

Abstract
Paired associative stimulation (PASLTP) of the human primary motor cortex (M1) can induce LTP-like plasticity by increasing corticospinal excitability beyond the stimulation period. Previous studies showed that two consecutive PASLTP protocols interact by homeostatic metaplasticity, but animal experiments provided evidence that LTP can be augmented by repeated stimulation protocols spaced by ~30min. Here we tested in twelve healthy selected PASLTP responders the possibility that LTP-like plasticity can be augmented in the human M1 by systematically varying the interval between two consecutive PASLTP protocols. The first PASLTP protocol (PAS1) induced strong LTP-like plasticity lasting for 30-60min. The effect of a second identical PASLTP protocol (PAS2) critically depended on the time between PAS1 and PAS2. At 10min, PAS2 prolonged the PAS1-induced LTP-like plasticity. At 30min, PAS2 augmented the LTP-like plasticity induced by PAS1, by increasing both magnitude and duration. At 60min and 180min, PAS2 had no effect on corticospinal excitability. The cumulative LTP-like plasticity after PAS1 and PAS2 at 30min exceeded significantly the effect of PAS1 alone, and the cumulative PAS1 and PAS2 effects at 60min and 180min. In summary, consecutive PASLTP protocols interact in human M1 in a time-dependent manner. If spaced by 30min, two consecutive PASLTP sessions can augment LTP-like plasticity in human M1. Findings may inspire further research on optimized therapeutic applications of non-invasive brain stimulation in neurological and psychiatric diseases.
Introduction
Motor rehabilitation after cerebral injury such as stroke depends on neural plasticity, including synaptic strengthening by long-term potentiation (LTP) [1–4]. Paired associative stimulation (PASLTP) of the human primary motor cortex (M1) can induce an increase in corticospinal excitability as measured by motor evoked potentials (MEPs) beyond the stimulation period [5], which resembles LTP as studied at the cellular level [6–9]. However, LTP-like plasticity induced by PASLTP is regulated by homeostatic metaplasticity [6, 10, 11], i.e., a higher-order form of plasticity, which keeps neuronal and network excitability in a physiological range [12–15]. This homeostatic regulation implies that repeated induction of LTP-like plasticity at short delays is suppressed, which may limit the therapeutic potential of PASLTP to increase corticospinal excitability.
Experiments in animals, however, show that the brain possesses powerful mechanisms, which permit continued synaptic strengthening in the context of prior LTP. One such mechanism is based on an N-methyl-D-aspartate receptor (NMDAR)-dependent form of metaplasticity by which continued synaptic strengthening is possible through activation of metabotropic glutamate receptors (mGluRs) [16]. In addition, inhibition of glycogen synthase kinase-3 beta (GSK3β) results in a ~60 min lasting blockade of subsequent induction of NMDAR-dependent long-term depression (LTD), because expression of LTD requires a high level of GSK3β activity [17]. These mechanisms ensure that information encoded by LTP is not erased during ongoing neural activity, but can be retained. On the system level of the human cortex it has been shown, that motor learning immediately following PASLTP is not suppressed, as would be expected in the framework of homeostatic metaplasticity, but rather facilitated [18, 19]. In addition, several other studies have occasionally reported non-homeostatic metaplasticity between two consecutive non-invasive brain stimulation protocols, mainly at short intervals of 30min or less [20–24], for review [25]. Even though these studies provide system-level evidence for non-homeostatic interactions between plasticity-inducing non-invasive brain stimulation protocols, and motor learning, respectively, the conditions favoring non-homeostatic vs. homeostatic metaplasticity in the human brain remain poorly understood.
The present study investigated the role of time between two consecutive PASLTP protocols for repetitive induction of LTP-like plasticity in M1 of healthy human subjects. We studied the interactions between two identical sessions of PASLTP spaced at inter-PASLTP intervals (IPI) of 10, 30, 60 and 180min. Findings show that metaplasticity in human M1 is expressed in a time-dependent manner with a window of non-homeostatic metaplasticity at an IPI of 30min. If spaced by 30min, two consecutive PASLTP sessions can augment LTP-like plasticity in human M1. These findings may inspire further research on optimized therapeutic applications of non-invasive brain stimulation techniques in a clinical setting.
Materials and Methods
Subjects
Written informed consent was obtained from all subjects prior to participation. None of the subjects had a history of neurological or psychiatric disease or was on CNS-active drugs at the time of the experiments and all subjects were checked for contraindications to transcranial magnetic stimulation (TMS) [26]. The study conformed to the Declaration of Helsinki and was approved by the ethics committee of the Medical Faculty of Goethe-University Frankfurt. Twenty-seven subjects were screened for a significant PASLTP-induced increase in motor evoked potential (MEP) amplitude ≥ 1.1 (ratio of mean MEP amplitude post-PASLTP / pre-PASLTP) [27, 28]. Sixteen subjects fulfilled this criterion (PASLTP responders) and were included into the study. Of those, four subjects withdrew consent after the first experiment for experiencing some discomfort by TMS. Thus, complete datasets were obtained in 12 subjects (6 females, mean (± SEM) age, 25.6 ± 1.4 years). All subjects were right-handed according to the Edinburgh handedness questionnaire [29].
Electromyography (EMG) recordings
Surface EMG recordings were obtained from the right abductor pollicis brevis (APB) muscle using Ag-AgCl electrodes in a belly-tendon montage. The raw EMG signal was amplified and band-pass filtered (20Hz–2kHz, Counterpoint Mk2 electromyograph, Dantec, Denmark), digitized at an A/D rate of 5kHz (Micro1401, Cambridge Electronic Design, UK) and stored in a laboratory computer for online display and offline analysis, using customized software (Spike2 for Windows, Version 3.05, Cambridge Electronic Design).
Transcranial magnetic stimulation (TMS)
Subjects were seated comfortably in a reclining chair. TMS was performed using a Magstim 200 magnetic stimulator (Magstim Company, UK) with a monophasic current waveform. Stimuli were applied to the hand area of left M1 through a figure-of-eight coil (inner diameter of each wing, 70mm) with the handle pointing backwards and 45° away from midline. The optimal coil position for eliciting MEPs in the right APB was marked with a soft-tipped pen on the scalp in order to ensure constant placement of the coil throughout the experiment.
At the beginning of each experiment the resting motor threshold (RMT) was measured, which was defined as the lowest intensity (indicated in percent of maximum stimulator output, MSO) that elicited small MEPs (>50 μV) in at least five out of ten consecutive trials in the resting APB. RMT was determined to nearest 1% of MSO. Thereafter, the intensity to elicit MEPs of, on average, 1mV peak-to-peak amplitude (SI1mV) was determined in the resting APB. This stimulus intensity was then kept constant throughout a given session of the main experiment (Fig 1).
Fig 1. Time line of experiments.
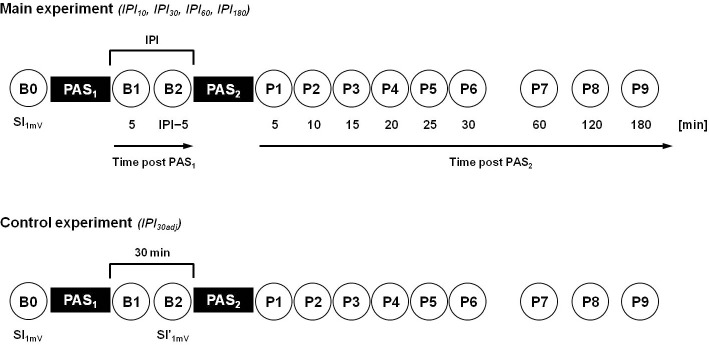
In the main experiment, motor-evoked potentials (MEPs) were recorded at all time points (circles; B0, B1-B2, P1-P9) with SI1mV, i.e. the stimulation intensity that evoked MEPs of, on average, 1mV peak-to-peak amplitude in the resting abductor pollicis brevis muscle at baseline (time point B0). Note that subjects took part in four experimental sessions in a crossover design with different intervals between the consecutive sessions of two identical LTP-like plasticity inducing PASLTP protocols (PAS1, PAS2): 10min (IPI10), 30min (IPI30), 60min (IPI60), and 180min (IPI180). In the control experiment (IPI30adj) SI1mV was readjusted at time point B2 to match baseline MEPs of 1mV peak-to-peak amplitude (SI'1mV). Otherwise, the control experiment was identical to the IPI30 condition of the main experiment.
Paired associative stimulation (PASLTP)
PASLTP was applied according to a protocol originally described by Stefan and colleagues [5] and slightly modified by our group [6, 10]. Briefly, electrical stimulation of the median nerve at the wrist of the right hand was applied through a bipolar electrode (cathode proximal), using constant-current square-wave pulses (duration, 1ms) at an intensity of three times the perceptual sensory threshold. Each stimulus was followed by single-pulse TMS of the left M1 at SI1mV. The interstimulus interval equaled the individual N20-latency of the median nerve somatosensory-evoked cortical potential plus 2ms (mean ± SEM, 21.3 ± 0.3ms). At this or similar intervals PAS induces an LTP-like increase of MEPs in the majority of subjects [5, 6, 27, 30–32]. PASLTP consisted of 225 stimulus pairs applied at a frequency of 0.25Hz. The level of attention, a significant modulator of PASLTP effects [33], was controlled and attention was maximized to the stimulated hand by a light emitting diode (LED) attached to the right wrist which flashed randomly (0.2–1Hz) during PASLTP. Subjects were requested to count and report the total number of flashes as correctly as possible at the end of PASLTP.
Experimental design
In the main experiment, all subjects took part in four different sessions in a pseudorandomized crossover design (Fig 1). Each session consisted of two identical, consecutive PASLTP protocols (PAS1, PAS2) with a specific inter-PASLTP interval (IPI). Intervals were 10min (IPI10), 30min (IPI30), 60min (IPI60) and 180min (IPI180). Between PAS1 and PAS2 subjects were requested to stay awake, keep seated and not to use their stimulated hand. The order of IPI conditions was pseudo-randomized across subjects and experimental sessions were separated by at least three days to avoid carry-over effects (mean individual minimum inter-session interval 7.6 days, range 3–14 days).
To test whether the significant PAS2-induced increase of MEP amplitude in the IPI30 condition could be attributed to the PAS1-induced increase in MEP amplitude at time point B2 immediately before PAS2 (see below and cf. Figs 2 and 3), we conducted a control experiment (IPI30adj), in which we readjusted MEP amplitudes at time point B2 by reducing the stimulation intensity (SI'1mV) in order to match baseline MEPs of, on average, 1mV peak-to-peak amplitude. The readjusted stimulation intensity SI'1mV was then used for PAS2 and all following MEP measurements. Otherwise the control experiment was identical to the IPI30 condition of the main experiment (Fig 1). Nine subjects (4 females, mean (± SEM) age, 26.1 ± 1.6 years) took part in the control experiment.
Fig 2. PAS1-induced increases in MEP amplitude.
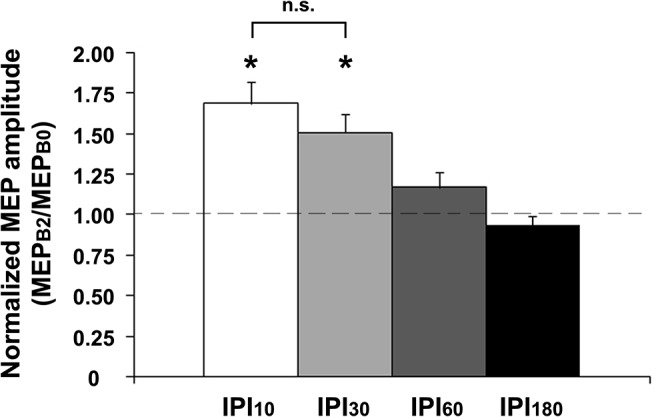
PAS1 resulted in comparable immediate MEP amplitude increases (MEPB1/MEPB0) in all IPI conditions (data not shown). At time point B2 (MEPB2/MEPB0), MEP amplitude increases were present 10min (IPI10) and 30min (IPI30) after PAS1, but no longer at 60min (IPI60) and 180min (IPI180) after PAS1. Asterisks indicate significant differences from 1 (P < 0.01; one-sample two-tailed t tests). Note that MEPB2/MEPB0 was not significantly different between conditions IPI10 and IPI30 (P > 0.09; paired two-tailed t tests). Data are means ± 1 SEM from twelve subjects.
Fig 3. PAS2-induced increases in MEP amplitude after PAS1-priming.

A: The PAS2-induced increase in MEP amplitude after PAS1-priming (MEPPmean/MEPB2) was significantly higher at IPI30 than IPI10 (#, P < 0.05; paired two-tailed t test). B: Time course of MEPPi/MEPB2 (i = 1,2,…,6) over 30min after PAS2 for IPI10 (open squares), IPI30 (filled squares), IPI60 (open circles), and IPI180 (filled circles). C: The cumulative effect of PAS1 and PAS2 on MEP amplitudes (MEPPmean/MEPB0) was significantly higher for IPI30 than for IPI60 and IPI180 (#, P < 0.05; paired two-tailed t tests), and showed a trend to be higher for IPI30 vs. IPI10 (P = 0.095; paired two-tailed t test). D: Time course of MEPPi/MEPB2 (i = 1,2,…,6) over 30min after PAS2 for IPI10 (open squares), IPI30 (filled squares), IPI60 (open circles), and IPI180 (filled circles). *, P < 0.05; one-sample two-tailed t tests. Data are means ± 1 SEM from twelve subjects.
Quantification of PASLTP effects
The PAS1 effect was quantified by comparing 20 single-trial peak-to-peak MEP amplitudes at SI1mV 5min after PAS1 (time point B1) with those at baseline immediately before PAS1 (time point B0; cf. Fig 1). The inter-trial interval was 10s ± 25% to limit anticipation of the next trial. If the PAS1 effect (ratio MEPB1/MEPB0) was < 1.1, the experiment was stopped, the data were discarded and the experiment was repeated on another day, to minimize intra-individual variability [34] and ensure PAS1-induced LTP-like plasticity in all experiments (overall, 12 repeated experiments in all subjects; 1.0 ± 0.4 repeated experiments per subject). Five minutes before the second PASLTP intervention (PAS2) another block of 20 MEPs was measured (time point B2; in case of IPI10 B1 was taken as B2). After PAS2 six blocks of MEP measurements were performed at intervals of 5min (time points P1–P6) to monitor PAS2 effects on MEPs for 30min after the end of PAS2. In six subjects, additional MEP measurements were conducted 60min (time point P7), 120min (P8), and 180min (P9) after PAS2 to determine the time course of the return of PAS2-induced MEP amplitude increases (see Results) back to baseline. All measurements were recorded in the resting APB at SI1mV, except for the control experiment (IPI30adj), in which the TMS intensity was readjusted to SI'1mV at time point B2 and then used for PAS2 and all following MEP measurements. Complete voluntary relaxation was monitored by audio-visual feedback of the EMG raw signal at high gain (at 50μV ⁄ division). Trials contaminated with voluntary EMG activity were excluded from analysis (<1% of all trials).
Statistical analyses
Statistical testing was performed with IBM SPSS Statistics (Version 20.0.0). To test for differences of RMT, SI1mV, and MEP amplitude at baseline (time point B0) independent one-way repeated measures analyses of variance (rmANOVAs) with the within-subject factor IPI (IPI10, IPI30, IPI60, IPI180) were conducted. To test for differences of the PAS1 effect one-way rmANOVAs with the within-subject factor IPI (IPI10, IPI30, IPI60, IPI180) were performed separately on mean MEP amplitudes at time points B1 and B2, respectively, normalized to the individual mean MEP amplitude at time point B0 (MEPB1/MEPB0; MEPB2/MEPB0). To test for the effects of IPI (IPI10, IPI30, IPI60, IPI180) and TIME (P1–P6) on PAS2 effects, two-way rmANOVAs were performed separately on mean MEP amplitudes at time points P1–P6 normalized to the individual mean MEP amplitude at time points B0 and B2, respectively (MEPPi/MEPB0, MEPPi/MEPB2, i = 1,2,…,6). Normalization to B2 provides information specifically on the effects of PAS2 while normalization to B0 provides information on the cumulative effects of PAS1 and PAS2.
Mauchly’s test was applied to test for non-sphericity and in case of violation of sphericity the Greenhouse-Geisser correction was used. Conditional on significant main effects or their interactions in the rmANOVAs, post hoc pairwise comparisons or one-sample two-tailed t tests were performed. The Bonferroni correction was applied to adjust for multiple comparisons.
To compare the time course of PAS1- and PAS2-induced changes in MEP amplitude, the MEP amplitude raw data after PAS1 and PAS2 were fitted independently with a linear mixed effects model with fractional polynomials at all IPIs. PAS1 data comprised mean MEP amplitudes measured at time points B0 (time: 0min) and B2 across IPI conditions (time: 10min at IPI10, 30min at IPI30, 60min at IPI60, and 180min at IPI180), whereas PAS2 data comprised mean MEP amplitudes measured at time points B2 (time: 0min), P2 (time: 10min), P6 (time: 30min), P7 (time: 60min), and P9 (time: 180min), respectively, for each IPI condition separately. Model functions for PAS1- and PAS2 effects on MEP amplitudes were compared using F- and t-statistics, respectively.
In all tests the significance level was set to P < 0.05. All data are expressed as means ± SEM.
Results
All subjects tolerated the experimental procedures well without any adverse effects.
Baseline excitability data (RMT, SI1mV, MEP)
The data are summarized in Table 1. There were no significant differences between IPI conditions on RMT (F 3,33 = 0.55, P = 0.53), SI1mV (F 3,33 = 0.46, P = 0.71), or MEP amplitude (F 3,33 = 1.07, P = 0.38) at baseline (time point B0).
Table 1. Summary of baseline excitability measures in the different IPI conditions.
| Condition | RMT | SI1mV | MEP |
|---|---|---|---|
| IPI10 | 40.1 ± 2.1 | 50.8 ± 2.2 | 0.97 ± 0.04 |
| IPI30 | 39.3 ± 1.6 | 51.4 ± 2.0 | 0.91 ± 0.05 |
| IPI60 | 38.9 ± 1.8 | 50.5 ± 2.4 | 0.93 ± 0.04 |
| IPI180 | 38.8 ± 1.4 | 49.6 ± 1.7 | 1.02 ± 0.05 |
Abbreviations: IPI, inter-PASLTP interval [in min, index]; MEP, motor-evoked potential [in mV]; RMT, resting motor threshold [in %maximum stimulator output, MSO]; SI1mV, stimulation intensity that induces MEPs of 1mV peak-to-peak amplitude on average [in %MSO].
PAS1 effects on MEP amplitudes (comparison of time points B1 and B2 vs. B0)
The one-way rmANOVA for the PAS1 effect at time point B1 (MEPB1/MEPB0) showed no significant difference between IPI conditions (F 3,33 = 0.71, P = 0.55), indicating similar PAS1-induced LTP-like plasticity across all IPI conditions. MEPB1/MEPB0 was > 1.0 for IPI10 (1.69 ± 0.13; t = 5.25, P = 0.0003), IPI30 (1.49 ± 0.10; t = 5.01, P = 0.0004), IPI60 (1.50 ± 0.07; t = 7.57, P < 0.0001), and IPI180 (1.53 ± 0.13; t = 4.12, P = 0.0017). In contrast, the one-way rmANOVA for the PAS1 effect at time point B2 (MEPB2/MEPB0) immediately before PAS2 showed a significant effect of IPI (F 3,33 = 11.74, P < 0.001). MEPB2/MEPB0 was > 1.0 for IPI10 (1.69 ± 0.13; t = 5.25, P = 0.0003) and IPI30 (1.51 ± 0.12; t = 4.32, P = 0.0012), but no longer for IPI60 (1.17 ± 0.09; t = 1.90, P = 0.084) or IPI180 (0.94 ± 0.05; t = -1.20, P = 0.26) (Fig 2). Of note, there was no significant difference of MEPB2/MEPB0 between conditions IPI10 and IPI30 (P > 0.9), indicating similar persistence of PAS1-induced LTP-like plasticity in these conditions at time point B2 immediately before PAS2.
PAS2 effects on MEP amplitudes (comparison of time points P1-P6 vs. B2)
The two-way rmANOVA for the PAS2 effect on MEP amplitudes at time points P1–P6 (MEPPi/MEPB2, i = 1,2,…,6) showed a significant effect of IPI (F 3,33 = 2.94, P = 0.048) and TIME (F 5,55 = 2.50, P = 0.041), whereas the interaction IPI with TIME was not significant (F 15,165 = 0.73, P = 0.75) (Fig 3A and 3B). MEPPmean/MEPB2 was > 1.0 for IPI30 (1.39 ± 0.15; t = 2.59, P = 0.025), but not for IPI10 (0.90 ± 0.06; t = -1.72, P = 0.11), IPI60 (1.20 ± 0.15; t = 1.34, P = 0.21) or IPI180 (1.23 ± 0.16; t = 1.48, P = 0.17). Post hoc testing showed that MEP amplitudes after PAS2 (MEPPmean/MEPB2) were significantly different between conditions IPI10 and IPI30 (P = 0.047), while all other pairwise comparisons were not significant (all P > 0.2).
It could be argued that the significant PAS2 effect on MEP amplitudes at IPI30 was due to the increased MEP amplitude at time point B2 immediately before PAS2 (cf. Fig 2). To address this issue, we conducted a control experiment (IPI30adj), in which we readjusted MEP amplitudes at time point B2 immediately before PAS2 by reducing the stimulation intensity (SI'1mV) in order to match baseline MEPs (0.99 ± 0.05mV at time point B0 with SI1mV = 45.8 ± 1.6%MSO; 1.01 ± 0.06mV at time point B2 with SI'1mV = 44.2 ± 1.6%MSO, t = -0.29, P > 0.7; paired two-tailed t test). Thus, whilst PASLTP induced similar MEP increases at time point B1 (MEPB1/MEPB0) in the control (IPI30adj, 1.31 ± 0.05; t = 6.62, P = 0.0002, one-sample two-tailed t test) and the main experiment (IPI30, 1.49 ± 0.10; t = 5.01, P = 0.0004, one-sample two-tailed t test; IPI30adj vs. IPI30: P > 0.15, unpaired two-tailed t test), MEPB2/MEPB0 was significantly different between the two experiments (IPI30adj: 1.02 ± 0.04, t = 0.38, P > 0.7; IPI30: 1.51 ± 0.12; t = 4.32, P = 0.0012, one-sample two-tailed t tests; IPI30adj vs. IPI30: P = 0.0025, unpaired two-tailed t test; Fig 4). However, PAS2 induced a similar increase in MEP amplitudes (MEPPmean/MEPB2) in the control (IPI30adj: 1.29 ± 0.09, t = 3.09, P = 0.015) compared to the main experiment (IPI30: 1.39 ± 0.15; t = 2.59, P = 0.025; one-sample two-tailed t tests; IPI30adj vs. IPI30: P > 0.5, unpaired two-tailed t test; Fig 4). This finding strongly suggested that the increased MEP amplitude at time point B2 at IPI30 was not relevant for the significant PAS2-induced increase of MEP amplitudes in this condition. This notion was further supported by the observation, that PAS2 had significantly different effects on MEP amplitudes at IPI30 vs. IPI10 in the main experiment (cf. Fig 3A and 3B), although MEP amplitudes were increased to a similar level immediately before PAS2 (MEPB2/MEPB0) in these two conditions (cf. Fig 2).
Fig 4. PAS2-induced increase in MEP amplitude after PAS1-priming in the control experiment (IPI30adj).
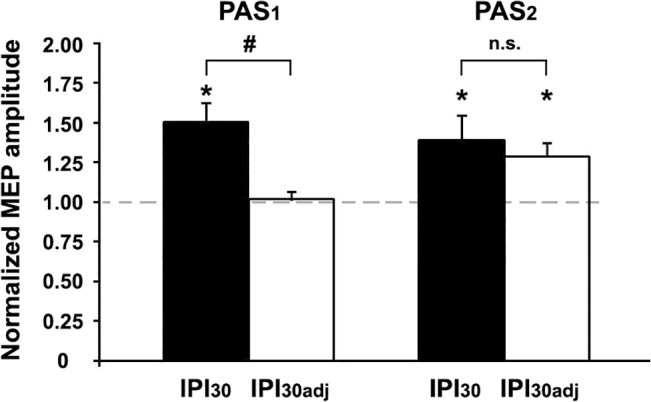
MEP amplitudes at time point B2 immediately before PAS2 were successfully readjusted by reducing the stimulation intensity (SI'1mV) to match baseline MEPs at time point B0 (PAS1, MEPB2/B0). Despite this readjustment, PAS2 induced a similar increase in MEP amplitudes in the control (IPI30adj) compared to the main experiment (IPI30) (PAS2, MEPPmean/MEPB2). *, P < 0.05, one-sample two-tailed t tests; #, P < 0.01, unpaired two-tailed t test. Data from the control experiment are from nine subjects, means ± 1 SEM.
Cumulative PAS1 and PAS2 effects on MEP amplitudes (comparison of time points P1-P6 vs. B0)
The two-way rmANOVA for the cumulative PAS1 and PAS2 effect on MEP amplitudes at time points P1–P6 (MEPPi/MEPB0, i = 1,2,…,6) showed a significant effect of IPI (F 3,33 = 11.36, P < 0.0001) and TIME (F 5,55 = 2.49, P = 0.042), whereas the interaction IPI with TIME was not significant (F 15,165 = 0.66, P = 0.82) (Fig 3C and 3D). MEPPmean/MEPB0 was > 1.0 for IPI10 (1.51 ± 0.14; t = 3.61, P = 0.0041), IPI30 (2.04 ± 0.23; t = 4.54, P = 0.0009), and IPI60 (1.28 ± 0.09; t = 2.98, P = 0.013), but not for IPI180 (1.12 ± 0.12; t = 0.97, P = 0.35). Post hoc testing showed significantly higher MEP amplitudes after PAS2 (MEPPmean/MEPB0) for IPI30 than for IPI60 (P = 0.009) and IPI180 (P = 0.01), and a trend for higher MEP amplitudes for IPI30 vs. IPI10 (P = 0.095). Notably, at IPI30 the MEP amplitude increase 30 minutes after PAS2 normalized to baseline (MEPP6/MEPB0 = 2.22 ± 0.25; Fig 3D) was significantly higher than the MEP amplitude increase 30 minutes after PAS1 in this condition (MEPB2/MEPB0 = 1.51 ± 0.12; P = 0.018, paired two-tailed t test), indicating that consecutive application of PAS1 and PAS2 at IPI30 induced significantly higher MEP increases than application of PAS1 alone.
Modelling of PAS1 and PAS2 effects on MEP amplitudes
Computational modelling of the PAS1 and PAS2 effects on the absolute MEP amplitude raw data as a function of time revealed highly significant differences between PAS1 and PAS2 MEP functions (P < 0.0001) (Fig 5). Post hoc testing showed significant differences between the PAS1 and PAS2 model functions for all IPI conditions (P < 0.0001 each), suggesting that PAS2 effects on MEP amplitudes were modulated by prior application of PAS1 in all IPI conditions. Specifically, at IPI10 PAS2 prolonged the MEP increase induced by PAS1. At IPI30, PAS2 induced an extra MEP increase, notably with a longer time course as compared to the PAS1-induced MEP increase. In contrast, at IPI60 and IPI180 there was no significant change in MEP amplitudes after PAS2.
Fig 5. Computational modeling of the time course of PAS1 and PAS2 effects.

MEP amplitudes as a function of time modeled from the experimental MEP raw data independently for PAS1 and PAS2 at all IPIs. PAS1 and PAS2 model functions, i.e. the time course of MEP amplitude changes after PAS1 vs. those after PAS2, were significantly different at all IPIs (P < 0.0001 each). In addition, PAS2 effects at IPI10 and IPI30 were significantly different from PAS2 effects at IPI60 and IPI180 (P < 0.0001), and PAS2 effects at IPI10 from PAS2 effects at IPI30 (P = 0.033), but not PAS2 effects at IPI60 from those at IPI180 (P > 0.5). Experimental data are shown as mean ± SEM. Note that data for PAS2 at time points 60min and 180min post PAS2 are from six subjects only, whereas all other data are from twelve subjects (see Material and Methods). Y-axis, logarithmic scaling.
In addition, the time courses of PAS2-induced MEP amplitude changes were significantly different between IPI conditions (P < 0.0001). Post hoc analysis showed that PAS2 effects at IPI10 and IPI30 were significantly different from PAS2 effects at IPI60 and IPI180 (all P < 0.0001), and PAS2 effects at IPI10 were significantly different from PAS2 effects at IPI30 (P = 0.033), whereas the comparison of PAS2 effects at IPI60 and IPI180 showed no significant differences (P > 0.5). These differences are explained by a prolonged MEP increase at IPI30 compared to all other conditions (Fig 5).
Discussion
We showed here that LTP-like plasticity can be augmented in human M1 when two consecutive PASLTP protocols are spaced by 30min. In contrast, at longer intervals (60-180min) we found a suppressive interaction between two consecutive PASLTP protocols. These findings support the notion of non-homeostatic and homeostatic metaplasticity, respectively, and will be discussed in detail below.
The PAS1-induced MEP increase lasted for 30-60min, in accord with the literature, and represents a form of plasticity resembling LTP as studied at the cellular level [5, 7, 9, 35]. In contrast, the effects of PAS2 on MEP amplitude depended critically on the interval to PAS1. Computational modelling of the time courses of MEP amplitude (Fig 5) revealed significant differences between PAS1 and PAS2 effects on MEP amplitude for all IPI conditions, indicating that priming M1 by PAS1 modulated the effects of a subsequent identical PAS2 protocol for at least three hours.
Metaplasticity constitutes a higher-order form of plasticity, which regulates the magnitude and duration of synaptic plasticity in an activity-dependent manner [12]. Importantly, it modulates the plasticity state of neurons and networks, i.e. the induction of LTP subsequent to the priming stimulation, in the absence of synaptic plasticity induced by the priming stimulation itself. Here, we demonstrated a homeostatic interaction between PAS1 and PAS2 at IPI60 and IPI180 (Fig 3) in the absence of any persistent PAS1-induced MEP increase at the time of PAS2 (Fig 2). Further, both magnitude and/or duration of PAS2-induced LTP-like plasticity were modulated by PAS1 (Fig 5). Thus, these findings support the notion that the interactions between PAS1 and PAS2 effects described in the present study represent a form of metaplasticity similar to the ones reported at the cellular level [12, 36].
Previous studies have reported on metaplasticity between two consecutive identical non-invasive brain stimulation protocols in the human M1 [10, 20–24, 37–40]. The predominant interaction was homeostatic metaplasticity, but several occasions of non-homeostatic metaplasticity have also been reported, predominantly at short IPIs of 3-20min (for review, [25]). This non-homeostatic metaplasticity resulted in late LTD- and LTP-like changes in corticospinal excitability that were prevented by pharmacological blockade of NMDA receptors by dextromethorphan [24] and that were resistant to de-depression interventions such as voluntary contraction of the target muscle [22]. These data are compatible with the finding in the present study that two consecutive PASLTP protocols, if spaced by 30min, resulted in non-homeostatic augmentation of LTP-like plasticity with a prolonged duration.
These time-dependent interactions between PAS1 and PAS2 may be explained in the framework of a cascade model of synaptic plasticity [41]. As synapses are modified during the course of LTP, they change between discrete mechanistic states [42–44]. For example, silent synapses, i.e. synapses containing NMDARs, but being devoid of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) move to a “recently silent state” (i.e. with inserted AMPARs), in which they initially cannot be further potentiated. Additional potentiation is possible only if they move to the “active state”, which occurs at around 30min after LTP induction [42, 45]. Recent results from organotypic entorhino-hippocampal slice cultures suggest that repetitive magnetic stimulation indeed may lead to an activation of silent synapses [46]. As PAS1 and PAS2 were identical in our study and thus presumably modified the same set of synapses, such a cascade model of synaptic plasticity states may explain why PAS2 induced significant LTP-like plasticity at IPI30, but not at IPI10. In addition, the MEP increase after PAS2 at IPI30 may have been enabled by LTP-induced suppression of subsequent LTD. In rat hippocampus NMDAR-dependent LTP inhibits GSK3β, resulting in a ~60 minutes lasting blockade of subsequent NMDAR-dependent LTD induction [17]. In contrast, the suppressive interactions between PAS1 and PAS2 at IPI60 and IPI180 are in line with homeostatic metaplasticity [12]. Evidence in support of the notion of a delayed onset of homeostatic metaplasticity comes from animal experiments that showed that an experience-dependent increase in the NR2A/NR2B NMDAR subunit ratio, which is associated with a rightward shift of the synaptic modification threshold that favors induction of LTD over LTP [47, 48], did not occur within the first ~60min after a priming protocol [49].
An alternative explanation for the augmenting PAS2 effect on MEPs at IPI30 may have been gating [50] by the increased level of cortical excitability immediately prior to PAS2 in this condition (time point B2, Fig 2). However, this explanation is rather unlikely, as (i) computational modelling of MEP time functions after PAS2 showed significant differences between conditions IPI10 and IPI30 (cf. Fig 5), although the PAS1-induced MEP increases immediately before PAS2 were comparable in these two conditions (cf. Fig 2), and (ii) a control experiment (IPI30adj), in which we readjusted MEP amplitudes after PAS1 immediately before PAS2 to match baseline values, found similar PAS2 effects as in the main experiment (IPI30), in which MEP amplitudes were not readjusted (Fig 4).
At IPI30, PAS2 induced significant MEP increases over and above those induced by PAS1. The cumulative PAS1 and PAS2 increase of MEP amplitude at IPI30 significantly exceeded the PAS1 effect alone, and the cumulative effects of PAS1 and PAS2 at IPI60 and IPI180. These results show that LTP-like plasticity can be augmented in human M1, if consecutive PASLTP protocols are properly spaced within a window of non-homeostatic metaplasticity.
In contrast, homeostatic metaplasticity, i.e. the suppressive interaction between consecutive PASLTP protocols at longer intervals (IPI60 and IPI180), may prevent runaway LTP, enabling the human motor network to maintain its modifiability within a useful dynamic range. In line with this notion, dysfunctional homeostatic metaplasticity can result in a deficient control of activity-dependent plasticity, as seems to be the case in task-dependent dystonia [51, 52]. The mechanisms of homeostatic metaplasticity at IPI60 and IPI180 are as of yet unclear, but may involve Ca2+ influx through voltage-gated Ca2+ channels [53], or intracellular Ca2+ stores [54].
Studies in experimental animals have provided evidence, that an mGluR-dependent form of LTP is retained in the context of NMDAR-dependent increased synaptic strength [16, 55]. Thus, continued experience leads to increased synaptic strength over time, although NMDAR-dependent LTP is occluded during continued neural activity. It could be speculated that LTP-like plasticity induced by PAS1 vs. PAS2 at IPI30 in our experiments was due to different underlying physiological mechanisms, i.e. an NMDAR- and an mGluR-dependent form of LTP-like plasticity, respectively. This idea is indirectly supported by the following observations: (i) LTP-like plasticity induced by a single PASLTP protocol can be blocked by an NMDAR antagonist [7]; (ii) the PAS1-induced LTP-like plasticity in the present study was likely saturated as the MEP amplitude increase of ≥1.5 is among the highest reported in the literature [30, 33, 56]; (iii) the time-course of PAS1- vs. PAS2-induced MEP changes at IPI30 in the present study was significantly different (cf. Fig 5). Thus, properly timed consecutive application of non-invasive brain stimulation protocols such as PASLTP may lead to augmentation of LTP-like plasticity through recruitment of different physiological mechanisms. Regardless of the underlying mechanisms, our results provide experimental evidence that consecutive applications of PASLTP can lead to significantly increased LTP-like plasticity as compared to a single PASLTP session.
At first sight, the present data are at variance with those from one previous study of our group where we found homeostatic metaplasticity between two consecutive PASLTP protocols if spaced by 30min [10]. However, the two studies differ in one important aspect: in this but not the previous study, we deliberately maximized LTP-like plasticity induced by PASLTP by including only those data with a PAS1-induced MEP increase ≥1.1, and by ensuring high attention towards the stimulated hand, which is known to facilitate PASLTP-induced LTP-like plasticity [33]. As a result, the PAS1–induced MEP increase in this study (time point B1, 1.49 ± 0.10, condition IPI30) was significantly higher than in the previous study (1.14 ± 0.12; t = 2.24, P = 0.036; unpaired two-tailed t test). Previous work in mice showed that the expression of non-homeostatic metaplasticity critically depended on the level of the priming activity: only during strong but not weak stimuli, potentiated synapses could be further potentiated, specifically by induction of a switch in NMDAR and mGluR properties [16, 55]. Thus, the significantly stronger LTP-like plasticity induced by PAS1 in this compared to our previous study [10] may explain why we observed augmentation of LTP-like plasticity in the present study only.
In summary, the present study demonstrated that LTP-like plasticity in the human M1 can be augmented by application of two consecutive identical PASLTP protocols if spaced by 30min, while homeostatic interaction occurred at intervals of 60-180min. These findings may inspire further research to optimize therapeutic applications of non-invasive brain stimulation in patients with neurological or psychiatric diseases to modify synaptic transmission in their disordered brain networks more effectively than hitherto possible.
Acknowledgments
We thank Prof. G.W. Thickbroom and Dr. P. Jedlicka for helpful comments on earlier versions of this manuscript.
Data Availability
All relevant data are within the paper.
Funding Statement
The authors received no specific funding for this work.
References
- 1. Cooke SF, Bliss TV. Plasticity in the human central nervous system. Brain. 2006;129(Pt 7):1659–73. . [DOI] [PubMed] [Google Scholar]
- 2. Murphy TH, Corbett D. Plasticity during stroke recovery: from synapse to behaviour. Nature reviews Neuroscience. 2009;10(12):861–72. 10.1038/nrn2735 . [DOI] [PubMed] [Google Scholar]
- 3. Rioult-Pedotti MS, Friedman D, Donoghue JP. Learning-induced LTP in neocortex. Science. 2000;290(5491):533–6. [DOI] [PubMed] [Google Scholar]
- 4. Rosenkranz K, Kacar A, Rothwell JC. Differential modulation of motor cortical plasticity and excitability in early and late phases of human motor learning. J Neurosci. 2007;27(44):12058–66. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stefan K, Kunesch E, Cohen LG, Benecke R, Classen J. Induction of plasticity in the human motor cortex by paired associative stimulation. Brain. 2000;123(Pt 3):572–84. [DOI] [PubMed] [Google Scholar]
- 6. Ziemann U, Ilic TV, Pauli C, Meintzschel F, Ruge D. Learning modifies subsequent induction of LTP-like and LTD-like plasticity in human motor cortex. J Neurosci. 2004;24(7):1666–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stefan K, Kunesch E, Benecke R, Cohen LG, Classen J. Mechanisms of enhancement of human motor cortex excitability induced by interventional paired associative stimulation. The Journal of physiology. 2002;543(Pt 2):699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stefan K, Wycislo M, Gentner R, Schramm A, Naumann M, Reiners K, et al. Temporary Occlusion of Associative Motor Cortical Plasticity by Prior Dynamic Motor Training. Cereb Cortex. 2006;16(3):376–85. . [DOI] [PubMed] [Google Scholar]
- 9. Müller-Dahlhaus F, Ziemann U, Classen J. Plasticity resembling spike-timing dependent synaptic plasticity: the evidence in human cortex. Front Syn Neurosci. 2010;2(34):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Müller JF, Orekhov Y, Liu Y, Ziemann U. Homeostatic plasticity in human motor cortex demonstrated by two consecutive sessions of paired associative stimulation. The European journal of neuroscience. 2007;25:3461–8. [DOI] [PubMed] [Google Scholar]
- 11. Pötter-Nerger M, Fischer S, Mastroeni C, Groppa S, Deuschl G, Volkmann J, et al. Inducing homeostatic-like plasticity in human motor cortex through converging cortico-cortical inputs. Journal of neurophysiology. 2009;102(6):3180–90. 10.1152/jn.91046.2008 [DOI] [PubMed] [Google Scholar]
- 12. Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nature reviews Neuroscience. 2008;9(5):387–99. 10.1038/nrn2356 [DOI] [PubMed] [Google Scholar]
- 13. Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends in neurosciences. 1996;19(4):126–30. . [DOI] [PubMed] [Google Scholar]
- 14. Bienenstock EL, Cooper LN, Munro PW. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci. 1982;2(1):32–48. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hulme SR, Jones OD, Abraham WC. Emerging roles of metaplasticity in behaviour and disease. Trends in neurosciences. 2013;36(6):353–62. Epub 2013/04/23. 10.1016/j.tins.2013.03.007 . [DOI] [PubMed] [Google Scholar]
- 16. Clem RL, Celikel T, Barth AL. Ongoing in Vivo Experience Triggers Synaptic Metaplasticity in the Neocortex. Science. 2008;319:101–4. 10.1126/science.1143808 [DOI] [PubMed] [Google Scholar]
- 17. Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron. 2007;53(5):703–17. . [DOI] [PubMed] [Google Scholar]
- 18. Teo JT, Swayne OB, Cheeran B, Greenwood RJ, Rothwell JC. Human Theta Burst Stimulation Enhances Subsequent Motor Learning and Increases Performance Variability. Cereb Cortex. 2011;21(7):1627–38. 10.1093/cercor/bhq231 [DOI] [PubMed] [Google Scholar]
- 19. Jung P, Ziemann U. Homeostatic and non-homeostatic modulation of learning in human motor cortex. J Neurosci. 2009;29:5597–604. 10.1523/JNEUROSCI.0222-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gamboa OL, Antal A, Laczo B, Moliadze V, Nitsche MA, Paulus W. Impact of repetitive theta burst stimulation on motor cortex excitability. Brain stimulation. 2011:in press. [DOI] [PubMed]
- 21. Goldsworthy MR, Pitcher JB, Ridding MC. The application of spaced theta burst protocols induces long-lasting neuroplastic changes in the human motor cortex. The European journal of neuroscience. 2012;35:125–34. 10.1111/j.1460-9568.2011.07924.x [DOI] [PubMed] [Google Scholar]
- 22.Goldsworthy MR, Müller-Dahlhaus F, Ridding MC, Ziemann U. Resistant Against De-depression: LTD-Like Plasticity in the Human Motor Cortex Induced by Spaced cTBS. Cereb Cortex. 2014. Epub 2014/02/04. 10.1093/cercor/bht353 . [DOI] [PubMed]
- 23. Monte-Silva K, Kuo MF, Liebetanz D, Paulus W, Nitsche MA. Shaping the optimal repetition interval for cathodal transcranial direct current stimulation (tDCS). Journal of neurophysiology. 2010;103(4):1735–40. 10.1152/jn.00924.2009 [DOI] [PubMed] [Google Scholar]
- 24. Monte-Silva K, Kuo MF, Hessenthaler S, Fresnoza S, Liebetanz D, Paulus W, et al. Induction of late LTP-like plasticity in the human motor cortex by repeated non-invasive brain stimulation. Brain stimulation. 2013;6(3):424–32. Epub 2012/06/15. 10.1016/j.brs.2012.04.011 . [DOI] [PubMed] [Google Scholar]
- 25. Müller-Dahlhaus F, Ziemann U. Metaplasticity in Human Cortex. The Neuroscientist: a review journal bringing neurobiology, neurology and psychiatry. 2015;21(2):185–202. 10.1177/1073858414526645 [DOI] [PubMed] [Google Scholar]
- 26. Keel JC, Smith MJ, Wassermann EM. A safety screening questionnaire for transcranial magnetic stimulation [letter]. Clin Neurophysiol. 2001;112:720 [DOI] [PubMed] [Google Scholar]
- 27. Müller-Dahlhaus JF, Orekhov Y, Liu Y, Ziemann U. Interindividual variability and age-dependency of motor cortical plasticity induced by paired associative stimulation. Experimental brain research Experimentelle Hirnforschung. 2008;187(3):467–75. 10.1007/s00221-008-1319-7 [DOI] [PubMed] [Google Scholar]
- 28. Heidegger T, Krakow K, Ziemann U. Effects of antiepileptic drugs on associative LTP-like plasticity in human motor cortex. The European journal of neuroscience. 2010;32:1215–22. 10.1111/j.1460-9568.2010.07375.x [DOI] [PubMed] [Google Scholar]
- 29. Oldfield RC. The assessment and analysis of handedness: the Edinburgh inventory. Neuropsychologia. 1971;9(1):97–113. [DOI] [PubMed] [Google Scholar]
- 30. Wolters A, Sandbrink F, Schlottmann A, Kunesch E, Stefan K, Cohen LG, et al. A temporally asymmetric Hebbian rule governing plasticity in the human motor cortex. Journal of neurophysiology. 2003;89(5):2339–45. [DOI] [PubMed] [Google Scholar]
- 31. Hamada M, Galea JM, Di Lazzaro V, Mazzone P, Ziemann U, Rothwell JC. Two distinct interneuron circuits in human motor cortex are linked to different subsets of physiological and behavioral plasticity. J Neurosci. 2014;34(38):12837–49. 10.1523/JNEUROSCI.1960-14.2014 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hamada M, Strigaro G, Murase N, Sadnicka A, Galea JM, Edwards MJ, et al. Cerebellar modulation of human associative plasticity. The Journal of physiology. 2012;590(10):2365–74. 10.1113/jphysiol.2012.230540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stefan K, Wycislo M, Classen J. Modulation of associative human motor cortical plasticity by attention. Journal of neurophysiology. 2004;92:66–72. . [DOI] [PubMed] [Google Scholar]
- 34. Fratello F, Veniero D, Curcio G, Ferrara M, Marzano C, Moroni F, et al. Modulation of corticospinal excitability by paired associative stimulation: reproducibility of effects and intraindividual reliability. Clin Neurophysiol. 2006;117(12):2667–74. . [DOI] [PubMed] [Google Scholar]
- 35. Ziemann U, Paulus W, Nitsche MA, Pascual-Leone A, Byblow WD, Berardelli A, et al. Consensus: Motor cortex plasticity protocols. Brain stimulation. 2008;1(3):164–82. 10.1016/j.brs.2008.06.006 [DOI] [PubMed] [Google Scholar]
- 36. Mockett BG, Hulme SR. Metaplasticity: new insights through electrophysiological investigations. Journal of integrative neuroscience. 2008;7(2):315–36. . [DOI] [PubMed] [Google Scholar]
- 37. Murakami T, Müller-Dahlhaus F, Lu MK, Ziemann U. Homeostatic Metaplasticity of Corticospinal Excitatory and intracortical Inhibitory Neural Circuits in Human Motor Cortex. The Journal of physiology. 2012;590(22):5765–81. Epub 2012/08/30. 10.1113/jphysiol.2012.238519 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mastroeni C, Bergmann TO, Rizzo V, Ritter C, Klein C, Pohlmann I, et al. Brain-derived neurotrophic factor—a major player in stimulation-induced homeostatic metaplasticity of human motor cortex? PloS one. 2013;8(2):e57957 Epub 2013/03/08. 10.1371/journal.pone.0057957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fricke K, Seeber AA, Thirugnanasambandam N, Paulus W, Nitsche MA, Rothwell JC. Time course of the induction of homeostatic plasticity generated by repeated transcranial direct current stimulation (tDCS) of the human motor cortex. Journal of neurophysiology. 2011;105(3):1141–9. 10.1152/jn.00608.2009 [DOI] [PubMed] [Google Scholar]
- 40. Hamada M, Terao Y, Hanajima R, Shirota Y, Nakatani-Enomoto S, Furubayashi T, et al. Bidirectional long-term motor cortical plasticity and metaplasticity induced by quadripulse transcranial magnetic stimulation. The Journal of physiology. 2008;586(16):3927–47. 10.1113/jphysiol.2008.152793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fusi S, Drew PJ, Abbott LF. Cascade models of synaptically stored memories. Neuron. 2005;45(4):599–611. [DOI] [PubMed] [Google Scholar]
- 42. Montgomery JM, Madison DV. Discrete synaptic states define a major mechanism of synapse plasticity. Trends Neurosci. 2004;27(12):744–50. . [DOI] [PubMed] [Google Scholar]
- 43. Emond MR, Montgomery JM, Huggins ML, Hanson JE, Mao L, Huganir RL, et al. AMPA receptor subunits define properties of state-dependent synaptic plasticity. J Physiol. 2010;588(Pt 11):1929–46. 10.1113/jphysiol.2010.187229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee MC, Yasuda R, Ehlers MD. Metaplasticity at single glutamatergic synapses. Neuron. 2010;66(6):859–70. 10.1016/j.neuron.2010.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Montgomery JM, Madison DV. State-dependent heterogeneity in synaptic depression between pyramidal cell pairs. Neuron. 2002;33(5):765–77. . [DOI] [PubMed] [Google Scholar]
- 46. Vlachos A, Müller-Dahlhaus F, Rosskopp J, Lenz M, Ziemann U, Deller T. Repetitive magnetic stimulation induces functional and structural plasticity of excitatory postsynapses in mouse organotypic hippocampal slice cultures. J Neurosci. 2012;32(48):17514–23. Epub 2012/12/01. 10.1523/JNEUROSCI.0409-12.2012 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu Z, Chen R-Q, Gu Q-H, Yan J-Z, Wang SH, Liu S-Y, et al. Metaplastic Regulation of Long-Term Potentiation/Long-Term Depression Threshold by Activity-Dependent Changes of NR2A/NR2B Ratio. J Neurosci. 2009;29(27):8764–73. 10.1523/JNEUROSCI.1014-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Philpot BD, Cho KK, Bear MF. Obligatory Role of NR2A for Metaplasticity in Visual Cortex. Neuron. 2007;53(4):495–502. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Quinlan EM, Philpot BD, Huganir RL, Bear MF. Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nature neuroscience. 1999;2(4):352–7. . [DOI] [PubMed] [Google Scholar]
- 50. Ziemann U, Siebner H. Modifying motor learning through gating and homeostatic metaplasticity. Brain stimulation. 2008;1(1):60–6. 10.1016/j.brs.2007.08.003 [DOI] [PubMed] [Google Scholar]
- 51. Quartarone A, Rizzo V, Bagnato S, Morgante F, Sant'angelo A, Romano M, et al. Homeostatic-like plasticity of the primary motor hand area is impaired in focal hand dystonia. Brain. 2005;128:1943–50. . [DOI] [PubMed] [Google Scholar]
- 52. Kang J-S, Terranova C, Hilker R, Quartarone A, Ziemann U. Deficient homeostatic regulation of practice-dependent plasticity in writer’s cramp. Cereb Cortex. 2011;21(5):1203–12. 10.1093/cercor/bhq204 [DOI] [PubMed] [Google Scholar]
- 53. Wankerl K, Weise D, Gentner R, Rumpf JJ, Classen J. L-type voltage-gated Ca2+ channels: a single molecular switch for long-term potentiation/long-term depression-like plasticity and activity-dependent metaplasticity in humans. J Neurosci. 2010;30(18):6197–204. 10.1523/JNEUROSCI.4673-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Maggio N, Vlachos A. Synaptic plasticity at the interface of health and disease: New insights on the role of endoplasmic reticulum intracellular calcium stores. Neuroscience. 2014;281C:135–46. 10.1016/j.neuroscience.2014.09.041 . [DOI] [PubMed] [Google Scholar]
- 55. Cheyne JE, Montgomery JM. Plasticity-dependent changes in metabotropic glutamate receptor expression at excitatory hippocampal synapses. Molecular and cellular neurosciences. 2008;37(3):432–9. 10.1016/j.mcn.2007.10.015 . [DOI] [PubMed] [Google Scholar]
- 56. Nitsche MA, Roth A, Kuo M-F, Fischer AK, Liebetanz D, Lang N, et al. Timing-Dependent Modulation of Associative Plasticity by General Network Excitability in the Human Motor Cortex. J Neurosci. 2007;27(14):3807–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.