined using the structure validation program MolProbity (28). Structure factors and final atomic coordinates have been deposited with the Protein Data Bank (entries: 4WSZ and 4WT0, respectively)
Structure determination of LiaR(DBD)D191N with DNA
The crystal used in the DBD LiaRD191N/26-bp DNA structure was crystallized with 0.08 M KCl, 0.02 M BaCl2, 0.04 M sodium cacodylate (pH 6.0), 43% MPD, 0.012 M spermine tetrachloride, 0.01 M proline, 0.01 M strontium chloride. Crystals of the DBD LiaRD191N–22 bp DNA complex were grown in 0.03 M Bis–Tris propane/3.4, 0.07 M citric acid, 18% PEG 3350, 0.05% N,N-dimethyldodecylamine N-oxide, 0.1 M sodium malonate. The single wavelength (0.9788 Å) data sets were collected at (ANL APS) beamline 21-ID-D on a MarMosaic 300 CCD detector. Molecular replacement method was used to determine the structures of DNA–protein complexes using the DBD LiaRD191N (PDB 4WT0) as a search model (Supplemental Information) (PDB: 4WUL and 4WU4).
RESULTS AND DISCUSSION
DNA footprinting analysis of the upstream regions of liaXYZ and liaFSR reveal a larger than expected LiaR recognition sequence
A comparison of the DNA sequences upstream of the liaXYZ and liaFSR operons and comparable operons from Bacillus subtilis and S. aureus (7,29) suggested that LiaR recognizes a T(X)4C(X)4G(X)4A consensus motif (5). The degeneracy of potential sequences upstream of other putative LiaR target operons also suggested that LiaR-mediated regulation might be substantially more nuanced than a simple single consensus sequence (30). DNase I Footprinting followed by Automated Capillary Electrophoresis (DFACE) was used to delineate the LiaR footprint and identify potential target sequences for further physicochemical analysis (Figure 2). Our initial binding studies suggested that unphosphorylated LiaR had no measureable affinity for either operon but that the adaptive mutant LiaRD191N had substantially increased affinity for the liaXYZ operon. We therefore used LiaRD191N as a proxy for activated LiaR in our footprinting experiments. DFACE analysis of the liaXYZ promoter region from −320 to +30 provided clear evidence for a contiguous protected region approximately 43 bp in length that extended from position −120 to −77. LiaRD191N binds to an extended region of the DNA that includes not only the predicted consensus region (−99 to −84) (5), but also the DNA sequences from -100 to -120. We refer to these protected regions outside the consensus sequence as ‘secondary sequences.’ In addition to the protected sequences, we observed a distinct DNase I hypersensitive site at position -83 next to the adenine (underlined) of the T(X)4C(X)4G(X)4A consensus sequence (Figure 2A). To assess the concentration-dependence of the LiaR footprint, DFACE experiments were performed at 0.5 and 5.0 μM LiaR. Overall, the amount of protection at each position increased modestly at 5.0 μM protein but the pattern did not vary significantly with the notable exceptions of A(−84) and the hypersensitive site T(−83) where protection and reactivity increased, respectively. Protein concentration-dependent DNaseI hypersensitivity at T(−83) suggests that LiaR binding may alter the structure or curvature of the DNA allowing more efficient DNaseI cleavage. Based on sequence homology to other response regulators, the active species of LiaR would be expected to dimerize and have a site size of ∼21 nucleotides. Our protection experiments identified a region of protection that was twice the size expected for the simple binding of an activated LiaR dimer to DNA.
Figure 2.
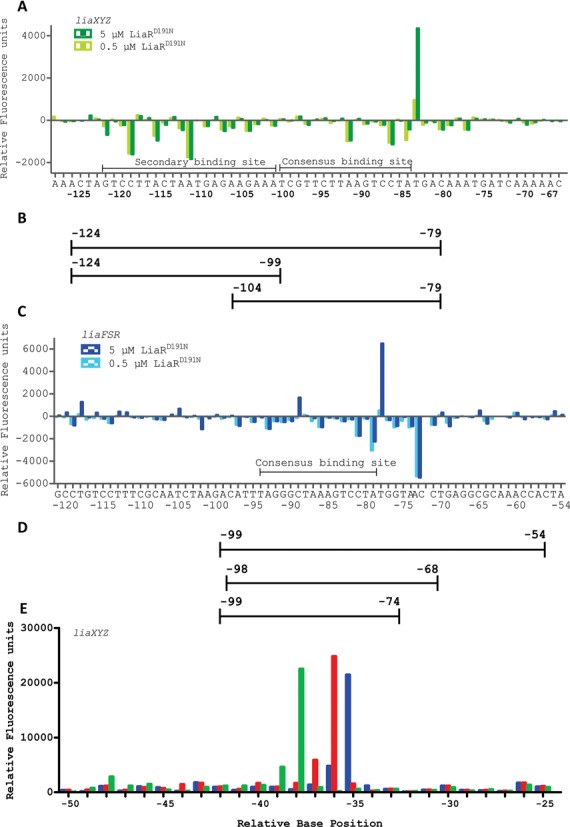
LiaR binds to extended region of DNA that includes sequences outside the proposed canonical consensus sequence. DNase I Footprinting followed by DFACE was used to identify the DNA sequences protected by LiaRD191N within the upstream regions of the liaXYZ (−320 to +30) and liaFSR (−367 to +30) operons of E. faecalis S613. Protection studies were performed at 0.5 and 5 μM LiaRD191N. DNaseI digestion patterns are shown as histograms (A, C, E) where negative changes in relative fluorescence units indicate regions of protection and positives changes indicate hypersensitivity. All DNaseI sensitivity data are relative to a no protein negative control. (A) LiaRD191N binding to the promoter region of the liaXYZ operon. Nucleotide positions refer to the region of LiaR binding (-120 to -77) on the DNA relative to the translation start site of LiaX. (B/D) Oligonucleotides used for DNA binding studies. (C) LiaRD191N binding to the promoter region of the liaFRS operon. Nucleotide positions refer to the region of LiaR binding (−97 to -68) on the DNA relative to the translation start site of LiaF. The AC bar indicates a compression artifact of two bases during electrophoresis seen in both the fragment pattern and the sequencing reaction results. (E) Superimposed electropherograms showing the shift of a hypersensitive location in the upstream region of the liaXYZ operon in response to increasing concentrations of LiaR (blue, 0 μM; red, 0.5 μM; green 5.0 μM). The hypersensitive position shifts from −35(T) to −36(C) to −38(C) in response to increasing protein concentration.
Previous studies on LiaR homologs had suggested that LiaR also regulates its own operon and inspection of the 5’ region of the liaFSR operon revealed two potential consensus sites. We used DFACE to establish the LiaR binding footprint and to observe whether LiaR binding to the liaFSR operon would produce an extended binding site comparable to that observed for the liaXYZ operon. We mapped LiaR binding over the region from −367 to +30 and observed that only the region from −97 to −68 had significant and contiguous protection when incubated with 0.5 μM LiaRD191N (Figure 2C). The potential second consensus binding site at position −258 to −243 did not show any protection (data not shown). There were no significant changes in the size of the footprint or degree of protection in the presence of 5 μM protein compared to 0.5 μM. As with the liaXYZ operon, we observed a strong hypersensitive site at T(−78) the nucleotide following the adenine (underlined) of T(X)4C(X)4G(X)4A consensus sequence (Figure 2C). It is interesting that the liaFSR hypersensitive site is located at the same relative position, e.g. one nucleotide 3’ of the consensus sequence suggesting that protein binding imposes a similar change in DNA backbone structure and accessibility of both the liaXYZ and liaFSR regulatory regions. The LiaR footprint on the liaFSR operon has an additional hypersensitive site at C (−89) that is not present in the LiaR:liaXYZ footprint (Figure 2C). Overall, the most striking difference in the protection patterns for the two operons is the length of the protected regions. The size of the protection site for liaFSR is about 29 bases while that of liaXYZ was 43 bases. Interestingly, LiaR protection for the liaFSR operon extends about 10 bases toward the downstream translation start site, while the liaXYZ protection pattern extends away from the consensus sequence in the upstream direction. A different orientation for the extended protection regions suggests that recognition sequences outside the consensus motif can be oriented in either direction and is consistent with a versatile regulatory architecture.
Phosphomimetic mutant LiaRD50E increases affinity for DNA
Phosphorylation of LiaR leads to activation of target operons to mitigate cell envelope stress (6). Based on a comparison of the E. faecalis LiaR sequence with that of other response regulators from NarL/FixJ subfamily (S. aureus VraR, B. subtilis LiaR, E. coli NarL, FixJ) we identified Asp-50 as a potential site for phosphorylation (31). We were unable to find in vitro conditions that led to quantitative and stable phosphorylation of E. faecalis LiaR using small-molecule phosphoryl donors, such as acetyl phosphate or phosphoryl-mimics such as beryllium fluoride (32). Therefore, in order to perform biochemical studies on the phosphorylated LiaR, we mutated the putative phosphorylation site Asp-50 to a glutamate to produce a potentially stable and constitutively active mutant.
We tested the affinity of LiaR and LiaRD50E for the region encompassing the consensus sequences by Microscale Thermophoresis (MST) (Figure 3). Since our DNA footprinting had clearly identified an extended region of protection that included the putative consensus sequences for the liaXYZ operon, we tested the affinity of LiaR for both the consensus and adjoining secondary sites separately as well as together to make an extended sequence that included the entire DNA footprint region (Figure 2B). Although wild type LiaR affinity for DNA was too low to estimate an accurate Kd, the putative phosphomimetic mutant, LiaRD50E, had substantially increased affinity (Table 1, Figure 3A). Our observed Kd of 4.13 ± 0.48 μM for the liaXYZ consensus sequence is consistent with the in vivo measurements made for the S. aureus homolog of LiaR, VraR suggesting regulation of relevant operons in the micromolar range (29,33). Given that the mutation of Asp-50 to glutamate is not identical to phosphorylation of aspartate, it may well be that the additional electronegative character of phospho-aspartate may further increase the Kd for specific DNA sequences. As expected, mutation of the putative phosphorylation site to alanine (LiaRD50A) decreased LiaR affinity for either sequence below detectable levels. These data are consistent with Asp-50 as the site of phosphorylation and the low affinity of unphosphorylated LiaR for target DNA.
Figure 3.
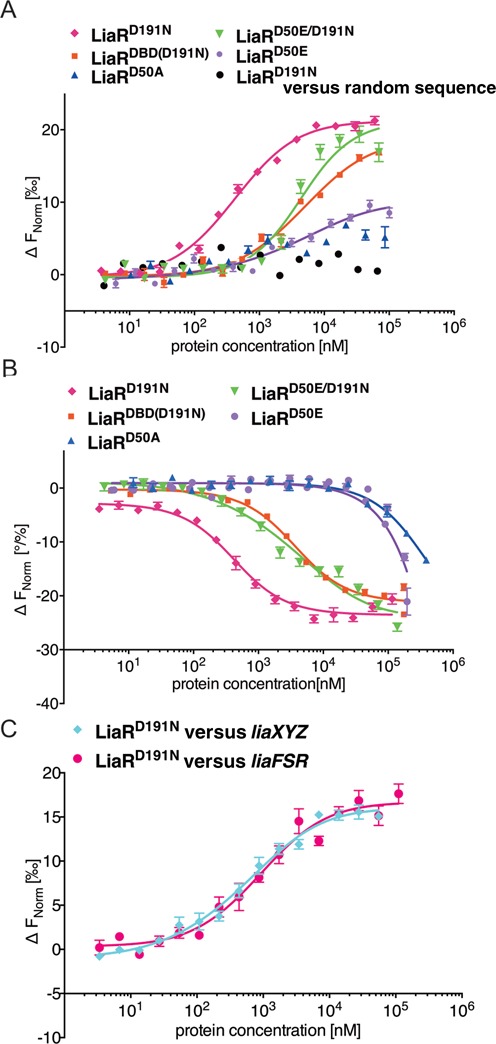
Adaptive mutant LiaRD191N that confers increased daptomycin resistance in E. faecalis S613 dramatically increases LiaR affinity for target DNA sequences. E. faecalis response regulator LiaR–DNA interactions were measured with MST. To determine the Kd, increasing concentrations of LiaRD191N was added to 40 nM of fluorescently labeled DNAs (Supplementary Information). Fnorm (normalized fluorescence) was plotted on the y-axis in per mil [‰] unit (meaning every 1000) against the total concentration of the titrated partner on a log10 scale on the x-axis (21). The resulting Kd values based on average from six independent MST measurements. Note, when the markers were increased in size for readability the error bars became covered in some cases. (A) The binding of LiaRD191N (magenta diamonds), LiaRD50E (purple circles), LiaRD50A (blue triangles), LiaRD50E/D191N (green triangles) and LiaRDBD(D191N) (red squares) to the consensus sequence within the liaXYZ operon or LiaRD191N with a random sequence (black circles). (B) The binding of LiaRD191N (magenta diamonds), LiaRD50E (purple circles), LiaRD50A (blue triangles), LiaRDBD(D191N) (red squares), LiaRD50E/D191N (green triangles) to the extended site of the liaFSR operon. (C) The binding of LiaRD191N to the entire contiguous protected regions (Figure 2B, D) of liaXYZ (cyan diamonds) and liaFSR (magenta circles).
Table 1. DNA binding affinity of E. faecalis S613 LiaR and variants.
| Protein | liaXYZ consensus Kd [μM] | liaXYZ secondary Kd [μM] | liaFSR consensus Kd [μM] | liaFSR extended Kd [μM] | liaXYZ entire protected Kd [μM] | liaFSR entire protected Kd [μM] |
|---|---|---|---|---|---|---|
| LiaR | N/Da | N/Da | N/Da | N/Da | ||
| LiaRD50E | 4.13 ± 0.48 | Weakb | Weakb | >250 | ||
| LiaRD191N | 0.39 ± 0.03 | 21.9 ± 4.5 | >150 | 0.37 ± 0.02 | 0.63 ± 0.04 | 0.88 ± 0.04 |
| LiaRD50A | N/Da | N/Da | N/Da | >250 | ||
| LiaRD50E/D191N | 3.63 ± 0.55 | 16.1 ± 2.10 | N/Da | 2.8 ± 0.30 | ||
| LiaRDBD | N/Da | N/Da | N/Da | N/Da | ||
| LiaR(DBD)D191N | 4.06 ± 0.76 | Weakb | >150 | 1.88 ± 0.08 |
aNo detectable binding my MST up to 466 μM LiaR, 224 μM LiaRD50A, 344 μM LiaRDBD, 138 μM LiaRD50E/D191N.
bAt the concentration range of this experiment setup, a small change in thermophoresis was observed, but it was not large enough to confidentially report a binding value.
Adaptive mutant LiaRD191N that confers increased daptomycin resistance in E. faecalis S613 dramatically increases LiaR affinity for target DNA sequences
In a previous study, we had recovered the mutant LiaRD191N that confers increased daptomycin resistance (5). In contrast to the weak, nearly non-detectable affinity of LiaR for the individual liaXYZ and liaFSR sites, the adaptive mutant LiaRD191N had dramatically increased affinity for all the DNA sequences we measured (Table 1, Figure 3). LiaRD191N had at least a 100-fold increase in the DNA binding affinity to either the liaXYZ or liaFSR consensus sequences compared to wild type LiaR and exhibited 10–500-fold higher affinity to the target sequences than the phosphomimetic mutant LiaRD50E. Binding to the liaXYZ secondary site was also strongly increased suggesting an overall increase in affinity for DNA. Surprisingly however, LiaRD191N affinity for the entire contiguous protected region identified by DNA footprinting for liaXYZ (46 nucleotides, see Figure 2B) was comparable to that for the consensus site alone (liaXYZ: Kd = 0.63±0.04 μM versus 0.39 ± 0.03 μM (Table 1)). These results reveal that despite being protected in our nuclease protection assays, the additional secondary region protected within the liaXYZ operon is not required for high affinity binding. Remarkably, the increased affinity of LiaRD191N for target DNA exceeded that of the phosphomimetic mutant LiaRD50E suggesting that the adaptive mutant is activated irrespective of its potential phosphorylation state. To test whether the mutations LiaRD50E and LiaRD191N functioned cooperatively or independently of each other, we made the double mutant LiaRD50E/D191N and measured its affinity for the liaXYZ and liaFSR sequences. The double mutant had affinity most like the single mutant LiaRD50E suggesting negative epistasis between the alleles. Our data is consistent with a model in which LiaRD191N is a constitutively activated or, perhaps, even a hyperactivated protein.
liaFSR consensus sequence is not sufficient for tight binding by LiaR
We tested the affinity of LiaRD191N for a 26 nucleotide DNA that contained only the putative liaFSR consensus sequence extending from −104 to −79. Surprisingly, wild type LiaR, adaptive mutant LiaRD191N, and phosphomimetic mutant LiaRD50E all had very low affinity for the sequence that was comparable in length and sequence to the high affinity site we had identified in the liaXYZ operon. The affinity of LiaRD191N for the liaFSR consensus was at least 400-fold lower than the liaXYZ consensus sequence (Table 1). DFACE had suggested that the footprint of LiaR on the liaFSR operon extended from the consensus sequence ∼10 bases toward the translation start site. Based upon this DNA protection data, we extended the DNA sequence an additional six nucleotides to include the entire protected region. LiaRD191N affinity for the extended 31 nucleotide sequence increased dramatically to 0.37 ± 0.02 μM making it nearly identical to the high affinity we observed for the liaXYZ consensus sequence (0.39 ± 0.03 μM) (See Figure 2B, D, Figure 3B and Table 1). When we scrambled the extended downstream site of liaFRS (11 nucleotides: ATGTACGAGCT) the LiaRD191N binding affinity decreased ∼3-fold (Kd = 1.35 ± 0.08 μM) but was still higher than for the consensus site alone (Kd > 150 μM) suggesting that both sequence and length of this additional protected region are critical for the binding affinity. Our findings suggest that the additional region protected within the liaFSR binding site (six nucleotides: ACCTGA) is likely important for LiaR binding and positioning relative to the transcriptional start site.
Importantly, our data suggests that the T(X)4C(X)4G(X)4A liaFSR consensus sequence, unlike that of liaXYZ, is not sufficient for high affinity binding and that other nucleotides within and proximal to the consensus sequence can have an important role in attenuating LiaR affinity for target DNA. That these extended DNA sequences have comparable affinities but different ways of realizing increased affinity suggests that each regulatory region is unique and contextualized to the specific operon.
Oligomerization of LiaR to tetramers suggests a potential mechanism for extended DNA target recognition
Studies of the related response regulator S. aureus VraR and the more widely characterized OmpR/PhoB winged-helix transcription factor response regulator subfamily have suggested that a phosphorylation-dependent oligomerization from monomer to dimer is frequently responsible for recognition of specific DNA target sequences (31,32,34). We were therefore surprised that analytical ultracentrifugation analysis (AUC) indicated that the best fit for data obtained on the full length LiaR was a dimer–tetramer equilibrium model with a Kd of 24 μM (Figure 4A, B). Mutation of the Asp-50 phosphorylation site to glutamic acid further increased the extent of tetramerization (Kd = 2 μM) suggesting that oligomer formation would be stimulated by phosphorylation. The correlation of tetramer formation to functional consequences for LiaR recognition of DNA is further reinforced by our observation that the adaptive mutant LiaRD191N is nearly entirely tetrameric. The extent of LiaRD191N tetramerization is so strong that we were unable to observe free dimer and could only estimate the Kd to be <0.06 μM—at least a 400-fold increase in tetramer formation compared to wild type LiaR. The best fits for all the LiaR variants were obtained with a model for dimer to tetramer equilibrium. Additionally, LiaR and its variants eluted during gel filtration as a broad peak with elution volumes, corresponding to the tetrameric and dimeric forms of LiaR, respectively with no evidence for either monomer or higher order oligomers (data not shown). The striking correlation of tetramer formation with increased DNA affinity from our MST studies suggests that E. faecalis LiaR does not follow the more canonical activation of a monomer to dimer species but rather is already present largely as a dimer at physiologically relevant concentrations and that upon phosphorylation, or mutation to a constitutively active analog like LiaRD191N, shifts the oligomeric state to an active tetramer able to bind extended DNA sequences. Concentration of LiaR in vivo has not been determined, but has been reported for the closest homolog of LiaR the S. aureus VraR (29). The concentration of response regulator VraR from S. aureus increased from 2.3 to 6 μM in the presence of cell wall stress (29), which is in good agreement with the maximum increase in vitro transcription product observed at 6 μM VraR-P (33). Note, that the concentration of VraR in the absence of stress is in agreement with those determined for other response regulators, such as OmpR (3500 molecules) (35) and CheY (6000 molecules) (36).
Figure 4.
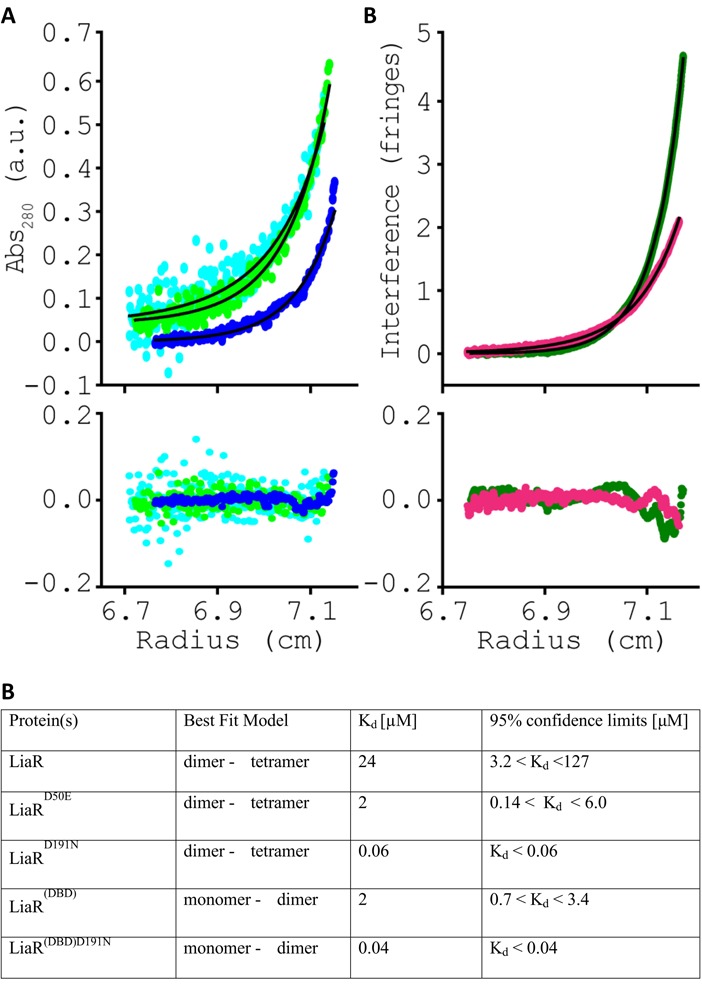
The adaptive mutation D191N mutation in LiaR promotes higher order complex formation. Sedimentation equilibrium analytical ultracentrifugation analysis for (A) LiaR (cyan), LiaRD50E (light green) or LiaRD191N (blue), and (B) LiaR(DBD) (dark green), LiaR(DBD)D191N (magenta). For simplicity, a representative dataset recorded at 14 000 rpm for LiaR, LiaRD50E and LiaRD191N or 36 000 rpm for LiaR(DBD) and LiaR(DBD)D191N are shown. Sedimentation equilibrium profiles for each protein were fitted to either a dimer↔tetramer (A) or a monomer↔dimer (B) self-association model, depicted by the black lines. The residuals for each fit are provided in the lower panel, below the experimental data.
Studies of the isolated LiaR DNA binding domain suggests that an increased affinity for DNA by the adaptive mutant LiaRD191N is correlated with increased oligomerization
To better understand the physicochemical basis for the daptomycin resistance conferred by mutation of position 191 to Asn, we measured the DNA binding affinity and oligomeric states of the isolated DNA binding domain (residues 140–206) of wild type LiaR, and the adaptive mutant LiaRD191N. Like the full length LiaR, the isolated DNA binding domain of LiaR (LiaR(DBD)) had undetectable levels of binding to liaXYZ and liaFSR sequences (Table 1) and, similar to the full length LiaRD191N, LiaR(DBD)D191N had strongly increased binding to liaXYZ (Kd = 4.06 ± 0.76 μM) and extended liaFSR consensus sequences (Kd = 1.88 ± 0.08 μM). Overall, LiaR(DBD)D191N binding to target DNA was ∼10-fold weaker than full-length protein and is consistent with a role for the receiver domain in protein–protein interactions that would bring two copies of LiaR together as an initial dimer that can then further oligomerize into a tetramer. Since the mutation at Asn-191 appears to constitutively activate LiaR in the absence of phosphorylation, the normal role of phosphorylation in stimulating oligomerization via the receiver domain may be effectively overruled by the D191N mutation.
We reasoned that since the adaptive mutation D191N is located in the C-terminal DNA binding domain and that oligomerization was well correlated to DNA recognition, that we could test the role of the adaptive mutation in driving the oligomeric state of LiaR by investigating the oligomerization properties of the isolated LiaR DNA binding domain. Using analytical ultracentrifugation, we observed that the wild type LiaR(DBD) has a modest monomer–dimer transition (Kd = 2 μM). So, despite not having a receiver domain for activation by phosphorylation or mediation of protein–protein contacts, the DNA binding domain of LiaR alone can establish a dimeric species in solution. In strong support for this additional role of the DNA binding domain in self-oligomerization, LiaR(DBD)D191N formed dimers much more strongly than wild-type LiaR with a dissociation constant of the Kd to be <0.04 μM (Figure 4). The association of LiaR(DBD)D191N to a dimer was so strong we could only estimate the Kd to be <0.04 μM, effectively making the DNA binding domain nearly entirely dimeric under these solution conditions. Additionally, gel filtration chromatography of the isolated DBD shows that DBD of LiaRD191N elutes at times consistent for a monomer to dimer transition (data not shown). The trend of increasing dimerization by LiaR(DBD)D191N parallels the trend to increased DNA binding that results from this mutation. Formation of a dimer comprised of the helix-turn-helix motifs in the LiaR DNA binding domain would be consistent with the strong and specific DNA binding we observed in our MST studies with both full-length LiaR and its isolated DNA binding domain. Our observation that LiaR(DBD)D191N has only a 50-fold higher propensity for oligomerization compared to the 400-fold increase in tetramer formation seen for LiaRD191N leads us to speculate that the mutation D191N in the DNA binding domain of LiaR may change the dynamics of the entire LiaR molecule such that a change in the DNA binding domain may propagate into the receiver domain to promote further dimerization contacts.
Structures of the LiaR(DBD)D191N with consensus and secondary DNA target sequences
To understand the atomic basis for specific DNA recognition by LiaR, we determined the structures of the DNA binding domains of wild-type LiaR (LiaR(DBD)), and LiaRD191N (LiaR(DBD)D191N) proteins alone as well as LiaR(DBD)D191N bound to DNA sequences derived from the liaXYZ consensus and secondary sequences by X-ray crystallography (Supplementary Table S1). The structures of LiaR(DBD) and LiaR(DBD)D191N are essentially the same with overall root mean square deviations (rmsd) of 0.4 Å (37). The domain architecture and quaternary arrangement is consistent with previously reported NarL/FixJ family DNA binding domain dimers, such as VraR, NarL, DosR and GerE in the activated state (38–41). As shown in Figure 5A and Supplementary Figure S1 the C-terminal DNA binding domain of LiaR is comprised of four α-helices where the two central helices α-3 from each promoter form a classic helix-turn-helix DNA binding motif. The dimer interface consists primarily of van der Waals contacts between helix-4 of protomer 1 and helix-4 of protomer 2 as well as several potential hydrogen bonds between interfacial residues. The orientation of helix-4 often varies among response regulators and is typically the product of specific but poorly conserved non-polar interactions across the dimer interface. Within the NarL/FixJ family the orientation of helix-4 from each protomer ranges from being almost parallel to one another in the case of NarL and VraR to projecting away from one another such that the two C-termini is spaced more widely than the two N-termini to make an almost V-shaped arrangement for GerE (38,39). In the LiaR–DNA structures, the dimerization helices are oriented almost parallel to each other, a case more similar to NarL and DosR than to GerE. Interestingly, both LiaR(DBD) and LiaR(DBD)D191N crystallize as dimers. This is consistent with our AUC studies suggesting a predominately dimeric quaternary structure. While position 191 is located near the dimer interface, a comparison of the main and side chain positions of Asp-191 from the LiaR(DBD) and Asn-191 of LiaR(DBD)D191N (Figure 6A) showed no significant changes in stereochemistry or potentially altered interactions through the δ-NH2 of Asn-191. The absence of significant structural changes is consistent with a more indirect effect on dimerization perhaps by stabilizing the folded state or the state that is most competent to forming the dimer. As shown in Figure 6B, there is a change in the electrostatic surface of the DNA binding surface due to the change from a negatively charged carboxylic acid group to a polar but uncharged carboxamide sidechain. We speculate that this electrostatic change (an overall less negatively charged surface) could explain both the observed increased DBD dimerization and tighter DNA binding.
Figure 5.
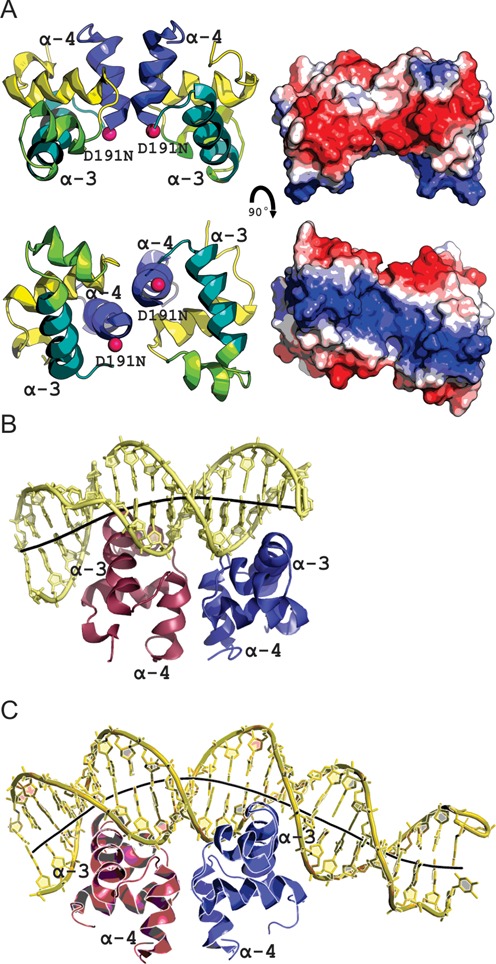
The structure of LiaR(DBD)D191N bound to DNA sequences derived from the liaXYZ consensus and secondary sequences. (A) Structural overview of the isolated DNA binding domain of LiaRD191N. The α4 helices of the LiaR(DBD) (blue) forms part of the molecular recognition surface responsible for formation of the functional dimer required for DNA binding. DNA-recognition helices (α3 from each promoter) are indicated in green. The two α3 helices in the dimer are positioned to create a large electropositive DNA-binding surface. (B, C) LiaR(DBD)D191N bound to DNA sequences derived from the liaXYZ consensus and secondary sites. The LiaR-DNA complex structure shows a strong bend in the DNA, as shown by its helical axis (gray). The helical axis calculated by the program CURVES+ (48) indicated an overall bend of 23.8° and 51.4° for the consensus (B) and secondary (C) sequences, respectively.
Figure 6.
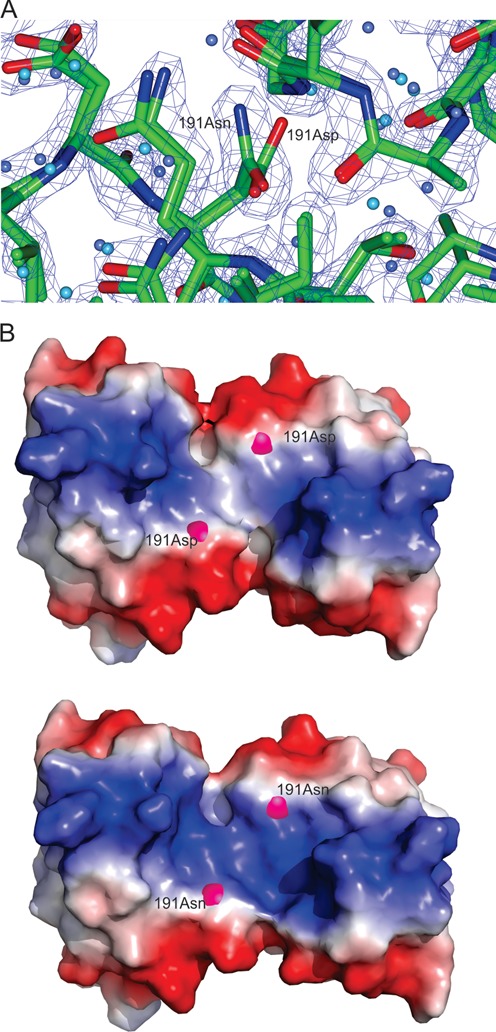
LiaR(DBD)D191N and LiaRDBD homodimer shown as both a cartoon and electrostatic surface representation. (A) Close up comparison of the main and side chain positions of Asp-191 from the LiaR(DBD) and Asn-191 of LiaR(DBD)D191N showed no significant changes in stereochemistry or potentially altered interactions. The (2FOFCWT-PH2FOFCWT) electron density map around Asn191 is contoured at 0.9 absolute value of electrons/Å3. (B). The electrostatic surface (charge surface) of the DNA binding domain of LiaR and LiaRD191N has been calculated. The magenta spheres highlight the position of amino acid 191. Red is negatively charged and blue is positive and there is a color gradient between the two (white being neutral). The DNA binding surface does become slightly more positively charged due to the D191N mutation and therefore more favorable for DNA binding.
In addition to structures of the LiaR(DBD) and LiaR(DBD)D191N, we determined the structure of LiaR(DBD)D191N bound to DNA sequences derived from the liaXYZ consensus and secondary sites (Figure 5B, C). The structure of the uncomplexed LiaR(DBD)D191N dimer is also similar to that of DNA complexes with an rmsd of 0.6 and 0.9 Å for the consensus and secondary sequence complexes. Furthermore, the buried surface areas within the dimerization interface for the two LiaR(DBD)D191N:DNA complexes are comparable to the uncomplexed LiaR(DBD)D191N dimer (∼440Å) suggesting that there are no large conformational changes within the LiaRDBD dimer upon DNA binding. The arrangement of the two helix-turn-helix motifs in the dimer creates a positively charged surface and positioning the two helices (α-3 from each promoter) at the appropriate spacing and relative orientation required for insertion into the major groove of DNA (Figure 5A), which has been observed previously for the DosR and NarL DNA complexes (38,40,41). DNA recognition by LiaR is comprised largely of residues from helix-3 (Figures 7A, B and 8). As might be expected from the DNA binding studies that showed the highest affinity for the liaXYZ consensus sequence, there are more base specific contacts to the DNA in the LiaR(DBD)D191N:liaXYZ consensus sequence complex than the LiaR(DBD)D191N:liaXYZ secondary sequence complex (Figure 8). In the LiaR(DBD)D191N:liaXYZ consensus sequence complex, Lys-174(A/B), Lys-177(A/B), Thr-178(B) are all in position to make specific H-bonds to bases of the consensus sequence (Figures 7A and 8, Supplementary Figure S2) while in the complex to the secondary sequence, only Lys-174(A/B) and Thr-178(A) have the potential make direct H-bonds (Figures 7B and 8, Supplementary Figure S2). While the consensus and secondary sequences have no discernable sequence homology, the molecular recognition surface and many of the comparable residues from LiaR that contact the DNA are largely the same.
Figure 7.
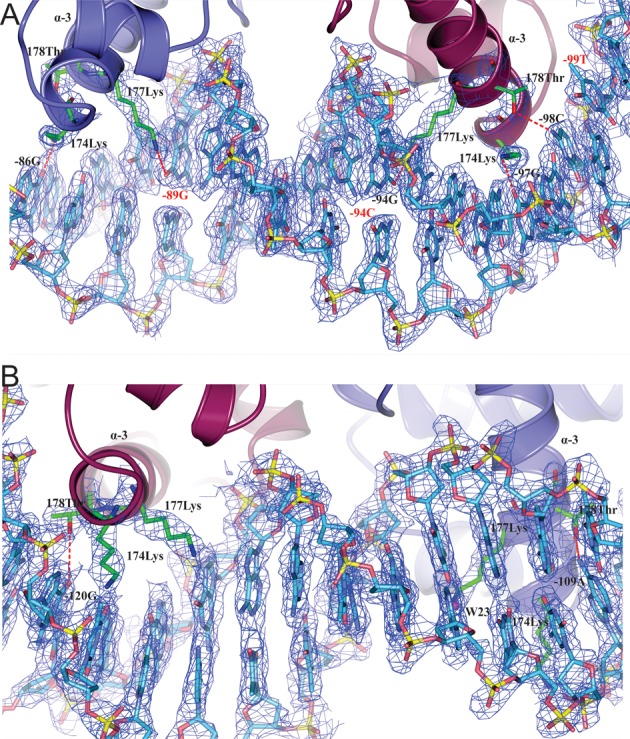
Expanded view of the binding interface of the of LiaR(DBD)D191N bound to DNA sequences derived from the liaXYZ consensus and secondary sites. (A, B) The FEMs (Feature Enhanced Maps) are modified 2mFobs-DFmodel σA-weighted maps computed using phenix to reduce the model bias and retain the existing features (26). The (FEM-PHIFEM) electron density map is contoured at 0.6 absolute value of electrons/Å3 to show how Lys174, Lys177 and Thr178 interact with DNA. The consensus sequence bases are indicated as red color.
Figure 8.
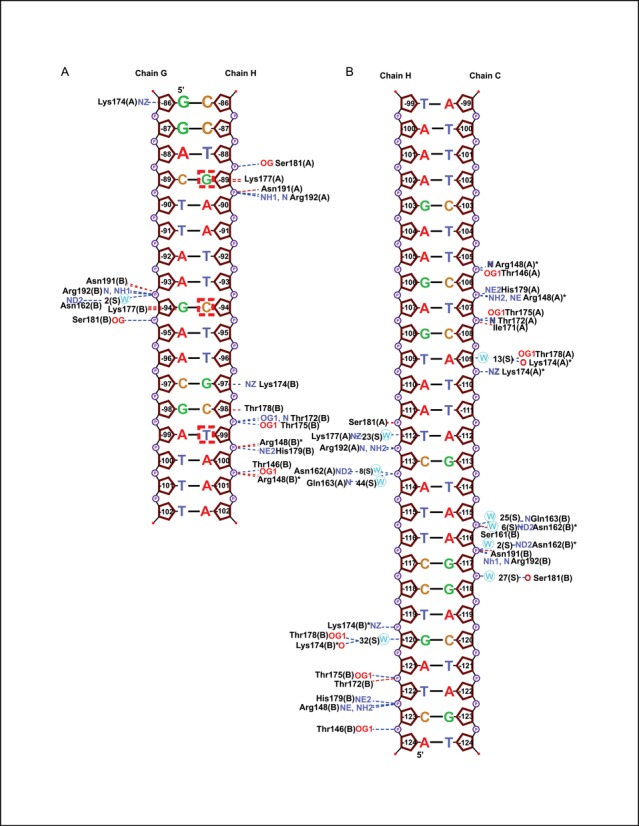
Schematic diagram of LiaR(DBD)D191N bound to DNA sequences derived from the liaXYZ consensus and secondary sites. Base numbers are relative to the LiaX translation start site. LiaR–DNA interactions and atoms are indicated as: hydrogen bonds (blue dotted lines); non-bonded contacts (red dotted lines); nitrogens (blue lettering); oxygens (red lettering); and waters (blue circles). The conserved consensus sequence nucleotides which we identified using in silico analysis G, C and T are boxed (T(X)4C(X)4G(X)4A).
Based on a structure-based sequence alignment of LiaR with the related response regulators DosR and NarL, LiaR Lys-177 is equivalent to Lys-188 of NarL and Lys-182 of DosR (40). In all three response regulators, different DNA sequences are accommodated by a flexible interaction to the major groove either directly or via bridging waters (40). Interestingly, Lys-177 of both chains (A, B) is involved in recognition of the liaXYZ consensus sequence, while there are essentially no contacts between Lys-177 and the DNA of the secondary site. Thus Lys-177 and the homologous residues in DosR and NarL appear to be be important for consensus sequence recognition. Within the context of the T(X)4C(X)4G(X)4A liaXYZ consensus sequence, LiaR makes base specific contacts from the Nϵ of Lys-177(B) to the O6 of G(−94) opposite the consensus sequence C(−94) (boxed) and from the Nϵ of Lys-177(A) to the O6 of G(-89) (boxed). Thus in both copies of the LiaR dimer, Lys177 makes the same interaction to guanines of the consensus sequence. Similarly, Lys-174(A) and (B) are poised to make H-bonds to the O6 positions of G(−86) and G(−97) respectively, though these are not conserved consensus sequence nucleotides. Thr-178(B) of helix-3 makes non-bonded close contacts (<3.5 Å) with C(−98) of the consensus sequence. In the LiaR(DBD)D191N:liaXYZ secondary sequence complex, Thr-178(B) also makes a non-bonded close contact with G(−120). LiaR Thr-178 is equivalent to Val-189 of NarL and Asn-183 of DosR, and in both structures the equivalent residue makes comparable close contacts to the DNA. Significant bending of the DNA is also observed for both LiaR:DNA complexes (40) and is consistent with our DFACE studies suggesting significant structural changes to the DNA upon LiaR binding (Figures 2 and 5B, C).
DNA sequences near the liaXYZ operon may be predisposed to bending
DNase I mapping of the liaXYZ -320 to +30 region in the absence of protein showed a strong hypersensitive position at T(−35) consistent with increased nuclease accessibility. Upon addition of LiaR to 0.5 μM the hypersensitive location shifts to C(-36), and at 5.0 μM protein the hypersensitive site shifts further to C(−38) suggesting that LiaR binding at −120 to −77 has a strong effect on the 3’ distal DNA structure (Figure 2E). While it is possible that the original hypersensitive site is the product of a unique DNA sequence context, it was striking that the position of the hypersensitive site shifts upon formation of the LiaR–DNA complex and that the position is within the putative footprint region of RNAP polymerase binding to canonical −10/−35 regions of DNA during the initiation of transcription. Taken together with proximal hypersensitive sites found for both the liaFSR and liaXYZ at the nucleotide following the adenine in the T(X)4C(X)4G(X)4A consensus sequences and the crystal structures of LiaR DNA binding domains bound to target DNA, it is apparent that LiaR binding imposes changes to DNA structure consistent with DNA bending.
A model for binding to extended DNA regulatory sequences by an activated LiaR tetramer
As shown in Figure 9, a LiaR tetramer would have four helix-turn-helix motifs potentially able to span ∼42 nucleotides. The observed protection site for the liaXYZ operon was 44 nucleotides. The protection site for the liaFSR operon appears smaller and is more consistent with three of the four potential DNA binding domains making strong interactions with the DNA. Our data suggests that the LiaR tetramer can be thought of as having four DNA reading heads that can each recognize and bind DNA depending on the organization of the upstream regulatory DNA sequences. In the liaXYZ operon, all four DNA reading heads are tightly associated with the DNA while in the case of the liaFSR operon three of the four reading heads are making strong contacts to the DNA leading to protection. Presumably since the fourth reading head is present, its interaction with DNA is too weak to produce detectable levels of DNA protection. Based upon the combination of these results we have constructed a model for how LiaR might bind to an extended DNA sequences through the two sets of helix-turn helix dimers that would be present in the LiaR tetramer (Figure 9). The orientation of the DNA binding domains from the crystallographic LiaR–DNA co-structures and increase in DNA hypersensitivity observed by our DFACE studies (Figure 2) suggest that the binding of LiaR will induce DNA bending. We speculate that increased DNA bending could recruit RNA polymerase through direct protein contacts and may favor formation of the pre-initiation bubble. Transcription factors (42) are divided into two classes based on their interactions with the RNA polymerase holoenzyme. Class I factors, such as BvgA (15) and OmpR (43) interact with the C-terminal domain of the subunit α-of RNAP while Class II factors, such as PhoB (44) and VanR (45) interact with the RNA polymerase sigma-subunit. The closest homolog of LiaR, VraR from S. aureus has been shown to interact with the sigma-subunit of RNA polymerase (29).
Figure 9.
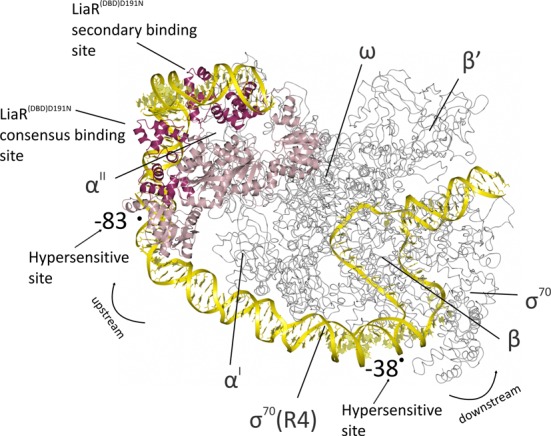
Model for the binding of the activated LiaR tetramer onto the regulatory sequences responsible for the LiaR-mediated cell envelope stress response. The LiaRD191N DNA binding domains bound to DNA sequences derived from the liaXYZ consensus and secondary sites (dark red; PDB: 4WUL and 4WU4) are modeled as an active tetramer using the protein alone tetramer structure of the S. aureus VraR receiver domains (pink; PDB: 4IF4). The structure of the E. coli RNA polymerase complex initiation complex (gray; PDB: 3IYD) was used to model the position of RNA polymerase and DNA. The combined model for the LiaR:DNA and RNA polymerase:DNA complexes (yellow) suggests a strong bend that is consistent with the crystal structures and DNaseI hypersensitive sites from our protection studies (arrows). Note that the RNA polymerase α’ and ω subunits are well poised for potential interactions with the LiaR tetramer.
To construct a potential model for LiaR-mediated regulation, we joined the DNA sequences of the E. coli RNA polymerase initiation complex (3IYD) with those of our LiaRD191N DNA binding domains complexed with the sequences derived from the liaXYZ consensus and secondary sites (dark red, PDB: 4WUL and 4WU4) modeled as an active tetramer using the protein alone available tetramer structure of the S. aureus VraR receiver domains (pink; PDB: 4IF4) (46). The resulting combined model is striking in its clear positioning of LiaR through DNA bending to make potential contacts with RNA polymerase. DNA bending has been observed frequently for other transcription factors and is consistent with enhanced recruitment of RNA polymerase to increase transcription. As shown in Figure 9, the LiaR tetramer is potentially well poised to recruit and interact with RNA polymerase at the promoter.
CONCLUSION
The cell envelope stress response of Gram-positive organisms is intimately associated with their ability to adapt to membrane damaging antibiotics such as daptomycin and other antimicrobial peptides. Mutations within the LiaFSR signaling pathway are often observed as part of the development of antibiotic resistance in the pathogens that are designated as high priorities for control by the CDC such as enterococci and S. aureus. Adaptive mutations such as LiaRD191N that lead to daptomycin resistance can activate the liaFSR regulon in a phosphorylation independent manner, by inducing the formation of the oligomeric states responsible for sequence specific DNA recognition (Figure 1). It is clear from our work, as well as that of others, that while there are some key similarities in the general principles describing LiaFSR and LiaFSR-like homologs for the regulation of target operons, there are important distinctions that can be used to enhance our understanding of these important systems. Strategies that would develop small molecules to inhibit the development of resistance should be cognizant of the differences between these systems in order to have the broadest efficacy.
In B. subtilis, E. faecalis, L. monocytogenes LiaR and S. aureus VraR there is clear evidence for consensus binding sites though there are significant variations within and among the organisms suggesting that a more complex and subtle DNA recognition strategy is common (6,30,47). Despite the fact that members of the NarL/FixJ response regulator family like E. faecalis LiaR and S. aureus VraR share a conserved domain structure and sequence specific DNA recognition via well conserved helix-turn-helix motifs and high overall sequence similarity, there are no obvious similarities to their DNA-binding consensus sequence (30). Presumably subtle changes in the interaction of the helix-turn-helix motifs with DNA targets are responsible for the wide range of recognition sequences across related response regulators. Even within the two operons we studied from E. faecalis S613 there are important differences in position and composition of the DNA regulatory elements. Our DNA binding studies indicate that for the liaXYZ operon, tight binding is achieved readily by the consensus sequence and that the extended secondary sequence, though protected in footprinting, does not contribute significantly to overall DNA affinity by LiaR (Table 1). In contrast, binding to the liaFSR operon consensus sequence is weak and that only upon including regions outside the canonical consensus sequence is high affinity achieved. Our DNA footprinting and analytical ultracentrifugation studies indicate an extended DNA recognition site consistent with the formation of an activated tetramer.
Interestingly, although solution studies indicate a phosphorylation-dependent monomer–dimer equilibrium for VraR, the activated protein crystallized as a tetramer, suggesting that there may be a role for higher-order oligomers. DNA footprinting studies show that, like E. faecalis LiaR, VraR has a larger than expected recognition site upstream of vraSR, upon phosphorylation, that would exceed the size protected by a single VraR dimer (30) and includes DNase I hypersensitive sites indicative of DNA bending. Activation of transcription may require significant DNA bending, involving 4 protein molecules (either as a pair of dimers or a single tetramer), necessary to recruit RNA polymerase (Figure 9) (30) to form the transcription initiation complex. The quaternary structure of LiaR and related cell envelope stress response pathways such as S. aureus VraR may share a common evolutionary monomer–dimer–tetramer equilibrium and that, over time, organisms have adapted both in the sequence of the DNA regulatory elements as well as the LiaR quaternary structure equilibrium to provide a well-adapted stress response. In principle, the equilibrium constants for a response regulator monomer–dimer and dimer–tetramer can be tuned readily to provide the appropriate level of response. E. faecium LiaR shares 84% identity with that of E. faecalis S613 and 60% with S. aureus VraR and therefore its solution binding properties and organization of the target operon regulatory elements could resemble either homolog and will have to await further study. So while there are some similarities between LiaR homologs of B. subtilis and S. aureus, the sequences recognized and the manner in which they are recognized and presumably regulated has changed to fit the specific ecological and evolutionary challenges that confront these organisms. Presumably the membrane stress response pathways of these organisms reflect that distinct evolutionary history.
ACCESSION NUMBERS
wwPDB: 4WSZ, 4WT0, 4WUH, 4WU4, 4WUL.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank the staff at ANL APS for their training and support during the data collection.
Authors contributions: M.D., C.A.A. and Y. S. designed research, analysed data, and wrote the paper; M.D., Y.S., T.A.J., P.G.L., and M.Z. performed the experiments. P.G.L., J.E.L. edited the manuscript.
FUNDING
National Institutes of Health Grant [R01AI080714 to Y.S.]; The Rice University Crystallographic Core Facility is supported by a Kresge Science Initiative endowment grant. Funding for open access charge: National Institutes of Health Grant [R01AI080714 to Y.S.].
Conflict of interest statement. Cesar A. Arias has received grant support, consulted for or provided lectures for Pfizer, Cubist, Bayer, Forest Pharmaceuticals, Novartis, and Theravance.
REFERENCES
- 1.Boucher H.W., Talbot G.H., Bradley J.S., Edwards J.E., Gilbert D., Rice L.B., Scheld M., Spellberg B., Bartlett J. Bad bugs, no drugs: no ESKAPE! an update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 2.Rice L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J. Infect. Dis. 2008;197:1079–1081. doi: 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- 3.Arias C.A., Murray B.E. The rise of the Enterococcus: beyond vancomycin resistance. Nat. Rev. Microbiol. 2012;10:266–278. doi: 10.1038/nrmicro2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arias C.A., Panesso D., McGrath D.M., Qin X., Mojica M.F., Miller C., Diaz L., Tran T.T., Rincon S., Barbu E.M., et al. Genetic basis for in vivo daptomycin resistance in enterococci. N. Engl. J. Med. 2011;365:892–900. doi: 10.1056/NEJMoa1011138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller C., Kong J., Tran T.T., Arias C.A., Saxer G., Shamoo Y. Adaptation of Enterococcus faecalis to daptomycin reveals an ordered progression to resistance. Antimicrob. Agents Chemother. 2013;57:5373–5383. doi: 10.1128/AAC.01473-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolf D., Kalamorz F., Wecke T., Juszczak A., Mader U., Homuth G., Jordan S., Kirstein J., Hoppert M., Voigt B., et al. In-depth profiling of the LiaR response of Bacillus subtilis. J. Bacteriol. 2010;192:4680–4693. doi: 10.1128/JB.00543-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jordan S., Junker A., Helmann J.D., Mascher T. Regulation of LiaRS-dependent gene expression in bacillus subtilis: identification of inhibitor proteins, regulator binding sites, and target genes of a conserved cell envelope stress-sensing two-component system. J. Bacteriol. 2006;188:5153–5166. doi: 10.1128/JB.00310-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCallum N., Meier P.S., Heusser R., Berger-Bachi B. Mutational analyses of open reading frames within the vraSR operon and their roles in the cell wall stress response of Staphylococcus aureus. Antimicrob. Agents Chemother. 2011;55:1391–1402. doi: 10.1128/AAC.01213-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schrecke K., Jordan S., Mascher T. Stoichiometry and perturbation studies of the LiaFSR system of Bacillus subtilis. Mol. Microbiol. 2013;87:769–788. doi: 10.1111/mmi.12130. [DOI] [PubMed] [Google Scholar]
- 10.Munita J.M., Panesso D., Diaz L., Tran T.T., Reyes J., Wanger A., Murray B.E., Arias C.A. Correlation between mutations in liaFSR of Enterococcus faecium and MIC of daptomycin: revisiting daptomycin breakpoints. Antimicrob. Agents Chemother. 2012;56:4354–4359. doi: 10.1128/AAC.00509-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Munita J.M., Tran T.T., Diaz L., Panesso D., Reyes J., Murray B.E., Arias C.A. A liaF codon deletion abolishes daptomycin bactericidal activity against vancomycin-resistant Enterococcus faecalis. Antimicrob. Agents Chemother. 2013;57:2831–2833. doi: 10.1128/AAC.00021-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palmer K.L., Daniel A., Hardy C., Silverman J., Gilmore M.S. Genetic basis for daptomycin resistance in enterococci. Antimicrob. Agents Chemother. 2011;55:3345–3356. doi: 10.1128/AAC.00207-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reyes J., Panesso D., Tran T.T., Mishra N.N., Cruz M.R., Munita J.M., Singh K.V., Yeaman M.R., Murray B.E., Shamoo Y., et al. A liaR deletion restores susceptibility to daptomycin and antimicrobial peptides in multidrug-resistant Enterococcus faecalis. J. Infect. Dis. 2014;211:1317–1325. doi: 10.1093/infdis/jiu602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park D.M., Akhtar M.S., Ansari A.Z., Landick R., Kiley P.J. The bacterial response regulator ArcA uses a diverse binding site architecture to regulate carbon oxidation globally. PLoS Genet. 2013;9:e1003839. doi: 10.1371/journal.pgen.1003839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boucher P.E., Maris A.E., Yang M.S., Stibitz S. The response regulator BvgA and RNA polymerase alpha subunit C-terminal domain bind simultaneously to different faces of the same segment of promoter DNA. Mol. Cell. 2003;11:163–173. doi: 10.1016/s1097-2765(03)00007-8. [DOI] [PubMed] [Google Scholar]
- 16.Griffith K.L., Grossman A.D. A degenerate tripartite DNA-binding site required for activation of ComA-dependent quorum response gene expression in Bacillus subtilis. J. Mol. Biol. 2008;381:261–275. doi: 10.1016/j.jmb.2008.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olekhnovich I.N., Kadner R.J. RNA polymerase alpha and sigma(70) subunits participate in transcription of the Escherichia coli uhpT promoter. J. Bacteriol. 1999;181:7266–7273. doi: 10.1128/jb.181.23.7266-7273.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saxer G., Krepps M.D., Merkley E.D., Ansong C., Deatherage Kaiser B.L., Valovska M.T., Ristic N., Yeh P.T., Prakash V.P., Leiser O.P., et al. Mutations in global regulators lead to metabolic selection during adaptation to complex environments. PLoS Genet. 2014;10:e1004872. doi: 10.1371/journal.pgen.1004872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zianni M., Tessanne K., Merighi M., Laguna R., Tabita F.R. Identification of the DNA bases of a DNase I footprint by the use of dye primer sequencing on an automated capillary DNA analysis instrument. J. Biomol. Tech. 2006;17:103–113. [PMC free article] [PubMed] [Google Scholar]
- 20.Joshi G.S., Zianni M., Bobst C.E., Tabita F.R. Further unraveling the regulatory twist by elucidating metabolic coinducer-mediated CbbR-cbbI promoter interactions in Rhodopseudomonas palustris CGA010. J. Bacteriol. 2012;194:1350–1360. doi: 10.1128/JB.06418-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jerabek-Willemsen M., Wienken C.J., Braun D., Baaske P., Duhr S. Molecular interaction studies using microscale thermophoresis. Assay Drug Dev. Technol. 2011;9:342–353. doi: 10.1089/adt.2011.0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vistica J., Dam J., Balbo A., Yikilmaz E., Mariuzza R.A., Rouault T.A., Schuck P. Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal. Biochem. 2004;326:234–256. doi: 10.1016/j.ab.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 23.Schuck P. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal. Biochem. 2003;320:104–124. doi: 10.1016/s0003-2697(03)00289-6. [DOI] [PubMed] [Google Scholar]
- 24.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kantardjieff K.A., Rupp B. Matthews coefficient probabilities: Improved estimates for unit cell contents of proteins, DNA, and protein-nucleic acid complex crystals. Protein Sci. 2003;12:1865–1871. doi: 10.1110/ps.0350503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.Chen V.B., Arendall W.B. 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belcheva A., Verma V., Korenevsky A., Fridman M., Kumar K., Golemi-Kotra D. Roles of DNA sequence and sigma A factor in transcription of the vraSR operon. J. Bacteriol. 2012;194:61–71. doi: 10.1128/JB.06143-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belcheva A., Verma V., Golemi-Kotra D. DNA-binding activity of the vancomycin resistance associated regulator protein VraR and the role of phosphorylation in transcriptional regulation of the vraSR operon. Biochemistry. 2009;48:5592–5601. doi: 10.1021/bi900478b. [DOI] [PubMed] [Google Scholar]
- 31.Belcheva A., Golemi-Kotra D. A close-up view of the VraSR two-component system. A mediator of Staphylococcus aureus response to cell wall damage. J. Biol. Chem. 2008;283:12354–12364. doi: 10.1074/jbc.M710010200. [DOI] [PubMed] [Google Scholar]
- 32.Leonard P.G., Golemi-Kotra D., Stock A.M. Phosphorylation-dependent conformational changes and domain rearrangements in Staphylococcus aureus VraR activation. Proc. Natl. Acad. Sci. U.S.A. 2013;110:8525–8530. doi: 10.1073/pnas.1302819110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steidl R., Pearson S., Stephenson R.E., Ledala N., Sitthisak S., Wilkinson B.J., Jayaswal R.K. Staphylococcus aureus cell wall stress stimulon gene-lacZ fusion strains: potential for use in screening for cell wall-active antimicrobials. Antimicrob. Agents Chemother. 2008;52:2923–2925. doi: 10.1128/AAC.00273-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stock A.M., Robinson V.L., Goudreau P.N. Two-component signal transduction. Annu. Rev. Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 35.Cai S.J., Inouye M. EnvZ-OmpR interaction and osmoregulation in Escherichia coli. J. Biol. Chem. 2002;277:24155–24161. doi: 10.1074/jbc.M110715200. [DOI] [PubMed] [Google Scholar]
- 36.Gegner J.A., Graham D.R., Roth A.F., Dahlquist F.W. Assembly of an MCP receptor, CheW, and kinase CheA complex in the bacterial chemotaxis signal transduction pathway. Cell. 1992;70:975–982. doi: 10.1016/0092-8674(92)90247-a. [DOI] [PubMed] [Google Scholar]
- 37.Krissinel E., Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 38.Maris A.E., Kaczor-Grzeskowiak M., Ma Z., Kopka M.L., Gunsalus R.P., Dickerson R.E. Primary and secondary modes of DNA recognition by the NarL two-component response regulator. Biochemistry. 2005;44:14538–14552. doi: 10.1021/bi050734u. [DOI] [PubMed] [Google Scholar]
- 39.Ducros V.M., Lewis R.J., Verma C.S., Dodson E.J., Leonard G., Turkenburg J.P., Murshudov G.N., Wilkinson A.J., Brannigan J.A. Crystal structure of GerE, the ultimate transcriptional regulator of spore formation in Bacillus subtilis. J. Mol. Biol. 2001;306:759–771. doi: 10.1006/jmbi.2001.4443. [DOI] [PubMed] [Google Scholar]
- 40.Wisedchaisri G., Wu M., Rice A.E., Roberts D.M., Sherman D.R., Hol W.G. Structures of Mycobacterium tuberculosis DosR and DosR-DNA complex involved in gene activation during adaptation to hypoxic latency. J. Mol. Biol. 2005;354:630–641. doi: 10.1016/j.jmb.2005.09.048. [DOI] [PubMed] [Google Scholar]
- 41.Maris A.E., Sawaya M.R., Kaczor-Grzeskowiak M., Jarvis M.R., Bearson S.M., Kopka M.L., Schroder I., Gunsalus R.P., Dickerson R.E. Dimerization allows DNA target site recognition by the NarL response regulator. Nat. Struct. Biol. 2002;9:771–778. doi: 10.1038/nsb845. [DOI] [PubMed] [Google Scholar]
- 42.Ishihama A. Protein-protein communication within the transcription apparatus. J. Bacteriol. 1993;175:2483–2489. doi: 10.1128/jb.175.9.2483-2489.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okamura H., Hanaoka S., Nagadoi A., Makino K., Nishimura Y. Structural comparison of the PhoB and OmpR DNA-binding/transactivation domains and the arrangement of PhoB molecules on the phosphate box. J. Mol. Biol. 2000;295:1225–1236. doi: 10.1006/jmbi.1999.3379. [DOI] [PubMed] [Google Scholar]
- 44.Makino K., Shinagawa H., Amemura M., Kimura S., Nakata A., Ishihama A. Regulation of the phosphate regulon of Escherichia coli. Activation of pstS transcription by PhoB protein in vitro. J. Mol. Biol. 1988;203:85–95. doi: 10.1016/0022-2836(88)90093-9. [DOI] [PubMed] [Google Scholar]
- 45.Depardieu F., Courvalin P., Kolb A. Binding sites of VanRB and sigma70 RNA polymerase in the vanB vancomycin resistance operon of Enterococcus faecium BM4524. Mol. Microbiol. 2005;57:550–564. doi: 10.1111/j.1365-2958.2005.04706.x. [DOI] [PubMed] [Google Scholar]
- 46.Hudson B.P., Quispe J., Lara-Gonzalez S., Kim Y., Berman H.M., Arnold E., Ebright R.H., Lawson C.L. Three-dimensional EM structure of an intact activator-dependent transcription initiation complex. Proc. Natl. Acad. Sci. U.S.A. 2009;106:19830–19835. doi: 10.1073/pnas.0908782106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fritsch F., Mauder N., Williams T., Weiser J., Oberle M., Beier D. The cell envelope stress response mediated by the LiaFSRLm three-component system of Listeria monocytogenes is controlled via the phosphatase activity of the bifunctional histidine kinase LiaSLm. Microbiology. 2011;157:373–386. doi: 10.1099/mic.0.044776-0. [DOI] [PubMed] [Google Scholar]
- 48.Blanchet C., Pasi M., Zakrzewska K., Lavery R. CURVES+ web server for analyzing and visualizing the helical, backbone and groove parameters of nucleic acid structures. Nucleic Acids Res. 2011;39:W68–W73. doi: 10.1093/nar/gkr316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.