

Abstract
Normal fusion of developing eyelids requires coordination of inductive signals from the eyelid mesenchyme with migration of the periderm cell layer and constriction of the eyelids across the eye. Failure of this process results in an eyelids open at birth (EOB) phenotype in mice. We have identified a novel spontaneous allele of Alx4 that displays EOB, in addition to polydactyly and cranial malformations.Alx4 is expressed in the eyelid mesenchyme prior to and during eyelid fusion in a domain overlapping the expression of genes that also play a role in normal eyelid development. We show that Alx4 mutant mice have reduced expression of Fgf10, a key factor expressed in the mesenchyme that is required for initiation of eyelid fusion by the periderm. This is accompanied by a reduced number of periderm cells expressing phosphorylated c-Jun, consistent with the incomplete ablation of Fgf10 expression. Together, these data demonstrate that eyelid fusion in mice requires the expression of Alx4, accompanied by the loss of normal expression of essential components of the eyelid fusion pathway.
Keywords: eyelid, Fgf10, Alx4, spontaneous mutant, mouse
Introduction
During development, the mouse eyelids close and fuse temporarily, reopening at approximately postnatal day 14. Similarly, human eyelids fuse at approximately week 9 of gestation, reopening prior to birth at around the 6-month time point. It is thought this event is important for the normal development of the eyelid margin, and failures of this process can result in a variety of eye defects in humans and mice (Zieske, 2004; Byun et al., 2011). Fusion of the eyelid is initiated, in part, by inductive signals from the eyelid mesenchyme that stimulate proliferation and migration of the periderm cell layer of the epithelium at approximately embryonic day 14.5 (E14.5). The periderm expands and migrates across the surface of the cornea, completing fusion by ∼E15.5-E16. The basal epithelial layer and mesenchymal core of each eyelid follow the periderm migration, arriving at the center of the eye in close proximity, but remaining completely separate. Failure of this process results in an EOB phenotype in neonatal mice. Because eyelid closure appears to be important for development of the cornea during gestation (Zieske, 2004), this frequently leads to corneal opacity and other defects in surviving adult mice (Toonen et al., 2012, for example) and more recently has been implicated in the normal development of the meibomian gland, tarsal muscles and other muscles of the eye (Meng et al., 2014). Induction and migration of the eyelid epithelium is thought to involve many of the same genes activated during wound healing in the adult (Grose, 2003; Li et al., 2003; Martin and Parkhurst, 2004; Xia and Kao, 2004), and thus represents a useful model for understanding regulation of wound healing pathways.
A number of signaling molecules and pathways regulate eyelid fusion, as revealed by mouse mutations that result in an EOB phenotype (Xia and Kao, 2004). These include growth factors and receptors such as Fgf10 (Tao et al., 2005), Fgfr2 (Li et al., 2001), Inhbb (Schrewe et al., 1994; Vassalli et al., 1994), Tgfa (Luetteke et al., 1993; Mann et al., 1993), and Egfr (Threadgill et al., 1995), and downstream effector genes and proteins such as OSR2 (Gao et al., 2009), Jun (c-Jun)(Li et al., 2003; Zenz et al., 2003), Map3k1 (MEKK) (Zhang et al., 2003) and Rock1 (Shimizu et al., 2005). This body of work supports a model where inductive signals expressed in the eyelid mesenchyme, such as Fgf10, activate signaling pathways in the adjacent epithelium at the distal tip of the developing lid (Tao et al., 2005). This in turn stimulates the periderm cell layer to proliferate and migrate across the surface of the cornea, which is followed by the progressive constriction of the eyelids themselves about the closure axis. Interestingly, recent work suggests this migration of the periderm cells is not strictly required for fusion; rather intercalation of the underlying epithelium drives closure. While much is known about the molecular signals that govern the later portion of this process, the early events that regulate inductive signals from the mesenchyme are poorly understood.
Alx4 is a member of the aristaless-like homeobox domain gene family, comprising transcription factors defined by a paired type homeobox domain and a conserved C-terminal “aristaless” domain. The mesenchyme of the developing craniofacial structures and limb buds display prominent expression of Alx4 and several family members, including Cart1 (Qu et al., 1997; Beverdam et al., 2001), which physically interacts with Alx4 and is required for transcriptional activation. Targeted null mutations of Alx4 result in highly penetrant preaxialpolydactyly, skull abnormalities resulting from defects inparietal bone formation and ventral body wall fusion defects (Qu et al., 1997). The spontaneous mutant Strong's luxoid (lst) (Forsthoefel, 1963) is caused by a deletion in the Alx4 gene, and displays a subset of the knockout phenotypes, indicating the range and penetrance of phenotypes is affected by genetic background (Qu et al., 1998). While the role of Alx4 in limb and skull development is well studied (Qu et al., 1997; Antonopoulou et al., 2004; Kuijper et al., 2005), no analysis of the eyelid fusion defect has been performed.
In this study, we explore the mechanisms that govern embryonic eyelid fusion, using a novel spontaneous mutant allele of Alx4. Mice harboring this mutation are born with an EOB phenotype. We find that ALX4 protein is expressed in the mesenchyme directly adjacent to the palpebral conjunctival epithelium, overlapping the expression domain of known mediators of eyelid fusion, including Fgf10. We show a clear downregulation of Fgf10 expression in the developing eyelids of Alx4 mutants indicating that Alx4 plays a critical role in stimulating Fgf10 expression during eyelid fusion.
Results
As part of a longstanding program to identify, characterize and positionally clone mouse mutants with craniofacial malformations, we identified Nm3768, a semi-dominant spontaneous mutation characterized by a short nose and frequent preaxialpolydactyly on one hind limb. Intercrosses of affected animals yielded litters with frequent perinatal lethality, but a few individual putatively homozygous adult survivors were noted. These mice display cataracts, short nose and wide-set eyes, dorsal alopecia, polydactyly on all four limbs and distinct abnormalities in the frontal bones of the skull (Figure 1A, B). Further inspection of newborns from intercrosses revealed a consistent phenotype at birth, including EOB, preaxialpolydactyly on all four limbs and large bulges at the anterior portion of the head, indicating defective formation of the calvaria (Figure 1C).Together, these data suggest mice with the short nose and single limb polydactyly are heterozygous for the Nm3768 mutation, while those that exhibit the EOB phenotype are homozygous for the mutation.
Figure 1.
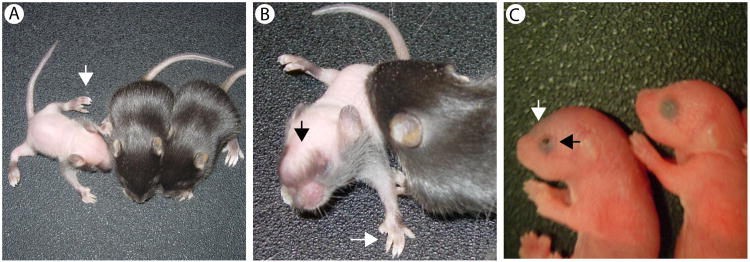
Phenotype of Nm3768 homozygous mice. Intercrosses of Nm3768 animals with a short nose and single limb polydactyly generated mutants with fewer than expected severely affected animals displaying dorsal alopecia (A, B), preaxial polydacyly on all four limbs (A, B), bulging anterior skull (B) and eye defects. In addition, the expected ratio of more severely affected pups is seen at postnatal day 0 (P0), with clear anterior skull defects and eyelids open at birth (C).
We mapped the causative gene of Nm3768 using the dominant phenotypes of a short nose and polydactyly. Nm3768 was found to reside on chromosome 2 (Mmu2) between markers D2Mit15 at 92 Mb and D2Mit386 at 94 Mb (genome build GRCm38/mm10). Several alleles of one candidate gene, Alx4, appeared to harbor a number of similar phenotypes in the homozygous state including preaxialpolydactyly, dorsal alopecia, EOB, and craniofacial abnormalities (Qu et al., 1997; Qu et al., 1998).
To test whether Nm3768 displayed any alterations in the Alx4 gene, we PCR amplified and sequenced fragments of the four exons from homozygous mutant samples. Although we identified no coding mutations, we were unable to amplify fragments from the 5′ portion of exon 1, including both the transcriptional and translational start sequences. This suggested that a deletion encompassing part of the first exon of Alx4 is the cause of the Nm3768 phenotype. We also failed to generate additional amplicons up to 30Kb 5′ of the first exon of Alx4, suggesting the presence of a large 30-35kb deletion that includes the transcription and translation start site. Southern Blot analysis using both an internal an external probe (Figure 2A), clearly show a band shift in mutants consistent with a 33.4 Kb deletion (Figure 2D). Based on these results we were able to design primers spanning the breakpoint, confirmed the precise deletion coordinates of Mmu2:93,612,724-93,646,101 (Figure 2B) and designed a genotyping assay to unambiguously distinguish all three genotype classes (Figure 2C).
Figure 2.

Nm3768 is caused by a large deletion in Alx4. (A) Allele structure of the mouse Alx4 gene. Genotyping primer positions and southern blot probe positions are indicated. Blue and red boxes indicate the proximal and distal ends of the Alx4lst-2J deletion (B), which was confirmed by sequencing. Allele-specific genotyping (C) confirms that severely affected mutant mice are homozygous for the deletion. Notably, some “unaffected” controls were shown to be heterozygous for the deletion, indicating the dominantly inherited short nose, single limb polydactyly phenotype is incompletely penetrant. Southern blot analysis (D) confirms the genomic structure of the deletion. The expected 3.8 Kb WT band from probe A is converted to a 8.4 Kb band in homozygous mutant animals, consistent with the predicted band size of the deletion, while no signal is detected in mutant mice for internal probe B.
To confirm Alx4 is the causative gene for Nm3768, we tested allelism using Strong's Luxoid Jackson (B6C3Fe a/a-Alx4lst-J/J; STOCK #000221), a spontaneous allele of Alx4 (Qu et al., 1998). The Alx4lst-J/J allele failed to complement the EOB and polydactyly phenotype of Nm3768 in compound double heterozygous mutants (data not shown), unambiguously establishing Nm3768 as a new allele of Alx4 (Alx4lst-2J/J).
To confirm that ALX4 protein expression was altered in Alx4lst-2J/J mice, we performed immunostaining on coronal sections of E14.5 embryo heads. Homozygotes show a clear lack of specific nuclear staining in mesenchymal cells along the medial aspect of the developing eyelid compared to littermate controls (Figure 3A, B). These results suggest Alx4lst-2J/J is a null allele of the Alx4 gene, resulting in loss of protein expression. In addition, Hematoxylin and Eosin (H&E) staining of E14.5 eyelids (Figure 3 C, D) suggests the initial formation and outgrowth of the eyelid core is unaffected in mutants as the size and shape appears identical to controls at this stage and at E15.5 (data not shown). Accordingly, no differences in proliferation or apoptosis in mutant eyelids were observed (data not shown).
Figure 3.

Expression of Alx4 in the mesenchyme of the developing eyelid. Immunostaining for Alx4 in control eyelids (A) is localized to the eyelid mesenchyme adjacent to the conjuntava. Alx4lst-2J mutants (B) show no specific Alx4 staining in either domain. H&E staining of control and mutant eyelids (C and D) at E14.5 shows normal development of the core eyelid structure at this stage. Scale bar is shown in A is 50 μm and applies to all panels.
Prior description of the Alx4 knockout phenotype noted open eyelids at birth, but no further phenotypic or mechanistic characterization was performed. Given the mesenchymal expression of the ALX4 protein, we reasoned that Alx4 must regulate the activation of the eyelid tip periderm in a cell nonautonomous fashion through signaling intermediates expressed in the adjacent mesenchymal compartment. A number of genes are expressed in a domain overlapping Alx4, including Osr2, and Fgf10.Null mutations in Osr2 and Fgf10 lead to the EOB phenotype (Tao et al., 2005; Gao et al., 2009), and Fgf10 has been shown to directly regulate activation of the eyelid tip periderm through engagement of the Fgfr2 receptor (Tao et al., 2005). In Alx4lst-2J/J mutants, there is a consistent reduction in Fgf10 staining in the eyelid mesenchyme, overlapping the Alx4 expression domain, while no differences in expression of Osr2 or Fgfr2 are noted (Figure 4). This would support a model where Alx4 regulates eyelid closure through the activity of Fgf10.
Figure 4.

Expression of markers of eyelid development in Alx4 mutant embryos. Section in situ hybridization of genes expressed in the developing eyelid mesenchyme and epithelium. Osr2 (A, D) show similar patterns of expression in the eyelid mesenchyme adjacent to the conjunctiva and in a domain overlapping normal Alx4 expression. Mutant embryos also display normal expression of Osr2 in the epithelial compartment. Fgfr2 expression (B, E) is restricted to the eyelid epithelium and is unchanged in mutant samples. Fgf10 (C, F) is weakly expressed in the mesenchyme of the developing eyelid, and displays a partial, but consistent, reduced level of expression in Alx4 mutant samples. All control samples are WT genotype. Scale bar is shown in A is 100 μm and applies to all panels.
Because Fgf10 is only weakly expressed, we confirmed our preliminary finding by quantitative RT-PCR (qPCR) on E14.5 whole eyelids. As shown in Figure 5, Fgf10 expression is significantly lower in Alx4 mutants, and there is an apparent, though not statistically significant, decrease in Alx4 heterozygotes, suggesting the relative dose of ALX4 protein is important in regulating Fgf10. Consistent with our in situ hybridization findings, Fgfr2 expression is unaffected, while Alx4 expression decreases with gene copy number, as expected. We did not observe any significant difference in the expression of putative downstream effectors of Fgf10 function, including Bmp4 (Huang et al., 2009), Ihnbb (Tao et al., 2005), and Shh (Tao et al., 2005) (Figure 5). However these genes are normally expressed in fairly limited domains, and therefore our assay might lack the sensitivity to detect small changes. This is also consistent with the moderate, but incomplete, loss of Fgf10 expression. No differences were noted in any targets (apart from Alx4) at E15.5 (data not shown), consistent with the model that Alx4 regulates the initiation of periderm outgrowth via Fgf10 expression in the mesoderm at E14.5.
Figure 5.

Quantitative expression of markers of eyelid development in isolated embryonic eyelids. Expression levels of Alx4, Bmp4, Fgf10, Inhbb, and Shh measured by qPCR using Taqman probes (Applied Biosystems) on mRNA from microdissected E14.5 eyelids. For each gene, one WT sample is arbitrarily set to 1 and relative fold change calculated for the remaining samples. Asterisk indicates a significant difference (p<0.05) for WT versus homozygous for Fgf10 expression, determined by student's t-test. Sample sizes: WT=8; Het=9; Hom=9.
To investigate the functional impact of the Alx4 mutation on the periderm, additional markers of periderm structure (KRT6) (Mazzalupo and Coulombe, 2001; Shimizu et al., 2005) and activation (phospho-c-Jun) were examined (Li et al., 2003; Zenz et al., 2003). As shown in Figure 6, KRT6 expression is detected in the periderm of both control and homozygous mutant samples, although the extent of staining is reduced in mutants reflecting the reduced number of total periderm cells. These data lead us to conclude that the periderm in Alx4 mutants forms properly, but fails to respond to the signals to expand and migrate across the cornea. Similarly, we see evidence of activation of the JNK signaling pathway, a known mediator of periderm cell migration, in both mutants and controls. However, as with KRT6, a reduced number of cells display activation of JNK as measured by phospho-c-Jun staining. These data suggest an overall reduction, but not complete ablation, of normal periderm activation during eyelid fusion in Alx4 mutants. This is consistent with a reduced, but not absent, level of Fgf10 expression and suggests a minimum threshold of periderm activation by Fgf10 is required for the migration and fusion of the eyelids.
Figure 6.

Expression of markers of eyelid periderm development and activation. Immunofluorescence indicates expression of KRT6 in the clusters of periderm cells at the tips of both WT control (A) or mutant (B) eyelids of E15.5 day embryos. Note the periderm cells in control embryos have expanded and extended partially across the cornea at this stage. (C, D) Expression of Phosph-c-Jun is noted by immunofluorescence in individual cells within the periderm of both mutant and control eyelids.
Discussion
In this study, we present a detailed analysis of the EOB phenotype of a novel allele of Alx4. While our new allele Alx4lst-2J, recapitulates many of the features of the original Strong's Luxoid and subsequent targeted mutations, including preaxialpolydactyly and, dorsal alopecia, defects in calvaria development and EOB(Qu et al., 1997; Qu et al., 1998; Qu et al., 1999; Antonopoulou et al., 2004), we did not observe any instances of omphalocele in severely affected mice. This could be attributed to lower penetrance in our model due to an accumulation of modifiers, which has been maintained as a closed colony for several generations. We have not examined neonates in sufficient numbers to rule out rare instances of omphalocele, which along with severe calvarial defects would explain the small number of viable adult homozygotes. We demonstrate that our allele is a clear null, like the targeted mutation, so our observation is unlikely to be due to a hypomorphic effect of the mutation.
Prior work examining the function Alx4 in vivo is focused on its role in the developing limb and skull (Qu et al., 1997; Qu et al., 1998; Qu et al., 1999; Antonopoulou et al., 2004). In the case of the former, Alx4 is thought to provide anterior identity to the developing limb bud, repressing the expression of posterior-specific genes. Of interest, Alx4-/- mutants display ectopic expression of Fgf4 in the anterior limb bud (Qu et al., 1997), suggesting Alx4 functions directly or indirectly to downregulate the Fgf4 gene. We observe the opposite effect on Fgf10 in the eyelid mesenchyme, where loss of Alx4 leads to a reduction on Fgf10 levels. This is consistent with other work that demonstrates Alx4 can function as a transcriptional activator (Qu et al., 1999). Interestingly, mice homozygous for a null mutation in Fgf10 lack limbs entirely.
Eyelid fusion in mice requires both autocrine signals within the periderm cell population at the eyelid tip and paracrine instructive signals from the adjacent mesenchyme. A complex array of signaling components and pathways has been implicated in this process, including Activin-beta-B (Innhb)/JNK/c-JUN, Tgfa/HB-EGF/ERK and BMP4/Smad. A consensus model has emerged in which expression of Fgf10 in the mesenchyme of the developing eyelid triggers activation of these pathways in the adjacent epithelium and periderm to initiate cell migration and ultimately eyelid fusion. While Fgf10 mice display an EOB phenotype (Sekine et al., 1999; Tao et al., 2005), it is distinct from that of the knockout of its receptor Fgfr2 (Li et al., 2001), in which the core of the eyelids fails to form well prior to the initiation of fusion, suggesting additional roles for Fgfr2 earlier in eyelid development. Our ability to detect phospo-c-Jun in a smaller number of periderm cells is consistent with the partial, but incomplete downregulation of Fgf10, and suggests there is a threshold of activation required for proper morphogenesis to proceed. The regulation of Fgf10 expression itself, however, is not understood. Alx4 is a transcription factor that binds, both independently and in a cooperative fashion with Cart1 (Qu et al., 1999), to a paired-type homeodomain consensus sequence of 5′-TAAT NNN ATTA-3′. Interestingly, two perfect consensus motifs lie ∼2.5Kb 5′ of the first exon of Fgf10, although we have not explored the potential for a direct effect of Alx4 on the transcriptional activity of the Fgf10 promoter. Consistent with this, our work in this study strongly suggests that Alx4 functions upstream of Fgf10 and is necessary for proper expression of this key regulator of eyelid fusion.
Experimental Procedures
Mice
Alx4lst-2J/J mice were identified as part of the Jackson Laboratory Craniofacial Mutant Resource program. The spontaneous mutation arose on a C57BL/6J background. A colony was established and maintained by brother/sister mating of affected heterozygote by unaffected littermate. B6C3Fe a/a-Alx4lst-J/J (STOCK #000221) mice were cryo recovered from the Jackson Laboratory Repository. Mice had access to bottled acidified water (pH 2.8-3.1) and NIH 31M feed pellets with 6% fat (Purina Mills Inc., Richmond, IN) ad libitum. The light:dark cycle was maintained at 12:12. For timed matings, two females were placed with single housed males after 4 PM and day 0 of pregnancy was defined as the mid point of the dark cycle prior to the appearance of a copulation plug. All procedures were approved by The Jackson Laboratory's Institutional Animal Care and Use Committee and performed in accordance with National Institutes of Health guidelines for the care and use of animals in research.
Genetic Mapping
Adult dominant carriers of the Alx4lst2J mutation were mapped with a linkage cross to A/J via a backcross strategy. Analysis of recombinants using a genome-wide panel of polymorphic microsatellite markers established linkage to Chr 2 with a minimum interval between D2Mit15 (92035835-92035976) and D2Mit386 (94018729-940188500) Mb.
Genotyping and Southern Blot
PCR amplicons from each exon were purified and sequenced to identify causative mutations. Primer pairs for exon one failed to generate bands in homozygotes, suggesting a 5′ deletion. Additional small amplicons were produced at various intervals 5′ and 3′ of exon 1 until amplification was noted in homozygous samples. We were then able to identify two primers, Alx4 F (5′-TTGACTCCCAAGGGACATTC-3′) and Mut R (5′-TCTCAGAGGTCCCTGGATTG-3′) that spanned the deletion breakpoint. A third primer (WT R: 5′-TAGGCCCATCCAGTGACTTC-3′) was designed within the deleted region to provide a genotyping assay. A 32P labeled 591 bp probe 5′ of the breakpoint (2:93612053-93612643) and 842bp probe 3′ of the breakpoint (2:93615034-93615875) were hybridized to HindIII digested genomic DNA of WT, heterozygous and mutant embryos as described (cit).
Histology and immunostaining
To generate homozygous embryos, affected carriers or genotype-confirmed carriers were intercrossed via timed mating, setting the midpoint of the dark cycle prior to the appearance of a copulation plug as day 0 of gestation. Embryos were dissected at the times indicated and fixed in 4% Parafomaldehyde overnight at 4°C. For histology and immunohistochemistry, embryos were washed in PBS, dehydrated through an ethanol series and embedded in paraffin. 5 micron coronal sections through the eye were affixed to Superfrost+ slides (Fisher), dewaxed and hydrated prior to staining with Hematoxylin and Eosin (H&E) or immunohistochemistry. Antibody staining was performed essentially as described (Murray and Gridley, 2006), using antibodies to ALX4 (Santa Cruz), KRT6 (Covance) and Phospho-c-Jun (Cell Signaling Technology) at a 1:100 dilution each. An HRP-conjugated secondary antibody (Invitrogen) was used for ALX4 and staining was visualized by DAB development (Sigma). For KRT6 and phospho-c-Jun, AlexaFluor488-conjugated (Invitrogen) secondary antibodies were used, and images captured by confocal microscopy.
In situ hybridization
Heads of embryos isolated and fixed as described above were equilibrated in a 30% sucrose gradient overnight at 4°C then frozen in OCT on dry ice. Twenty micron frozen sections through the eye were affixed to Superfrost+ slides and air dried briefly prior to processing. Sections were then fixed, permeabilized with proteinase K, postfixed, dehydrated then hybridized overnight at 70°C using a 50% formamide hybridization solution and 200-500μg/microliters of digoxigenin-labeled antisense RNA probe. Following a series of SSC washes, the slides were blocked in embryo powder/sheep serum, then incubated overnight at 4°C with an alkaline phosphatase-conjugated anti-digoxygenin antibody (Roche). After washing, the slides were developed using BM purple (Roche) substrate, mounted and imaged. Fgf10 and Fgfr2 cDNA clones were obtained from the Mammalian Gene Collection (http://mgc.nci.nih.gov/), Osr2 was a gift from Dr. Rulang Jaing.
Quantitative PCR analysis
Individual eyelids from embryos were isolated by microdissection, stored in RNA later (Ambion, Austin, TX) per manufacturer's instructions and homogenized in RLT Buffer from an RNeasy Micro Kit (Qiagen, Valencia, Ca.). Total RNA was isolated according to manufacturer's protocols (Qiagen) including the optional DNase treatment step, and quality was assessed using a 2100 Bioanalyzer instrument and RNA 6000 Pico LabChip assay. 500μg of total RNA was then reverse transcribed with random decamers and M-MLV RT using the Message Sensor RT Kit (Ambion, Austin, TX).
A portion of the cDNA was then used in a PCR reaction containing Taqman Univeral PCR Master Mix (Applied Biosystems, Carlsbad, Ca) which includes AmpliTaq Gold DNA Polymerase, AmpErase UNG, dNTPs with dUTP, Passive Reference 1 and other buffer components. The gene specific primers and probe sets were obtained from Applied Biosystem Assay on Demand service and used according to manufacturer's protocols. Real-Time PCR was performed in an AB PRISM® 7900HT Sequence Detection System. (Applied Biosystems) with the standard protocol of 50°C for 2 min, 95°C 10 min, 40 cycles of 95°C for 15 sec, and 60° for 1 min. The threshold cycle (Ct) and relative quantitation (RQ) was determined using the Sequence Detection System software (SDS2.2). For each gene, one WT sample was arbitrarily set to 1, and all other samples are calculated as relative fold change.
Acknowledgments
This work is supported by NIH grants EY015073 (to L.R.D) and DE020052 (to S.M. and L.R.D.). The authors would like to thank Harold Coombs for assistance with colony maintenance and Aimee Picard and Kevin Peterson for their review and editing of the manuscript.
Grant sponsor: National Institutes of Health grants EY015073 (to L.R.D) and DE020052 (to S.M. and L.R.D.)
References
- Antonopoulou I, Mavrogiannis LA, Wilkie AO, Morriss-Kay GM. Alx4 and Msx2 play phenotypically similar and additive roles in skull vault differentiation. Journal of anatomy. 2004;204:487–499. doi: 10.1111/j.0021-8782.2004.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beverdam A, Brouwer A, Reijnen M, Korving J, Meijlink F. Severe nasal clefting and abnormal embryonic apoptosis in Alx3/Alx4 double mutant mice. Development. 2001;128:3975–3986. doi: 10.1242/dev.128.20.3975. [DOI] [PubMed] [Google Scholar]
- Byun TH, Kim JT, Park HW, Kim WK. Timetable for upper eyelid development in staged human embryos and fetuses. Anatomical record. 2011;294:789–796. doi: 10.1002/ar.21366. [DOI] [PubMed] [Google Scholar]
- Forsthoefel PF. The Embryological Development of the Effects of Strong's Luxoid Gene in the Mouse. Journal of morphology. 1963;113:427–451. doi: 10.1002/jmor.1051130307. [DOI] [PubMed] [Google Scholar]
- Gao Y, Lan Y, Ovitt CE, Jiang R. Functional equivalence of the zinc finger transcription factors Osr1 and Osr2 in mouse development. Developmental biology. 2009;328:200–209. doi: 10.1016/j.ydbio.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grose R. Epithelial migration: open your eyes to c-Jun. Current biology : CB. 2003;13:R678–680. doi: 10.1016/s0960-9822(03)00607-9. [DOI] [PubMed] [Google Scholar]
- Huang J, Dattilo LK, Rajagopal R, Liu Y, Kaartinen V, Mishina Y, Deng CX, Umans L, Zwijsen A, Roberts AB, Beebe DC. FGF-regulated BMP signaling is required for eyelid closure and to specify conjunctival epithelial cell fate. Development. 2009;136:1741–1750. doi: 10.1242/dev.034082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijper S, Feitsma H, Sheth R, Korving J, Reijnen M, Meijlink F. Function and regulation of Alx4 in limb development: complex genetic interactions with Gli3 and Shh. Developmental biology. 2005;285:533–544. doi: 10.1016/j.ydbio.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Li C, Guo H, Xu X, Weinberg W, Deng CX. Fibroblast growth factor receptor 2 (Fgfr2) plays an important role in eyelid and skin formation and patterning. Developmental dynamics : an official publication of the American Association of Anatomists. 2001;222:471–483. doi: 10.1002/dvdy.1205. [DOI] [PubMed] [Google Scholar]
- Li G, Gustafson-Brown C, Hanks SK, Nason K, Arbeit JM, Pogliano K, Wisdom RM, Johnson RS. c-Jun is essential for organization of the epidermal leading edge. Developmental cell. 2003;4:865–877. doi: 10.1016/s1534-5807(03)00159-x. [DOI] [PubMed] [Google Scholar]
- Luetteke NC, Qiu TH, Peiffer RL, Oliver P, Smithies O, Lee DC. TGF alpha deficiency results in hair follicle and eye abnormalities in targeted and waved-1 mice. Cell. 1993;73:263–278. doi: 10.1016/0092-8674(93)90228-i. [DOI] [PubMed] [Google Scholar]
- Mann GB, Fowler KJ, Gabriel A, Nice EC, Williams RL, Dunn AR. Mice with a null mutation of the TGF alpha gene have abnormal skin architecture, wavy hair, and curly whiskers and often develop corneal inflammation. Cell. 1993;73:249–261. doi: 10.1016/0092-8674(93)90227-h. [DOI] [PubMed] [Google Scholar]
- Martin P, Parkhurst SM. Parallels between tissue repair and embryo morphogenesis. Development. 2004;131:3021–3034. doi: 10.1242/dev.01253. [DOI] [PubMed] [Google Scholar]
- Mazzalupo S, Coulombe PA. A reporter transgene based on a human keratin 6 gene promoter is specifically expressed in the periderm of mouse embryos. Mechanisms of development. 2001;100:65–69. doi: 10.1016/s0925-4773(00)00489-5. [DOI] [PubMed] [Google Scholar]
- Meng Q, Mongan M, Carreira V, Kurita H, Liu CY, Kao W, Xia Y. Eyelid closure in embryogenesis is required for ocular adnexa development. Investigative ophthamology and visual science. 2014;55:7652–7661. doi: 10.1167/iovs.14-15155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray SA, Gridley T. Snail family genes are required for left-right asymmetry determination, but not neural crest formation, in mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:10300–10304. doi: 10.1073/pnas.0602234103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu S, Niswender KD, Ji Q, van der Meer R, Keeney D, Magnuson MA, Wisdom R. Polydactyly and ectopic ZPA formation in Alx-4 mutant mice. Development. 1997;124:3999–4008. doi: 10.1242/dev.124.20.3999. [DOI] [PubMed] [Google Scholar]
- Qu S, Tucker SC, Ehrlich JS, Levorse JM, Flaherty LA, Wisdom R, Vogt TF. Mutations in mouse Aristaless-like4 cause Strong's luxoid polydactyly. Development. 1998;125:2711–2721. doi: 10.1242/dev.125.14.2711. [DOI] [PubMed] [Google Scholar]
- Qu S, Tucker SC, Zhao Q, deCrombrugghe B, Wisdom R. Physical and genetic interactions between Alx4 and Cart1. Development. 1999;126:359–369. doi: 10.1242/dev.126.2.359. [DOI] [PubMed] [Google Scholar]
- Schrewe H, Gendron-Maguire M, Harbison ML, Gridley T. Mice homozygous for a null mutation of activin beta B are viable and fertile. Mechanisms of development. 1994;47:43–51. doi: 10.1016/0925-4773(94)90094-9. [DOI] [PubMed] [Google Scholar]
- Sekine K, Ohuchi H, Fujiwara M, Yamasaki M, Yoshizawa T, Sato T, Yagishita N, Matsui D, Koga Y, Itoh N, Kato S. Fgf10 is essential for limb and lung formation. Nature genetics. 1999;21:138–141. doi: 10.1038/5096. [DOI] [PubMed] [Google Scholar]
- Shimizu Y, Thumkeo D, Keel J, Ishizaki T, Oshima H, Oshima M, Noda Y, Matsumura F, Taketo MM, Narumiya S. ROCK-I regulates closure of the eyelids and ventral body wall by inducing assembly of actomyosin bundles. The Journal of cell biology. 2005;168:941–953. doi: 10.1083/jcb.200411179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao H, Shimizu M, Kusumoto R, Ono K, Noji S, Ohuchi H. A dual role of FGF10 in proliferation and coordinated migration of epithelial leading edge cells during mouse eyelid development. Development. 2005;132:3217–3230. doi: 10.1242/dev.01892. [DOI] [PubMed] [Google Scholar]
- Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC, et al. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science. 1995;269:230–234. doi: 10.1126/science.7618084. [DOI] [PubMed] [Google Scholar]
- Toonen J, Liang L, Sidjanin DJ. Waved with open eyelids 2 (woe2) is a novel spontaneous mouse mutation in the protein phosphatase 1, regulator (inhibitor) subunit 13 like (Ppp1r13l) gene. BMC Genetics. 2012;13:76. doi: 10.1186/1471-2156-13-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassalli A, Matzuk MM, Gardner HA, Lee KF, Jaenisch R. Activin/inhibin beta B subunit gene disruption leads to defects in eyelid development and female reproduction. Genes & development. 1994;8:414–427. doi: 10.1101/gad.8.4.414. [DOI] [PubMed] [Google Scholar]
- Xia Y, Kao WW. The signaling pathways in tissue morphogenesis: a lesson from mice with eye-open at birth phenotype. Biochemical pharmacology. 2004;68:997–1001. doi: 10.1016/j.bcp.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Zenz R, Scheuch H, Martin P, Frank C, Eferl R, Kenner L, Sibilia M, Wagner EF. c-Jun regulates eyelid closure and skin tumor development through EGFR signaling. Developmental cell. 2003;4:879–889. doi: 10.1016/s1534-5807(03)00161-8. [DOI] [PubMed] [Google Scholar]
- Zhang L, Wang W, Hayashi Y, Jester JV, Birk DE, Gao M, Liu CY, Kao WW, Karin M, Xia Y. A role for MEK kinase 1 in TGF-beta/activin-induced epithelium movement and embryonic eyelid closure. The EMBO journal. 2003;22:4443–4454. doi: 10.1093/emboj/cdg440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zieske JD. Corneal development associated with eyelid opening. The International journal of developmental biology. 2004;48:903–911. doi: 10.1387/ijdb.041860jz. [DOI] [PubMed] [Google Scholar]