


Abstract
The conventional model for splicing involves excision of each intron in one piece; we demonstrate this inaccurately describes splicing in many human genes. First, after switching on transcription of SAMD4A, a gene with a 134 kb-long first intron, splicing joins the 3′ end of exon 1 to successive points within intron 1 well before the acceptor site at exon 2 is made. Second, genome-wide analysis shows that >60% of active genes yield products generated by such intermediate intron splicing. These products are present at ∼15% the levels of primary transcripts, are encoded by conserved sequences similar to those found at canonical acceptors, and marked by distinctive structural and epigenetic features. Finally, using targeted genome editing, we demonstrate that inhibiting the formation of these splicing intermediates affects efficient exon–exon splicing. These findings greatly expand the functional and regulatory complexity of the human transcriptome.
INTRODUCTION
Introns occupy most of the length of human genes, and most intronic RNA is removed co-transcriptionally (1,2). Initially regarded as ‘junk’, introns are now understood to regulate gene expression through alternative splicing (3) and ‘transcriptional delay’ (4). Much of what we know about intron splicing has been obtained from analyses of short introns (5); however, ∼3400 human introns are longer than 50 kb, and ∼1200 more than 100 kb (6). The current model for splicing involves co-transcriptional excision of the whole intron as one contiguous piece (Figure 1) and occurs after the splice-acceptor site has been transcribed (1,5). In the case of long introns, e.g. a 100-kbp one transcribed in ∼30 min by an RNA polymerase processing at ∼3 kb/min (7,8), it has been assumed they survive intact, without hydrolytic cleavage or degradation, until the splice acceptor site is copied. However, results obtained in Drosophila (9,10) suggest an alternative fate for long introns. Here, the 3′ end of an exon is joined successively to segments within the following intron before splicing to the 5′ end of the next exon. Such ‘recursive splicing’ (Figure 1) results in the stepwise degradation of intronic sequences, involves a defined sequence motif, and leaves no trace in the mature transcript (10). As in silico analyses do not find the fly motif enriched in human introns (11,12), we investigated whether functionally similar but compositionally discrete sites occur therein.
Figure 1.
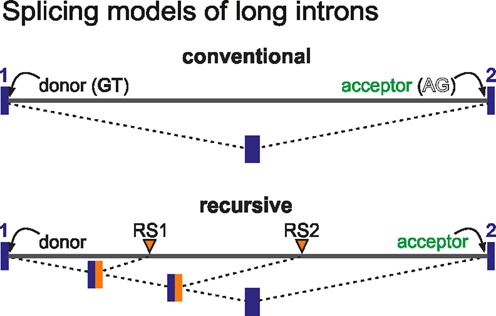
Mechanisms potentially involved in long intron splicing. Top: Conventional splicing: a donor site at the 3′ end of exon 1 (blue) is joined in one step to the acceptor site at the 5′ end of exon 2; this requires the polymerase to have made the acceptor site at exon 2. Bottom: Recursive splicing: the donor site is joined successively to sites within the intron (RS1 and RS2) before the final splice to the 5′ end of exon 2. Both splices may occur before the polymerase reaches exon 2. The dinucleotide immediately 3′ of RS1 becomes the donor used for splicing to RS2. Thus, an RS site must act as both an acceptor and a donor in different contexts.
Analysis of splicing intermediates is complicated by their short half-lives, making them difficult to detect. For example, a recent genome-wide screen of 1.2 billion human RNA sequences uncovered just ∼850 unique splicing lariats; interestingly, ∼70 of these appeared to be produced from splicing involving sites within introns (13). To increase our chances of detecting splicing events within long introns, we employed a powerful gene switch. Tumor necrosis factor α (TNFα) is a pro-inflammatory cytokine that robustly and rapidly activates transcription of many genes (7,14). SAMD4A is one of the first genes to respond to this signaling cascade; it spans 221 kb and contains a 134-kb first intron. After stimulation, hybrid RNAs containing the 5′ end of SAMD4A exon 1 joined to sites positioned progressively more 3′ within the same intron appear (and disappear) in a spatio-temporal order that coincides with progressive transcription of the long intron. Moreover, each of these hybrids is found at ∼1/5 the levels of nascent RNA. Genome-wide transcriptome profiling reveals that similar hybrids can be found in >60% of active genes, and that the respective junctions involve conserved sites within introns. Finally, targeted genome editing (using the CRISPR–Cas9n system; (15)) verified the involvement of these intronic sites in the pathway leading to efficient exon-exon splicing. Our findings greatly expand the complexity of the human transcriptome, and add an extra layer where mRNA production may be regulated.
MATERIALS AND METHODS
Cell culture
Human umbilical vein endothelial cells (HUVECs) from pooled (or single; Supplementary Figure S2B) donors were grown to 80–90% confluence in Endothelial Basal Medium 2-MV with supplements (EBM; Lonza) and 5% fetal bovine serum (FBS), ‘starved’ for 16–18 h in EBM + 0.5% FBS, treated with TNFα (10 ng/ml; Peprotech), and harvested at different times post-stimulation; where necessary, 100 ng/ml spliceostatin A (16) or 50 μM DRB (5,6-dichloro-1-β-d-ribo-furanosyl-benzimidazole) was added before harvesting.
Reverse-transcriptase PCR and real-time quantitative RT-PCR
Total RNA was isolated using TRIzol (Invitrogen) from 106 cells, treated with RQ1 DNase (1 unit DNase/μg total RNA; 37°C, 45 min; Promega), and nascent RNA amplified using the One-Step RT-PCR/qRT-PCR kit (Invitrogen) as per manufacturer's instructions, on a PTC-200 (MJ Research) or Rotor-Gene 3000 cycler (Corbett). For lariat detection, RNase R (Epicentre) was used as per manufacturer's instructions at 37°C to deplete samples of linear RNAs. Amplimers were analyzed by gel electrophoresis (and melting curve analysis where qPCR was used), and their identities confirmed by sequencing (Geneservices, Oxford). Reactions in which Platinum Taq polymerase (Invitrogen) replaced RTase/Taq were performed to ensure amplimers did not result from residual genomic DNA. For Supplementary Figure S1B, HUVECs were grown in 2 mM 5-ethynyl-uridine (EU; Invitrogen) for 5 min, biotin ‘clicked’ on to EU-RNA, and now-biotinylated RNAs selected using streptavidin-coated magnetic beads (M280; Life Technologies); after washing, elution and DNase-treatment, enrichments relative to RNU6, 5.8S or GAPDH RNA were determined by qRT-PCR.
Next-generation RNA sequencing (RNA-seq) and custom analysis
HUVECs were stimulated with TNFα for 15 or 45 min, total RNA was isolated and DNase-treated as above, rRNA was depleted (using the RiboZero kit; Epicentre), RNA was chemically fragmented to ∼350 nucleotides, and cDNA generated using random hexamers as primers (True-seq protocol; Illumina). Poly(A)+-enriched samples were prepared from cells treated with TNFα for 60 min (True-seq protocol following selection on an oligo-dT column). Adapters were then ligated to cDNA molecules, and libraries sequenced (Illumina HiSeq2000 platform; 100-bp paired-end reads; one sample per lane). Approximately 180 × 106 read pairs/sample of total RNA, and 120 × 106 of poly(A)+-selected RNA, were mapped to the human genome (hg19) using ‘stampy’ (17) allowing 1 bp mismatch/40 bp (allowing 0 or 2 mismatches changed the number of uniquely-mapped reads by ±3%) via two sequential rounds of mapping as follows (using custom Perl scripts, available on request). First, a sequence file comprising all CCDS human exons (http://www.ncbi.nlm.nih.gov/CCDS/CcdsBrowse.cgi) was compiled. Then, paired-end reads were mapped to this file, and reads that mapped across 5′ or 3′ termini of any exon to produce a 25–75 nt overhang were selected for further analysis. The overhanging sub-sequence from each mapped read was excised and remapped to the human genome (hg19) to determine its origin. We classified results in three categories: (i) read-through (the overhang mapped to the region immediately following or preceding the original exon), (ii) conventional splicing (the overhang mapped to a 5′ or 3′ end of a different annotated exon), and (iii) unconventional (the overhang mapped to a region not currently annotated as an exon). The last class includes: recursive (RS) splicing events, rarer connections between 5′ exon ends and points in upstream introns (derived by splicing these ends to RS sites in previous introns), and products arising from trans-splicing (18) and exon circularization (19).
RNA fluorescence in situ hybridization
RNA FISH was performed as previously described (20), using sets of five 50-mers (Gene Design, Japan) as probes targeting regions c–e of SAMD4A intron 1. In each 50-mer, roughly every tenth thymine residue was substituted by an amino-modifier C6-dT coupled to Alexa Fluor 488 or 555 reactive dyes (Invitrogen). Note that previous work (7,20) has shown that (i) essentially all HUVECs contain only two SAMD4A alleles and are synchronized in the G0 phase of the cell cycle by the serum-starvation used, (ii) probes can detect different single intronic RNAs with comparable efficiency, (iii) colocalization results from targets copied from the same allele (as spot area is so small compared to nuclear area that a green focus will overlap by chance a red one copied from a different allele in <1 nucleus/1000, assuming random distributions) and (iv) ∼30% SAMD4A alleles in the population are being transcribed by pioneering polymerases at any one time post-stimulation.
Oligonucleotides and siRNAs
PCR primers were designed using ‘qPCR’ settings in Primer 3 Plus (http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi, i.e. for an optimal primer length of 20–22 nt, a melting temperature of 62°C, and amplimers of 100–300 bp). Primer sequences are available on request. For the RRP40 knock-down, previously tested siRNAs (21) were introduced into HUVECs by electroporation using the GenePulser XCell device (Bio-Rad) at 250 mV with 1 × 20-ms square pulse (‘KD1’), or 350 mV with 1× exponential pulse at 350 μF (‘KD2’), on ∼1 million cells/ml per hit in 4-mm cuvettes; cells were then transferred to 15-cm plates and grown for ∼36 h before RNA was harvested.
Chromatin immunoprecipitation (ChIP)
Approximately 107 HUVECs were crosslinked in 1% paraformaldehyde (10 min; 20°C) at the appropriate times after TNFα induction. Chromatin was prepared, fragmented, washed, and eluted using the ChIP-It-Express kit (Active motif). Immunoprecipitations were performed on aliquots of ∼25 μg chromatin using a rat monoclonal (3E10) against phospho-Ser2 in the C-terminal domain of the largest subunit of RNA polymerase II (22), a mouse monoclonal against U2AF65 (U4758, Sigma), and rabbit polyclonals recognizing tri-methylated lysine 36 of histone H3 (ab9050, Abcam) or histone H3 (sc-10809X, Santa Cruz Biotechnology). DNA was purified using a MicroElute Cycle-Pure kit (Omega BioTek) prior to qPCR analysis using a Rotor-Gene 3000 cycler (Corbett) and Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen). Following incubation at 50°C for 2 min to activate the qPCR mix, and 95°C for 5 min to denature templates, reactions were for 40 cycles at 95°C for 15 s, and 60°C for 50 s. The presence of single amplimers was confirmed by melting-curve analysis, and data were analyzed to obtain enrichments relative to input.
Statistical analysis
P values (two-tailed) from unpaired Student's t-tests and Fisher's exact tests were calculated using GraphPad online (http://www.graphpad.com), and were considered significant when <0.05. Pearson's correlation coefficients were calculated in MS Excel.
Consensus analysis
Consensus sequence plots were generated using 'WebLogo 3' applying default parameters (23); ‘conventional’ 5′ and 3′ splice-site plots used ∼135 000 donor and acceptor intron–exon junctions, respectively, retrieved from all active genes in our total RNA-seq dataset.
RNA secondary-structure analysis
200-bp segments surrounding each splice site were extracted from the genome sequence and RNA structure analyzed using ‘Vienna’ (24). Extracted sequences were folded at 37°C and the Boltzmann probability distribution for each nucleotide in the sequence averaged to generate the mean base-pair probability for any given base in the set of analyzed sequences.
Nucleosome positioning and correlation analysis
Genome-wide correlations of nucleosome positioning around RS sites were performed using MNase-seq data previously generated in HUVECs stimulated with TNFα for 0 or 30 min (25). The rest of the correlations used publicly-available data generated by (i) the ENCODE project (ChIP-seq performed in HUVECs; CTCF—GSM733716; H3K36me3—GSM733757; H3K9ac—GSM733735; H3K27ac—GSM733691; H3K27me3—SM733688; H4K20me1—GSM733640; (26)), (ii) Zarnack et al. (for U2AF65 CLIP-seq performed in HeLa; (27)), and (iii) Papantonis et al. (RNA polymerase II ChIP-seq performed in HUVECs 30 min post-stimulation; (28)). For correlation analyses, occupancies of protein factors and histone modifications at RS sites were compared (after converting .BED files to ‘tag directories’). Resulting frequencies were averaged over a 10-bp fixed-size window and plotted using R. Chromatin signatures of RS sites were compared to those around canonical 5′ donor, 3′ acceptor, and random intronic sites from the same, active, genes.
CRISPR/Cas9n genome editing
Human embryonic kidney HEK293A cells were grown in Dulbecco's modified Eagle's medium (DMEM) + 10% FBS at 37°C, and editing at/around three RS sites was performed using the CRISPR–Cas9n system, where the bacterially-derived Cas9 nickase mutant is guided to its genomic target via a 20-nt single-guide RNA (sgRNA) to induce single-strand breaks in the target site at substantially higher levels than found off-target (15). For each RS site, a pair of sgRNAs was designed that targeted positions up- and down-stream of the site; henece, two vectors were co-transfected simultaneously using Lipofectamine 3000 as per manufacturer's instructions (Invitrogen). The following sgRNAs were designed using the http://tools.genome-engineering.org online tool: ‘dR2-For’: GCAATTCAGGTGATGGGAAG; ‘dR2-Rev’: AAAGACACAGACTTAATTTG; ‘eR9-For’: CTTTGCCCGGACGGTCTAGG; ‘eR9-Rev’: GTCACTTCACATGCAGCCCC; ‘x3-For’: TATCACTCTGCAGGTCAAGA; ‘x3-Rev’: AGAGAGAAGAACATGGACTC. These were annealed to complementary oligos and cloned into the pSpCas9n(BB)-2A-GFP (#48140; Addgene) or pSpCas9n(BB)-2A-Puro vector (#48141; Addgene) at BbsI restriction sites as described (15). Control transfections involved vectors carrying no sgRNA sequence. As these vectors carry a green fluorescent protein (GFP) and a puromycin-resistance gene, respectively, HEK293A cells were grown in 1 μg/ml puromycin for 3–4 days post-transfection, and monitored for GFP fluorescence. Thisselection allowed identification of small colonies which were then isolated and re-grown. Total RNA and DNA was isolated from these clones 0 and 45 min post-stimulation, the identity of mutations in RS sites in each clone was verified by DNA sequencing (Beckman-Coulter Genomics, Germany), and splicing defects by qRT-PCR on cDNA generated using random hexamers (Roche). .
RESULTS
The 5′end of SAMD4A intron 1 is degraded before exon 2 is transcribed
Under conditions used here, SAMD4A is essentially silent in HUVECs; TNFα addition then rapidly switches on the gene (7). Thus, when total RNA from unstimulated HUVECs is applied to a tiling microarray spanning SAMD4A, little signal is detected. However, 15 min after adding TNFα, transcripts indicative of rapid and synchronous initiation are seen at the transcription start site (TSS) (Figure 2A illustrates data from (7); see also Supplementary Figure S1A). Subsequently, pioneering polymerases transcribe at ∼3 kb/min to reach the terminus after 80 min (Figure 2A). This generates what we will call the ‘traveling’ wave of (mainly-intronic) RNA. ChIP analysis confirms that RNA polymerase II is present in this traveling wave during the time-course of the experiment (Supplementary Figure S1B). Between 30 and 82.5 min, signal is also seen around the TSS; this ‘standing’ wave is generated by following polymerases that abort soon after initiation (Figure 2A; see also (15)). In the conventional model, an intron can only be removed once the polymerase transcribes the next exon to produce the acceptor site (Figure 1). Three independent lines of evidence indicate this inaccurately describes splicing of SAMD4A intron 1.
Figure 2.
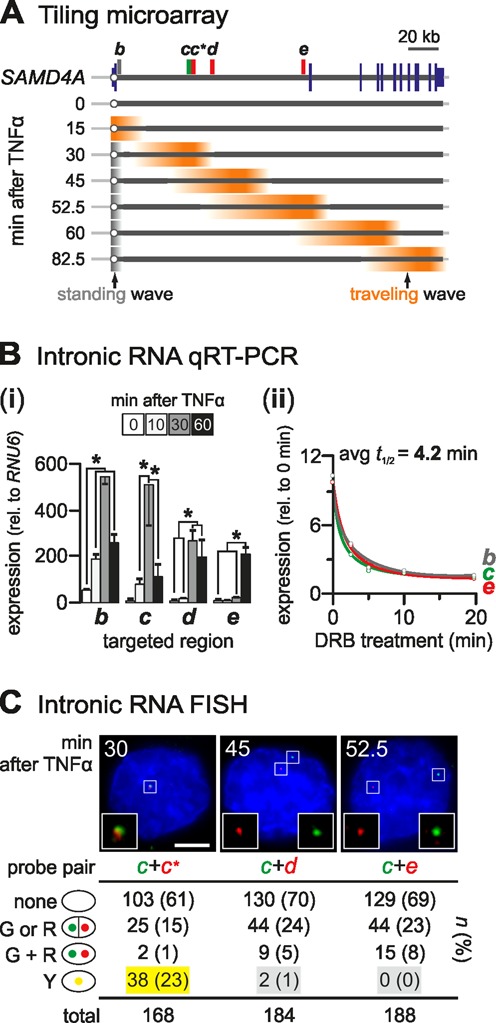
RNA at the 5′ end of SAMD4A intron 1 is degraded before exon 2 is transcribed. (A) Cartoon illustrating previous microarray results (from (7)). Total RNA from HUVECs stimulated with TNFα was isolated at the times indicated and applied to a tiling microarray spanning 221-kbp SAMD4A (map: blue lines – exons; circle – TSS; rectangles – probes used for RNA FISH and RT-PCR). Before stimulation (0 min), background signal is detected. After 15 min, signal (orange) appears at the TSS indicative of synchronous initiation; subsequently, signal generated by pioneering polymerases sweeps down the gene (‘traveling wave’) to reach the terminus after 82.5 min. After 30 min, TSS signal (gray) is also seen, generated by following polymerases that abort soon after initiation (‘standing wave’). Between 30 and 52.5 min, a widening trough develops between the two waves. (B) Quantitative reverse-transcriptase PCR (qRT-PCR). (i) Total RNA was isolated 0–60 min post-stimulation, DNase-treated, intronic RNA amplified, and levels (±SD; n = 3) normalized relative to those of RNU6 RNA. *: significantly different (P < 0.01; two-tailed unpaired Student's t-test). (ii) Half-lives of intronic RNAs copied from segments b, c and e. TNFα treatments were for 30 (b, c) or 60 min (e), and 50 μM DRB added 2.5, 5, 10, or 20 min before harvesting. Levels (± SD; n = 3) are normalized relative to those obtained without TNFα or DRB. The average half-life is 4.2 min, or <1/15 the time taken to transcribe intron 1. (C) RNA FISH. After hybridization with a pair of probes targeting intron 1, nuclei were stained with DAPI, and images collected on a wide-field microscope; typical images are shown (left: a yellow focus resulting from colocalizing foci marks nascent RNAs copied from one allele; middle/right: distinct red and green foci mark non-colocalizing transcripts at different alleles). Nuclei were counted (n; % in parentheses) and categorized as having zero (‘none’), one or two foci of a single colour (‘G or R’), or non-overlapping (‘G + R’) or overlapping (‘Y’; those where ≥ 30% pixels with red signal also contain green signal, or vice versa) green and red foci. Probes targeting RNA copied from c and c* serve as a control and yield 23% yellow foci (yellow highlight). If cotranscriptional removal of intron-1 segments does not occur, a transcript containing d (or e) must also contain c to give colocalizing (yellow) foci; despite detection of many red and green foci, there are almost no yellow foci (gray highlights; P < 10−4 in both cases; Fischer's exact test).
First, inspection of Figure 2A reveals a ‘trough’ in RNA abundance indicative of RNA degradation between the standing and traveling waves. If introns are removed in a single contiguous piece no trough should exist. In other words, all RNA molecules encoding region d (or e) should also encode c (as d/e and c lie in intron 1) and be equally as abundant. Similar troughs have been seen in four other TNFα-responsive genes with long introns (7).
Second, quantitative RT-PCR shows that RNAs from the 5′ end of intron 1 disappear before sufficient time has elapsed for the pioneering polymerases to reach exon 2. For example, intronic RNA from region c peaks after 30 min, and falls significantly by 60 min (when pioneers reach the 3′ end of the intron; Figure 2B, i). An analogous profile is seen when nascent RNA (labeled with 5-ethynyl-uridine) is selectively purified and analyzed (Supplementary Figure S1C). Moreover, half-lives of segments b, c and e are ∼4 min (Figure 2B, ii)—comparable to those of other introns (8,29). As the pioneering polymerases take more than fifteen such half-lives (i.e. >60 min) to transcribe this long intron, the 5′ ends of these intronic transcripts must be degraded well before the exon 2 acceptor is made.
Third, RNA FISH shows that distant segments of intron 1 are rarely found in the same (nascent) RNA molecule. This is shown using one probe targeting intronic RNA from region c (34 kb into intron 1) and another targeting RNA from d or e (separated from c by 11 and 94 kb, respectively; Figure 2C). If the conventional model applies, an RNA encoding d (or e) should always also encode c, and probe pairs targeting d (or e) and c should always colocalize. However, they rarely do (Figure 2C, gray highlights). This contrasts with significant colocalization given by a control (i.e. c+c*) involving targets lying only ∼1 kb apart (Figure 2C, yellow highlight). [All probes have been exhaustively tested (20); they work with comparable efficiencies, and give reproducible numbers of red and green foci (Figure 2C and 'Materials and Methods' section)].
Multiple splicing intermediates are detected in SAMD4A intron 1
As the premature disappearance of the 5′-proximal segments of intron 1 is difficult to reconcile with conventional splicing, we sought to determine if a mechanism involving some variant of recursive splicing (10) underlies this. We used RT-PCR to screen for hybrid RNAs encoding the 3′ end of exon 1 joined to sequences within intron 1. We paired in turn a (forward) primer targeting exon 1 with each of >100 (reverse) primers targeting sites positioned at ∼1 kb intervals along intron 1. Figure 3A displays a subset of results obtained with reverse primers cR1–8. Many bands appear following stimulation, but only one (Figure 3A, yellow arrowhead) results from joining the 3′ end of exon 1 with a segment within intron 1. Pre-treatment of cells with a splicing inhibitor, spliceostatin A (SSA; (16)), prevents the formation of this product which indicates it is produced by splicing (Figure 3A, gray box). The other sequenced bands contained off-target amplimers of contiguous RNA. Using this tiling approach, five additional analogous splicing products were identified in this intron (Supplementary Figure S2A); the formation of one was also verified in RNA preparations from HUVECs derived from two individual donors (Supplementary Figure S2B). Additionally, another example was found in the first long intron of the 312 kb-long EXT1 TNFα-responsive gene (Supplementary Figure S2C).
Figure 3.
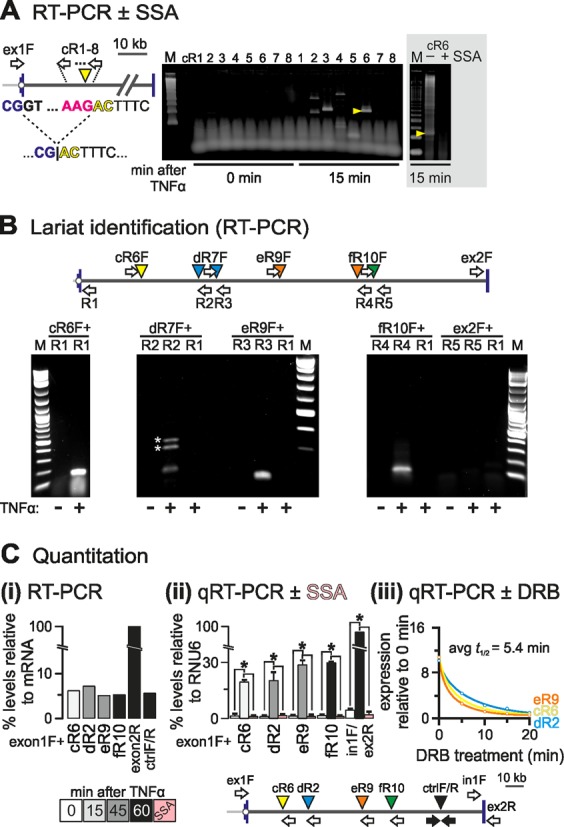
Detection of exon–intron product in SAMD4A intron 1. HUVECs were treated with TNFα for different times, total RNA isolated and DNase-treated, and hybrid RNAs or lariats detected by RT-/qRT-PCR. (A) Identification strategy. The map (left) shows SAMD4A intron 1 (exons 1/2: blue vertical lines) and primers used (white arrows; forward primer ‘ex1F’ targets exon 1 and is used successively with reverse primers cR1–8 targeting intron 1 at ∼1 kb intervals). The dotted line illustrates recursive splicing between the exon 1 donor (exonic sequence: blue; intronic: black) and an RS site (cR6, yellow arrowhead; acceptor sequence in red). The product loses the (canonical) donor GT to gain a (non-canonical) AC. Right: RT-PCR products were resolved by gel electrophoresis, gels stained and imaged (typical images shown; M: size marker), and all bands detected after stimulation (but not before) sequenced. One band (yellow arrowhead) possessed the hybrid exon–intron sequence (…CG|ACTTTC…) consistent with formation of a splicing intermediate. Gray box: pre-treatment (3 h) with 100 ng/ml spliceostatin A (SSA) abolishes the indicated band. (B) Lariat detection by inverse PCR. The map (top) shows the primers used for lariat detection. Each forward primer (e.g. ‘eR9F’) is used with a reverse one (e.g. ‘R3′) to amplify across the A nucleotide at the junction in the lariat; as controls, RNA from unstimulated HUVECs and pairing of forward primers with a reverse one at the 5′ end of intron 1 (‘R1’) were also assayed. *: spurious bands amplified using ‘dR6F + R2’. M: size marker. (C) Quantitation of RS products. The map (bottom) shows primers used for RT-/qRT-PCR. ‘Ex1F’ (targeting exon 1) was used with the reverse primers indicated, whilst the pairs ‘ctrlF/R’ (amplifying an intronic region with the Drosophila motif), ‘ex1F/ex2R’ (amplifying across the exons 1–2 steady-state junction), and ‘in1F/ex2R’ (amplifying across the intron 1/exon 2 boundary) serve as controls. (i) Levels of selected RS intermediates. Amplimers were resolved by gel electrophoresis, gels stained and imaged, and intensities of bands measured and expressed relative to that given by primers targeting fully-spliced mRNA. All intermediates are as abundant as the ‘ctrlF/R’ segment, and present at 4–7% the level of mRNA. (ii) RNA levels assessed by qRT-PCR and expressed relative to those given by RNU6 RNA (±SD; n = 3). RS hybrids are ∼20% the levels of primary transcripts and their formation is SSA-sensitive (pink bars). *: significantly different from 0 min (P < 0.01; two-tailed unpaired Student's t-test). (iii) Half-lives of RS intermediates. TNFα treatment was for 15 (cR6), 30 (dR2) or 45 min (eR9), and 50 μM DRB added 5, 10, 15 or 20 min before harvesting. Levels (±SD; n = 3) are normalized relative to those obtained without TNFα or DRB. The average half-life is 5.4 min.
A hallmark of splicing is the production of a lariat by juxtaposition of the ends of an intron. These can be detected using inverse PCR across the adenosine at the branch point in the lariat. To determine if the hybrid RNAs we identified by RT-PCR associated with the production of lariats, we used inversely-positioned primers in the intronic regions flanking the relevant donor and acceptor sites. This analysis identified lariats found only after stimulation with TNFα, while no conventional lariats (formed by joining conventional donor and acceptor sites at the extreme ends of intron 1), or intermediate ones involving the conventional donor and an unconventional site other than the first could be detected at the relevant time point (Figure 3B).
To determine if these novel hybrids are present at biologically-meaningful levels, we estimated the relative abundance of four by both semi-quantitative and quantitative RT-PCR. All four were present at 5–7% mature (spliced) transcript levels (Figure 3C, i) and at ∼20% primary transcript levels; all were produced by splicing, and all disappeared following SSA treatment (Figure 3C, ii). Analysis of the half-lives of three of these hybrids revealed that they are relatively long-lived, with half-lives comparable to those of nascent intronic fragments (compare Figures 2B and 3C, iii).
Intronic SAMD4A splice sites partially resemble canonical ones
We compared the sequence motifs found at the points that were used to generate these hybrid exon–intron junctions with the motif used during recursive splicing in Drosophila (i.e. YAG|GTRAGT, where the splice junction is indicated by vertical line; (10)). None of the sites in intron 1 of SAMD4A encode this consensus. However, a YAG|GTRAGV (V = A/G/C) motif is found at nine other points in the intron (Supplementary Figure S2A), but RT-PCR (applied as in Figure 3A using primers targeting these sites) gave no products indicative of recursive splicing (not shown).
Although the identified hybrid RNAs do not stem from sites carrying the Drosophila recursive-splicing motif, they do possess features characteristic of canonical splice acceptors (Supplementary Figure S3A–C). These include (i) an AG at positions −1 and −2, (ii) a T/C-rich tract between −3 and −16, (iii) a consensus branch-point sequence (5′-YTNAY-3′; (30)) between −5 and −50 (also confirmed by lariat formation; Figure 3B), (iv) a high ‘maximum entropy’ score given by a splicing-prediction algorithm (31), (v) two splicing-associated marks, histone H3 tri-methylated at lysine 36 (32) and bound splicing-cofactor U2AF65 (27), and (vi) the presence of a positioned nucleosome at the junction. Thus, they closely resemble canonical splice-acceptor sites, despite their unusual location away from exon ends.
Exon-intron hybrids are typical of many human genes
To determine the genome-wide occurrence of such intermediate exon–intron hybrids, we performed total RNA sequencing (RNA-seq). HUVECs were treated with TNFα for 15 and 45 min before total RNA was isolated, depleted of rRNA, and sequenced. Each library yielded >180 million (100 bp) read pairs that were analyzed via a custom pipeline (Figure 4A). Individual reads were mapped to a sequence database of annotated human exons (hg19). Reads mapping across the 5′ or 3′ termini of exons to produce overhangs were selected, the overhanging sub-sequences were excised and remapped to the entire genome. These mapped overhangs fell into 3 classes: (i) those mapping to the region immediately following/preceding an exon (representing unspliced nascent RNA, or ‘read-through’); (ii) those mapping to the 5′ or 3′ end of another exon (a conventionally-spliced mRNA); (iii) those mapping to a region not currently defined as exonic; this category includes exon–intron products (see 'Materials and Methods' section for a discussion of other products).
Figure 4.
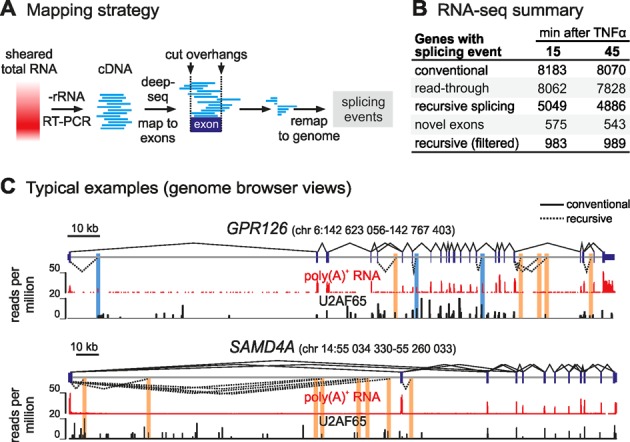
Exon–intron products detected genome-wide by RNA-seq. (A) Overview of the custom pipeline. Reads were mapped to an ensemble of human exons, those extending beyond an exon boundary selected, and overhangs re-mapped to the entire human genome. This allowed different splicing events and unspliced RNA to be quantified. (B) Summary of splicing events identified in each RNA-seq library. (C) Typical browser views. RefSeq gene models (hg19), conventional (solid lines), and recursive splicing (unfiltered; dotted lines) are shown above tracks for poly(A)+-selected RNA (60 min after TNFα; red) and U2AF65 binding (HeLa, from (27); black). Segments involving novel exons (with poly(A)+ signal) and RS junctions (lacking poly(A)+ signal) are highlighted blue and orange, respectively.
We focused on the ∼8100 active genes in each sample (defined as those with ≥100 reads spanning ≥1 exon junction; Figure 4B). Many reads were derived from steady-state mRNA, and most mapped junctions encoded expected exon-exon connections (Figure 4B, ‘conventional’). Within this subset, we defined genes engaged in transcription at the moment of harvest—those with ≥1 read spanning an exon-intron boundary, indicative of the polymerase reading through the exon-intron junction. This left ∼8000 genes per sample (Figure 4B, ‘read-through’). Levels of nascent RNA were ∼2% those of mature transcripts, estimated by comparing exon-intron to exon–exon reads (i.e. 579 000 and 521 000 out of 30 and 28 million reads for the 15- and 45-min samples, respectively). Exon–intron products were identified as reads encoding the 3′ end of an exon joined to a downstream intronic segment; these were not by-products of conventional splicing as no trace of their use, including micro-exons (33), is detected in poly(A)+ RNA. Reads from >60% selected genes in each sample contained such products. We will call these ‘recursive splicing’ (RS) products despite their sequence deviation from the fly motif (Figure 4B; examples in Figure 4C). These hybrids were present in significant numbers (i.e. 83 000 and 76 000 reads in the 15- and 45-min samples, respectively), and at ∼15% the levels of ‘read-through’ transcripts.
To exclude products resulting from splicing at intronic sites close to (but not quite at) canonical acceptor/donor sites, we stringently excluded reads mapping within 2 kb of annotated exon boundaries (as 99.9% annotated exons are <2 kb long; Supplementary Figure S4A). Also, as reads encoding known exons joined to hitherto-unannotated ones (within introns) could be interpreted as intermediate splicing products, we additionally excluded reads involving such ‘novel’ exons. To do this, we purified poly(A)+ RNA, sampled 120 million read pairs (at 60 min post-stimulation, to allow time for nascent transcripts to mature), called any poly(A)+ RNA peak lying within an annotated intron a putative ‘novel’ exon, and discarded hybrid reads encoding such novel exons from our list. We do not wish to speculate here on the functional significance of these novel exons, but they are numerous (∼1000 in >500 different genes per sample) and have a size distribution typical of annotated exons (Supplementary Figure S4A). The combination of these two stringent filters resulted in a final list of 2389 unique sites in total (representing >25% of unfiltered recursive-splicing events; Supplementary Table S1). This can be broken down to 1425 and 1400 sites harbored in 983 and 989 genes of the 15- and 45-min sample, respectively (with 436 sites in 342 genes shared by both samples; Figure 4B, ‘recursive splicing (filtered)’).
Seven sites were recorded (after filtering) in intron 1 of SAMD4A, and their respective products were present at ∼11% the levels of nascent transcripts (defined by read-through at the intron 1/exon 2 boundary; Supplementary Figure S3D). As four of these seven products were missed by our tiling approach, we manually confirmed the presence of two using RT-PCR (Supplementary Figure S2D). Moreover, we verified three more such RS hybrids in three other genes (i.e. HDLBP, UBR5 and KALRN) in the absence of serum starvation (to exclude the possibility they might arise from unforeseen starvation effects; Supplementary Figure S2E).
Distinctive features of RS sites genome-wide
Just like the sites identified in SAMD4A, RS sites seen genome-wide have both sequence (an AG dinucleotide and T-rich tract; Figure 5A) and structural features (a tightly-positioned nucleosome and U2AF bound immediately upstream the junction; Figure 5B) characteristic of canonical splice acceptors (27,34). Interestingly, the two bases immediately following the acceptor site are a GN dinucleotide in 44% of sites (Figure 5A, inset); this is significantly higher than expected if dinucleotide composition at these sites was random (expected 6.25% and 25% respectively; P < 0.001). In silico analysis of the RNA encoded by these sites points to the sequence preceding the splice junction being less structured than that following it, and that the +1 nucleotide is likely to be paired and sequestered in a secondary structure (Figure 5A, blue dotted lines)—both properties of canonical donors. These sites are also marked by a unique combination of acetylation at lysine 27 of histone H3 (H3K27ac) and methylation at lysine 20 of histone H4 (H4K20me1)—unlike canonical donors or acceptors. CTCF and ‘active’ chromatin features also mark many RS sites (Supplementary Figure S4B and C). Also, re-performing correlations after categorizing RS sites according to nucleotides at positions +1 and +2 (as these would act subsequently as donors in any recursive splicing pathway) revealed little variation in epigenetic features (Supplementary Figure S4D).
Figure 5.
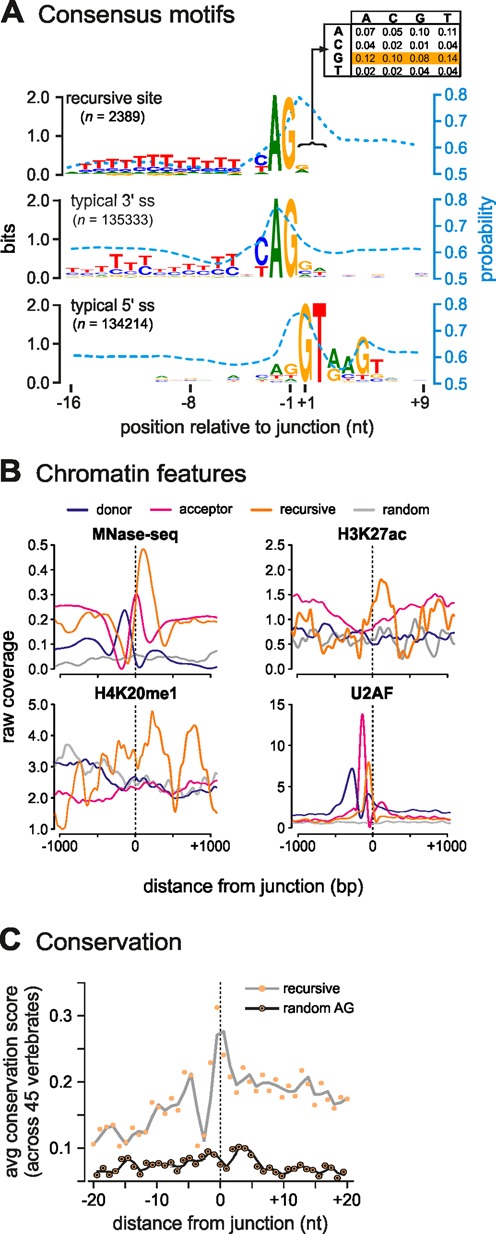
Features of filtered RS sites (combined results from 15- and 45-min datasets). (A) Intron acceptor sites consensus motifs resemble those of canonical acceptors. RS-donor dinucleotides (position +1/+2) are biased towards GN (44% of all dinucleotides; inset, yellow). The mean base-pair probability of each base to be sequestered in a secondary structure is indicated by a blue dotted line. (B) Epigenetic features of sites (orange) compared to canonical donors (blue) and acceptors (red); randomly-selected (but actively-transcribed) regions from the same introns as exon-intro sites provide a control (gray). ‘Raw coverage’ refers to reads/million. Exon–intron acceptor sites tend to have a well-positioned nucleosome immediately downstream (HUVEC Mnase-seq data; 30 min post-stimulation) carrying H3K27ac and H4K20me1 (HUVEC ENCODE data, from (26)), and U2AF bound to the nucleosome-free region immediately upstream (HeLa data, from (27)). (C) Conservation at detected sites. Plot showing ‘running means’ (gray line) of conservation scores for 40 bp around all 2389 filtered sites (junction at 0; hg19 coordinates in Supplementary Table S1) computed using ‘phyloP’ (48) from the PHAST package (http://compgen.bscb.cornell.edu/phast/) for 45 vertebrate genomes. As a control, an equal number of randomly-selected AG sites from the same introns were tested (black line).
In Drosophila, recursive splicing is associated with the facilitation of long-intron splicing (10), but our genome-wide analysis revealed no strong correlation of recursive-splice sites with intron length. However, upon closer inspection of 100 most frequently-observed RS events, we found that they were over-represented amongst genes of >100 kb in length (Supplementary Figure S5A). Moreover, these genes associated with particular GO terms (e.g. ‘inflammatory signaling’, ‘regulation’, ‘transcription factor’; Supplementary Figure S5B). Finally, given the numbers and abundance of RS sites in the human genome, we sought to determine whether these sites were conserved more broadly across vertebrates. Computing the mean nucleotide conservation score surrounding each site, and comparing it to an equal number of randomly-selected AG sites from the same introns, revealed that despite being located deep within introns RS sites are markedly conserved across 45 vertebrates (Figure 5C and Supplementary Figure S5C).
RS sites are required for efficient SAMD4A exon–exon splicing
Successive splicing intermediates appear in SAMD4A intron 1 coincidentally with transcription of the respective RS site by the pioneering polymerase. As these are also distributed approximately every ∼20 kb (Figure 3), it is attractive to assume that the mature message is built by their successive use (i.e. by recursively splicing the 3′ end of exon 1 first to one part of the intron, then to a second, and so on, before splicing to the 5′ end of exon 2; Figure 1). However, the mere existence of a hybrid product does not constitute formal proof for an ordered precursor-product relationship. To address this, we turned to human embryonic kidney HEK293A cells that are more amenable to transfection than HUVECs.
First, we verified that SAMD4A is switched on using TNFα (albeit at lower levels than in HUVECs, most probably due to poorer synchrony), and that three exemplary RS hybrids are detected after stimulation (Figure 6A–C). We chose to focus on a ‘non-GT’ (dR2) and a ‘GT’ RS site (eR9)—both discovered using our tiling RT-PCR approach (Supplementary Figure S2A) – and on a ‘GT’ site (x3) discovered using our RNA-seq approach (Supplementary Figure S2D). We then applied CRISPR-Cas9n technology (15) to mutate each site independently. Mutated clones were selected (using resistance to puromycin), grown to confluence, stimulated with TNFα for 45 min, and total cell RNA or DNA isolated (see ‘Materials and Methods’). DNA sequencing revealed that each of the three clones (Δ1–3) carried a particular set of mutations. Thus, in Δ1, the dR2 branch-point adenosine was mutated and part of the branch point and T-tract sequence deleted. In Δ2, the whole branch point and T-tract sequences, as well as part of the eR9 acceptor site were removed. In Δ3, the insertion of an adenosine exactly at the RS junction creates an (apparently) non-permissive ‘AG’ RS-donor (Figure 6A and Supplementary Figure S6). These mutations did not affect nascent transcript levels close to the exon1-intron 1 boundary (region b), but profoundly reduced exon 1–2 splicing. Thus, mutating each of three different RS sites, affects ‘conventional’ splicing (Figure 6D and E). Intriguingly, the splicing of the following two exons, exons 2 and 3, was also affected. Similarly, nascent RNA copied from further into intron 1 (∼34 and 94 kb, regions c and e, respectively) is differentially affected, pointing to some feedback loop linked to SAMD4A RNA degradation once RS splicing fails (Figure 6E; but note that this may not involve the exosome, as knock-down of the RRP40 subunit has little effect on the levels of RS products; Supplementary Figure S2F). Overall, this analysis points to RS sites being involved in the generation of the fully-spliced (mature) mRNA.
Figure 6.
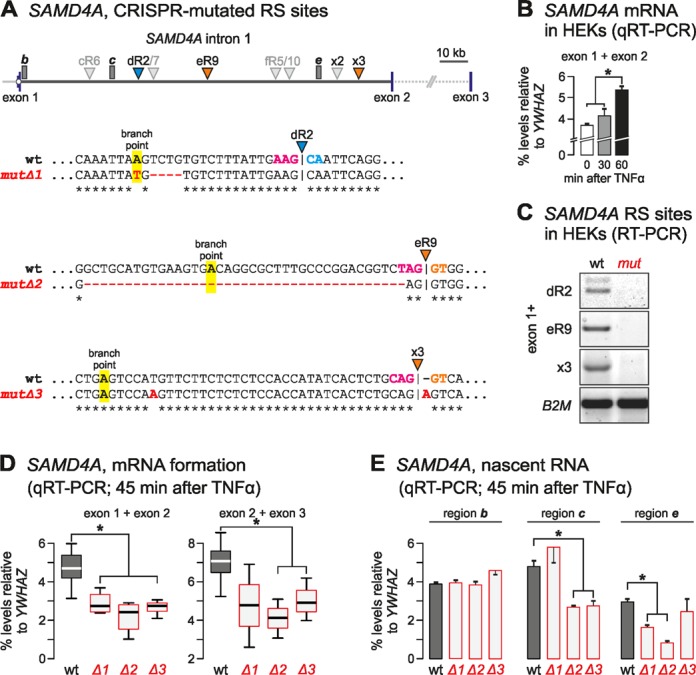
Mutations in RS sites in HEK293A cells reduce levels of mature exon–exon products (*: significantly different from wild-type levels; P < 0.05; two-tailed unpaired Student's t-test). (A) Top: map of SAMD4A intron 1 with RS sites targeted using CRISPR/Cas9 (i.e. dR2, eR9, and x3; coloured arrowheads); segments b, c, and e (where nascent RNA levels are measured) are also marked (gray rectangles). Bottom: wild-type (wt) and mutated RS sequences in mutΔ1–3 (branch-point adenosines, RS acceptors and RS donors are bold and highlighted magenta, blue or orange; point mutations, insertions, and deletions are red). (B) Responsiveness of SAMD4A to TNFα. Levels (±SD, n = 2) of the exons 1 and 2 spliced product were assessed using qRT-PCR, and normalized relative to those of a constitutively-expressed exon-exon junction in YWHAZ mRNA. (C) The mutations eliminate production of the RS product (assessed by RT-PCR using a forward primer in exon 1 with reverse primers after dR2, eR9, and x3 junctions—as in Figure 3A). Constitutively-expressed B2M mRNA provides a loading control. (D) Mutations reduce the levels (±SD; n = 3) of conventional exons 1 and 2 junctions (assessed using qRT-PCR 45 min after stimulation; levels normalized relative to those of a constitutively-expressed exon-exon junction in YWHAZ mRNA). (E) Mutations have little effect on intronic RNA levels from region b, but reduced region c levels (assessed as in (D) but n = 2).
DISCUSSION
Splicing is thought to involve joining of the 3′ end of an exon directly to the 5′ end of the next exon; here, we provide evidence showing that many human introns are removed via the use of intermediate intronic sites. This prompts the question: are RS products simply ‘dead-ends’ arising due to mis-splicing? Their very levels make this unlikely, as RS products are found genome-wide at ∼15% the levels of the primary transcripts, and—in the case of just one of the 12 introns in SAMD4A—there are so many products found at each of the seven RS sites in it that little, if any, primary transcript would ever survive to give the final product where intron 1 is joined to intron 2. Moreover, individually mutating three RS sites in this intron reduces levels of the final product—which points to RS production directly affecting the productive pathway.
Given that RS hybrids appear to be so prevalent in HUVECs, what role might it play in transcript metabolism? It is attractive to suppose that they can be used to modulate mRNA levels (even if all were non-productive), but is there a temporal order of usage of RS sites? Although the SAMD4A data point to a consecutive, ordered (from 5′ to 3′) usage, another report documents a non-ordered pattern (35). We favor a model that resembles the ‘zero-length exon’ and ‘dual-specificity splice-site’ models (36,37), and is based on the idea that splicing occurs stochastically (38) at any site carrying splice-acceptor properties. Which of several acceptors is then used will depend on the local concentration of relevant factors (e.g. U1, U2AF), epigenetic marks (e.g. a positioned nucleosome carrying marks like H3K27ac and H4K20me1), and the temporal availability of RS sites (39). Competition between sites would then lead to a given exon–exon junction being produced from the successive use of different sites. In one case this might involve one set of RS sites, while in another it might use a different, perhaps overlapping, set. Moreover, just like in the case of the ‘intraexon’ in the vertebrate 4.1-family genes (40), RS sites can also act to increase physical proximity of two exons. Of course, to ensure correct exon-exon joining, this model—like the conventional one—requires ‘exon definition’ (41) so that splicing to a downstream exon is distinguished from intermediate ones.
If RS products lie on the productive pathway, then the dinucleotide located immediately 3′ of an RS junction will subsequently be used as a splice donor (Figure 1). In the conventional case, this is typically a GT. We find GN dinucleotides in only ∼44% RS sites; however, we note that these can be used with ‘strong’ acceptors if splicing enhancers are present (42,43). But, how might non-GT dinucleotides be recognized? We can suggest two possibilities. First, conventional splicing relies on an AC in U1 snRNA base-pairing with a GT in the donor site; however, mispairing permits use of a wider range of donors (44). Second, we now know that the human genome encodes many U1 variants able to base pair with non-GT dinucleotides (45,46) – including that in the minor spliceosome (47). Therefore, we checked to see whether variant U1s and U11s (of the minor spliceosome) were expressed in HUVECs, and found that the ones that were could allow base-pairing with at least half of RS donor dinucleotides (Supplementary Figure S7). Therefore we suggest that these minor variants play more important roles than hitherto thought.
In summary, our results indicate that splicing pathways in man are much more complicated than imagined hitherto, with RS sites providing additional regulatory points in transcript production. Obviously, further work will be required to show that these novel splicing intermediates do indeed lie on the pathway to leads to a mature, translatable, message.
ACCESSION NUMBER
RNA-seq data generated here have been deposited at the Sequence Read Archive under accession number SRX487433.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Jon Bartlett and Athanasia Mizi for help with experiments, Dirk Eick for the 3E10 antibody, Minuro Yoshida for spliceostatin A, Evgenia Ntini for the RRP40-siRNA sequences, and the High-Throughput Genomics Group at the Wellcome Trust Centre for Human Genetics (Oxford, UK) and TGAC sequencing facility (Norwich, UK) for RNA sequencing.
Footnotes
Present address: Sarah Diermeier, Cold Spring Harbor Laboratory, 1 Bungtown Road, Cold Spring Harbor, NY 11724, USA.
FUNDING
Biotechnology and Biological Sciences Research Council [to P.R.C.]; Federal Ministry for Education and Research [to G.L.] via the ERASysBio+/FP7 initiative; Leverhulme Trust [to S.K.]; Collaborative Research Centre SFB960 grant [to S.D., G.L.]; James Martin Stem Cell Institute [to D.O.]; CMMC intramural funding [to T.G., A.P.]; Köln Fortune program [to A.Z.]. Funding for open access charge: Center for Molecular Medicine, Cologne (intramural funding).
Conflict of interest statement. None declared.
REFERENCES
- 1.Ameur A., Zaghlool A., Halvardson J., Wetterbom A., Gyllensten U., Cavelier L., Feuk L. Total RNA sequencing reveals nascent transcription and widespread co-transcriptional splicing in the human brain. Nat. Struct. Mol. Biol. 2011;18:1435–1440. doi: 10.1038/nsmb.2143. [DOI] [PubMed] [Google Scholar]
- 2.Tilgner H., Knowles D.G., Johnson R., Davis C.A., Chakrabortty S., Djebali S., Curado J., Snyder M., Gingeras T.R., Guigó R. Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res. 2012;22:1616–1625. doi: 10.1101/gr.134445.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luco R.F., Allo M., Schor I.E., Kornblihtt A.R., Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell. 2011;144:16–26. doi: 10.1016/j.cell.2010.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swinburne I.A., Miguez D.G., Landgraf D., Silver P.A. Intron length increases oscillatory periods of gene expression in animal cells. Genes Dev. 2008;22:2342–2346. doi: 10.1101/gad.1696108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pandya-Jones A., Black D.L. Co-transcriptional splicing of constitutive and alternative exons. RNA. 2009;15:1896–1908. doi: 10.1261/rna.1714509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bradnam K.R., Korf I. Longer first introns are a general property of eukaryotic gene structure. PLoS One. 2008;3:e3093. doi: 10.1371/journal.pone.0003093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wada Y., Ohta Y., Xu M., Tsutsumi S., Minami T., Inoue K., Komura D., Kitakami J., Oshida N., Papantonis A., et al. A wave of nascent transcription on activated human genes. Proc. Natl. Acad. Sci. U.S.A. 2009;106:18357–18361. doi: 10.1073/pnas.0902573106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh J., Padgett R.A. Rates of in situ transcription and splicing in large human genes. Nat. Struct. Mol. Biol. 2009;16:1128–1133. doi: 10.1038/nsmb.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hatton A.R., Subramaniam V., Lopez A.J. Generation of alternative Ultrabithorax isoforms and stepwise removal of a large intron by resplicing at exon-exon junctions. Mol. Cell. 1998;2:787–796. doi: 10.1016/s1097-2765(00)80293-2. [DOI] [PubMed] [Google Scholar]
- 10.Burnette J.M., Miyamoto-Sato E., Schaub M.A., Conklin J., Lopez A.J. Subdivision of large introns in Drosophila by recursive splicing at nonexonic elements. Genetics. 2005;170:661–674. doi: 10.1534/genetics.104.039701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ott S., Tamada Y., Bannai H., Nakai K., Miyano S. Intrasplicing – analysis of long intron sequences. Pac. Symp. Biocomput. 2003;339:50. doi: 10.1142/9789812776303_0032. [DOI] [PubMed] [Google Scholar]
- 12.Shepard S., McCreary M., Fedorov A. The peculiarities of large intron splicing in animals. PLoS One. 2009;4:e7853. doi: 10.1371/journal.pone.0007853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taggart A.J., DeSimone A.M., Shih J.S., Filloux M.E., Fairbrother W.G. Large-scale mapping of branchpoints in human pre-mRNA transcripts in vivo. Nat. Struct. Mol. Biol. 2012;19:719–721. doi: 10.1038/nsmb.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smale S.T. Selective transcription in response to an inflammatory stimulus. Cell. 2010;19:833–844. doi: 10.1016/j.cell.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaida D., Motoyoshi H., Tashiro E., Nojima T., Hagiwara M., Ishigami K., Watanabe H., Kitahara T., Yoshida T., Nakajima H., et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat. Chem. Biol. 2007;3:576–583. doi: 10.1038/nchembio.2007.18. [DOI] [PubMed] [Google Scholar]
- 17.Lunter G., Goodson M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011;21:936–939. doi: 10.1101/gr.111120.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gingeras T.R. Implications of chimaeric non-co-linear transcripts. Nature. 2009;461:206–211. doi: 10.1038/nature08452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salzman J., Gawad C., Wang P.L., Lacayo N., Brown P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 2012;7:e30733. doi: 10.1371/journal.pone.0030733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larkin J.D., Papantonis A., Cook P.R., Marenduzzo D. Space exploration by the promoter of a long human gene during one transcription cycle. Nucleic Acids Res. 2013;41:2216–2227. doi: 10.1093/nar/gks1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ntini E., Järvelin A.I., Bornholdt J., Chen Y., Boyd M., Jørgensen M., Andersson R., Hoof I., Schein A., Andersen P.R., et al. (2013) Polyadenylation site-induced decay of upstream transcripts enforces promoter directionality. Nat. Struct. Mol. Biol. 20:923–928. doi: 10.1038/nsmb.2640. [DOI] [PubMed] [Google Scholar]
- 22.Chapman R.D., Heidemann M., Albert T.K., Mailhammer R., Flatley A., Meisterernst M., Kremmer E., Eick D. Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science. 2007;318:1780–1782. doi: 10.1126/science.1145977. [DOI] [PubMed] [Google Scholar]
- 23.Crooks G.E., Hon G., Chandonia J.M., Brenner S.E. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorenz R, Bernhart S.H., Höner Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L. Vienna RNA Package 2.0. Algorithms Mol. Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diermeier S., Kolovos P., Heizinger L., Schwartz U., Georgomanolis T., Zirkel A., Wedemann G., Grosveld F., Knoch T.A., Merkl R., et al. (2014) TNFα signaling primes chromatin for NF-κB binding and induces rapid and widespread nucleosome repositioning. Genome Biol. 15:536. doi: 10.1186/s13059-014-0536-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoffman M.M., Ernst J., Wilder S.P., Kundaje A., Harris R.S., Libbrecht M., Giardine B., Ellenbogen P.M., Bilmes J.A., Birney E., et al. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013;41:827–841. doi: 10.1093/nar/gks1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zarnack K., König J., Tajnik M., Martincorena I., Eustermann S., Stévant I., Reyes A., Anders S., Luscombe N.M., Ule J. Direct competition between hnRNPC and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell. 2013;152:453–466. doi: 10.1016/j.cell.2012.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papantonis A., Kohro T., Baboo S., Larkin J.D., Deng B., Short P., Tsutsumi S., Taylor S., Kanki Y., Kobayashi M., et al. TNFα signals through specialized factories where responsive coding and miRNA genes are transcribed. EMBO J. 2012;31:4404–4414. doi: 10.1038/emboj.2012.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hicks M.J., Yang C.R., Kotlajich M.V., Hertel K.J. Linking splicing to Pol II transcription stabilizes pre-mRNAs and influences splicing patterns. PLoS Biol. 2006;4:e147. doi: 10.1371/journal.pbio.0040147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao K., Masuda A., Matsuura T., Ohno K. Human branch point consensus sequence is yUnAy. Nucleic Acids Res. 2008;36:2257–2267. doi: 10.1093/nar/gkn073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeo G., Burge C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004;11:377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 32.Kim S., Kim H., Fong N., Erickson B., Bentley D.L. Pre-mRNA splicing is a determinant of histone H3K36 methylation. Proc. Natl. Acad. Sci. U.S.A. 2011;108:13564–13569. doi: 10.1073/pnas.1109475108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Irimia M., Weatheritt R.J., Ellis J.D., Parikshak N.N., Gonatopoulos-Pournatzis T., Babor M., Quesnel-Vallières M., Tapial J., Raj B., O'Hanlon D., et al. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell. 2014;159:1511–1523. doi: 10.1016/j.cell.2014.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen W., Luo L., Zhang L. The organization of nucleosomes around splice sites. Nucleic Acids Res. 2010;38:2788–2798. doi: 10.1093/nar/gkq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suzuki H., Kameyama T., Ohe K., Tsukahara T., Mayeda A. Nested introns in an intron: evidence of multi-step splicing in a large intron of the human dystrophin pre-mRNA. FEBS Lett. 2013;587:555–561. doi: 10.1016/j.febslet.2013.01.057. [DOI] [PubMed] [Google Scholar]
- 36.Grellscheid S.N., Smith C.W. An apparent pseudo-exon acts both as an alternative exon that leads to nonsense-mediated decay and as a zero-length exon. Mol. Cell. Biol. 2006;26:2237–2246. doi: 10.1128/MCB.26.6.2237-2246.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang C., Hastings M.L., Krainer A.R., Zhang M.Q. Dual-specificity splice sites function alternatively as 5′ and 3′ splice sites. Proc. Natl. Acad. Sci. U.S.A. 2007;104:15028–15033. doi: 10.1073/pnas.0703773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melamud E., Moult J. Stochastic noise in splicing machinery. Nucleic Acids Res. 2009;37:4873–4886. doi: 10.1093/nar/gkp471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takahara T., Tasic B., Maniatis T., Akanuma H., Yanagisawa S. Delay in synthesis of the 3′ splice site promotes trans-splicing of the preceding 5′ splice site. Mol. Cell. 2005;18:245–251. doi: 10.1016/j.molcel.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 40.Parra M.K., Gallagher T.L., Amacher S.L., Mohandas N., Conboy J.G. Deep intron elements mediate nested splicing events at consecutive AG dinucleotides to regulate alternative 3′ splice site choice in vertebrate 4.1 genes. Mol. Cell. Biol. 2012;32:2044–2053. doi: 10.1128/MCB.05716-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Conti L., Baralle M., Buratti E. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip. Rev. RNA. 2013;4:49–60. doi: 10.1002/wrna.1140. [DOI] [PubMed] [Google Scholar]
- 42.Thanaraj T.A., Clark F. Human GC-AG alternative intron isoforms with weak donor sites show enhanced consensus at acceptor exon positions. Nucleic Acids Res. 2001;29:2581–2593. doi: 10.1093/nar/29.12.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dewey C.N., Rogozin I.B., Koonin E.V. Compensatory relationship between splice sites and exonic splicing signals depending on the length of vertebrate introns. BMC Genomics. 2006;7:311. doi: 10.1186/1471-2164-7-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roca X., Akerman M., Gaus H., Berdeja A., Bennett C.F., Krainer A.R. Widespread recognition of 5′ splice sites by noncanonical base-pairing to U1 snRNA involving bulged nucleotides. Genes Dev. 2012;26:1098–1109. doi: 10.1101/gad.190173.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kyriakopoulou C., Larsson P., Liu L., Schuster J., Söderbom F., Kirsebom L.A., Virtanen A. U1-like snRNAs lacking complementarity to canonical 5′ splice sites. RNA. 2006;12:1603–1611. doi: 10.1261/rna.26506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Reilly D., Dienstbier M., Cowley S.A., Vazquez P., Drozdz M., Taylor S., James W.S., Murphy S. Differentially expressed, variant U1 snRNAs regulate gene expression in human cells. Genome Res. 2013;23:281–291. doi: 10.1101/gr.142968.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turunen J.J., Niemelä E.H., Verma B., Frilander M.J. The significant other: splicing by the minor spliceosome. Wiley Interdiscip. Rev. RNA. 2013;4:61–76. doi: 10.1002/wrna.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S., et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.