

Abstract
Background
The morphogenesis of single-celled cotton fiber includes extreme elongation and staged cell wall differentiation. Designing strategies for improving cotton fiber for textiles and other uses relies on uncovering the related regulatory mechanisms. In this research we compared the transcriptomes and metabolomes of two Gossypium genotypes, Gossypium barbadense cv Phytogen 800 and G. hirsutum cv Deltapine 90. When grown in parallel, the two types of fiber developed similarly except for prolonged fiber elongation in the G. barbadense cultivar. The data were collected from isolated fibers between 10 to 28 days post anthesis (DPA) representing: primary wall synthesis to support elongation; transitional cell wall remodeling; and secondary wall cellulose synthesis, which was accompanied by continuing elongation only in G. barbadense fiber.
Results
Of 206 identified fiber metabolites, 205 were held in common between the two genotypes. Approximately 38,000 transcripts were expressed in the fiber of each genotype, and these were mapped to the reference set and interpreted by homology to known genes. The developmental changes in the transcriptomes and the metabolomes were compared within and across genotypes with several novel implications. Transitional cell wall remodeling is a distinct stable developmental stage lasting at least four days (18 to 21 DPA). Expression of selected cell wall related transcripts was similar between genotypes, but cellulose synthase gene expression patterns were more complex than expected. Lignification was transcriptionally repressed in both genotypes. Oxidative stress was lower in the fiber of G. barbadense cv Phytogen 800 as compared to G. hirsutum cv Deltapine 90. Correspondingly, the G. barbadense cultivar had enhanced capacity for management of reactive oxygen species during its prolonged elongation period, as indicated by a 138-fold increase in ascorbate concentration at 28 DPA.
Conclusions
The parallel data on deep-sequencing transcriptomics and non-targeted metabolomics for two genotypes of single-celled cotton fiber showed that a discrete developmental stage of transitional cell wall remodeling occurs before secondary wall cellulose synthesis begins. The data showed how lignification can be transcriptionally repressed during secondary cell wall synthesis, and they implicated enhanced capacity to manage reactive oxygen species through the ascorbate-glutathione cycle as a positive contributor to fiber length.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1708-9) contains supplementary material, which is available to authorized users.
Keywords: Ascorbate, Cell elongation, Cell wall synthesis, Cotton fiber development, Gossypium, Lignification, Reactive oxygen species, Metabolomics, RNA Seq transcriptomics
Background
Cotton fibers are single cells that elongate from the epidermis of the cotton ovule beginning near the day of anthesis. Cotton fiber morphogenesis includes initiation, elongation via primary cell wall synthesis, transitional cell wall remodeling, secondary cell wall (SCW) synthesis, and maturation. The fibers used for yarn and textile manufacturing contain 90 to 95 % cellulose in a secondary wall surrounded by a thin cuticulated primary wall. Cell wall differentiation is a major driver of cotton fiber morphogenesis and the final quality of cotton fiber [1]. The staged development of this single cell offers unprecedented opportunities to use comprehensive transcriptomics and metabolomics to gain insight into the molecular and biochemical regulation of cell elongation, cell wall synthesis, and fiber quality.
To explore the mechanisms regulating cotton fiber morphogenesis and quality, we analyzed two genotypes of modern commercial cotton with higher and lower fiber quality. Specifically, we analyzed a modern cultivar from each of the two allotetraploid (AD genome) species, Gossypium hirsutum (Gh; AD1) and Gossypium barbadense (Gb; AD2). The commercial allotetraploids derived from a single polyploidization event about 1 to 2 million years ago involving two diploid ancestors similar to extant G. raimondii (D genome) and G. arboreum/G. herbaceum (A genome). Both A and D genome diploid ancestral cottons had seed hairs (cotton fiber), with those in extant wild A genome species most closely resembling AD commercial fiber [2]. Independent natural selection and domestication trajectories from one ancestral allotetraploid led to modern Gh and Gb cotton, which has superior fiber properties [3, 4]. Gh (or ‘Upland’ cotton) is predominantly grown for its high yield in more varied environments, whereas Gb (or ‘Pima’ cotton) is grown to provide longer, stronger, and finer fiber for the manufacture of higher quality textiles [5].
Several studies have compared paired accessions of Gb and Gh fibers to look for the controls of cotton fiber quality. These studies have uncovered differences related to the biosynthesis of flavonoids, lignin-related phenolics, cell wall molecules, and proteins, as well as protein degradation [6–9]. Considered together, these comparisons included variable times between 5 to 34 DPA for fiber from different cultivars and growing conditions, both of which can shift the timing of fiber development and comparative fiber quality. In addition, these prior transcriptomic comparisons were based on 454 sequencing and microarray technology, creating a need for thorough comparative analysis using ultra-deep Illumina sequencing. Limited metabolite comparisons had been done before, showing higher concentrations of flavonoids including naringenin and dihydrokaempherol in 5 and 10 DPA Gh fiber as compared to Gb fiber [9].
Here we report transcriptomics based on Illumina deep sequencing and non-targeted metabolomics on the same fiber samples of two genotypes at 10, 15, 18, 21, and 28 DPA. The cultivars analyzed here were Gh cv Deltapine 90 and Gb cv Phytogen 800. They exhibit the expected fiber quality differences when grown in parallel under controlled growth conditions and have well characterized fiber developmental profiles [10]. Specifically, the fibers of both genotypes had similar: (a) rates of elongation between 10 to 22 DPA; (b) timing of transitional cell wall remodeling (17 to 21 DPA); and (c) timing of the onset (22 DPA) and termination (45 DPA) of SCW thickening. At 28 DPA, SCW cellulose synthesis was ongoing in both fiber genotypes, but late elongation was continuing only in Gb fiber. Using this background knowledge, we were able to compare the data from Gh and Gb fiber at each DPA in a highly similar physiological context while also looking for potential regulators of the sustained elongation of Gb fiber. Below, we briefly review the context for the major results.
Molecular regulation of cell wall synthesis
Cell walls compose our most abundant renewable biomaterials, so the regulation of their synthesis is of general interest. The staged development of single-celled cotton fiber can reveal regulatory mechanisms with less ambiguity than for other cell types that are differentiating within complex tissues. Elongating cotton fiber has typical primary walls [11]. Near the beginning of secondary wall synthesis, a thin ‘winding’ cell wall layer is deposited that contributes substantially to cotton fiber strength [12–14]. Most fiber thickening depends on the synthesis of a SCW composed of 90–95 % cellulose, and this occurs through use of a genetic and biochemical program similar to sclerenchyma cells [15]. As shown by numerous studies, genes encoding cell wall polymer synthases, structural proteins, and regulators are abundant within the cotton fiber transcriptome [6, 8, 16]. Here we focused our comparative analyses on genes encoding: (a) cellulose synthases (CESAs), which support the synthesis of strong cellulose fibrils within both primary and secondary walls [17]; (b) cellulose-synthase-like enzymes, which generate the backbones of diverse cell wall matrix polymers [18]; (c) transcription factors that regulate the onset of SCW synthesis in sclerenchyma cells [19, 20]; and (d) enzymes within the flavonoid and lignin biosynthetic pathways [21, 22].
Defining the transition between primary and secondary cell wall synthesis
Transitional cell wall layers between the primary and secondary wall are increasingly recognized as important for determining the mechanical properties of cotton fiber and wood [13, 14, 23]. A ‘transition stage’ occurs at the onset of cotton fiber secondary wall synthesis, as reviewed previously [1]. Metabolic and cellular changes at this time include a transient reduction in fiber respiration rate and lowering of glucose and fructose concentrations [24]. The ‘winding’ cell wall layer is deposited with a polymer composition similar to the primary wall. This thin transitional layer is synthesized while the content of crystalline cellulose in the fiber cell wall increases to about 25 % and both microtubules and cellulose fibrils shift to shallow helical orientations [11, 12]. The cotton fiber middle lamella (CFML), which previously joined the fibers together during earlier elongation, is degraded in parallel with changes in the size and composition of cell wall matrix polysaccharides [10, 11, 25–28].
Management of reactive oxygen species
Reactive oxygen species (ROS) including singlet oxygen, superoxide, hydrogen peroxide (H2O2), and hydroxyl radicals are generated as normal byproducts of aerobic respiration and also act as signals in development, defense, and stress responses [29]. ROS have several roles in cotton fiber development. Fiber initiation was increased by exogenous H2O2 in a fuzzless mutant [30]. Early cotton fiber elongation requires high levels of superoxide [31]. Similarly, adding H2O2 to 5 DPA cotton ovule cultures stimulated fiber elongation, coincident with the potential for maintaining ROS homeostasis through the increased expression and activity of cytosolic ascorbate peroxidase (APX) [32]. Over-expressing chloroplast-targeted pea APX was associated with increased fiber length in one Gh cotton line compared to its segregating null line in field experiments, but there was no difference compared to the wild-type line and no rationale provided for an effect on fiber [33]. The transition to SCW synthesis depends on a spike in H2O2 [34, 35], with potential downstream effects including the dimerization of cellulose synthases [36]. Several transcriptomic studies have implicated ROS management in the improvement of fiber quality during domestication [37–39], and relevant enzymes have been identified through proteomics [40]. For example, certain isoforms of dehydroascorbate reductase (DHAR) and APX were more abundant in domesticated Gh with longer fiber compared to its wild progenitor [41].
Despite their developmental roles, prolonged exposure to ROS in high concentrations can damage cells, causing oxidation of lipids and proteins and inducing programmed cell death in extreme cases [29, 42]. Sulfur-containing cysteine or methionine residues are commonly reversibly oxidized, and irreversible carbonyl derivatives of other amino acids can be produced. Either process can change protein conformation and function, along with activation of proteolysis or autophagy to remove damaged proteins [42–44]. Therefore, cells use chemical and enzymatic processes to control ROS concentrations. Common ROS-scavenging chemical reductants include: ascorbate (Vitamin C), glutathione, tocopherol, carotenoids, and phenolic compounds such as flavonoids [29, 45, 46]. The ascorbate-glutathione cycle is a major contributor to enzymatic ROS scavenging in plants [29], and transgenic experiments support similar effects in cotton fiber. Although the mechanism was not completely understood due to targeting of transgenes to the plastid, the fineness (mass per unit length) and bundle strength of Gh cotton fiber were increased through over-expression of APX or glutathione reductase (GSR, also abbreviated GR) [33]. Differences in ROS management in Gh and Gb cotton fibers will be discussed later.
Overview of the experimental approach and major results
We analyzed two cultivars of Gb and Gh fiber grown in parallel at 10 and 15 DPA (during rapid primary wall synthesis supporting elongation), 18 and 21 DPA (during transitional cell wall remodeling), and 28 DPA (during SCW cellulose synthesis, plus the last stage of elongation in Gb fiber). The metabolome was analyzed in fiber extracts using ion-optimized mass spectrometry and gas chromatography to reveal 206 total compounds, with 205 of them present in the fiber of both genotypes, and no unidentified compounds. For transcriptomic analysis, more than 2 billion high quality, 100-bp, paired-end reads were generated on an Illumina HiSeq2000 and mapped to a reference set derived from D-genome diploid cotton, G. raimondii (Gr; [47]) and expressed sequence tags from A-genome G. arboreum (Ga; [48]).
The transcriptomes and metabolomes were compared within the two genotypes across DPA and between genotypes at the same DPA to reach several novel conclusions. Previously, we highlighted how the well-characterized fiber development profiles in the two cultivars analyzed here allow us to meaningfully interpret the data. Major conclusions include: defining a discrete stage of transitional cell wall remodeling; showing how cotton fiber has modified an ancient transcription factor network regulating SCW synthesis to repress lignification; and providing a biochemical pathway perspective on ROS management in cotton fiber that reveals differences leading to less oxidative stress in a longer form of cotton fiber. This difference in ROS management, along with sustained transcription of elongation-associated genes, correlated with the prolonged elongation period of Gb cv Phytogen 800 fiber as compared to Gh cv Deltapine 90 fiber. Collectively, the data provide clues about future testable strategies for increasing fiber length in short-fibered Gossypium genotypes. We reanalyzed the microarray data from another pair of Gh and Gb cultivars [7], finding consistency with our data. Nonetheless, in this paper we use the abbreviations Gh and Gb for the concise indication of the genotypes studied without implying any generalization of the results to other accessions in the two species. We anticipate that the data will stimulate further tests of these findings in cotton diversity panels and through various biotechnology and breeding strategies.
Results and discussion
Overall transcriptomic and metabolic outcomes
Illumina sequencing of the transcriptomes of 10, 15, 18, 21, and 28 DPA Gh and Gb fiber yielded 2,015,378,704 high quality, multiplexed, 100-bp, paired end reads with quality scores ≥ 33 (deposited at NCBI Sequence Read Archive (SRA); study project number SRP049330). Nearly all (98 %) of the 77,267 transcripts in the Gr genome-scale reference set were detected. Not all of these transcripts are likely to be biologically relevant to cotton fiber development, so we analyzed only 41,566 total unique transcripts from the fiber of both genotypes with reads per kilobase per million reads mapped (RPKM) values ≥ 2. These represented 42 % of the total detected transcripts (Additional files 1 and 2). The number of transcripts expressed in Gb or Gh fiber (37,936 or 38,337) was nearly the same.
Fiber metabolites from the same samples were profiled by gas and liquid chromatography followed by mass spectrometry and identified by reference to approximately 3000 standards. All signals could be matched to standards and 205 of 206 total metabolites were identified in the fiber of both genotypes. One dipeptide (gamma-glutamylglutamate) was identified only in Gb fiber. Of the 206 total metabolites, 152 were curated in the KEGG database (www.genome.jp/kegg/) (Additional file 3).
Excluding seven broad pathways, between 1–20 metabolites fell into 73 more specific KEGG pathways (Additional file 4). Fifteen pathways (inclusive of 69 metabolites) were overrepresented in cotton fiber based on the number of metabolites in the pathway (Table 1), reflecting a strong demand for energy, amino acids, and carbohydrates. The sugar-related pathway, ‘galactose metabolism’ was populated by 11 metabolites: glucose, glucose-6-phosphate, UDP-glucose, fructose, sucrose, UDP-galactose, galactinol, glycerol, myo-inositol, raffinose, and stachyose. These represent precursors of carbohydrate and cell wall synthesis as well as end products (raffinose and stachyose oligosaccharides). The ‘glutathione metabolism’ pathway related to amino acid metabolism and ROS management was populated by 10 metabolites: glutamate, glycine, reduced and oxidized glutathione, ascorbate, cysteine, putrescine, spermidine, 5-oxoproline, and dehydroascorbate. These key aspects of cotton fiber metabolism will be discussed further below.
Table 1.
Overrepresented KEGG pathways in cotton fiber implicated by metabolites or enzyme-encoding transcripts. Entries reflect the combined data from Gh and Gb fiber
| Metabolome | Transcriptome |
|---|---|
| Alanine, aspartate and glutamate metabolism | Alanine, aspartate and glutamate metabolism |
| Aminoacyl-tRNA biosynthesis | Aminoacyl-tRNA biosynthesis |
| Ascorbate and aldarate metabolism | Carbon fixation in photosynthetic organisms |
| beta-Alanine metabolism | Citrate cycle (TCA cycle) |
| Butanoate metabolism | Cysteine and methionine metabolism |
| Citrate cycle (TCA cycle) | Flavonoid biosynthesis |
| Cyanoamino acid metabolism | Glycerolipid metabolism |
| Galactose metabolism | Glycolysis/Gluconeogenesis |
| Glutathione metabolism | One carbon pool by folate |
| Glycine, serine and threonine metabolism | Other glycan degradation |
| Glyoxylate and dicarboxylate metabolism | Oxidative phosphorylation |
| Oxidative phosphorylation | Phenylalanine, tyrosine, tryptophan biosynthesis |
| Pantothenate and CoA biosynthesis | Phosphatidylinositol signaling system |
| Taurine and hypotaurine metabolism | Purine metabolism |
| Valine, leucine and isoleucine biosynthesis | Starch and sucrose metabolism |
| Valine, leucine and isoleucine biosynthesis |
The 41,566 transcripts with RPKM ≥ 2 were analyzed with Blast2GO to identify homologs of 913 unique known enzymes (1E−10, Additional file 5), of which 692 mapped to KEGG pathways (Additional file 6). As inferred from transcripts, sixteen pathways were potentially overrepresented (Table 1, Additional file 7), and five of these related to energy and amino acids were also overrepresented in the metabolome: alanine aspartate and glutamate metabolism; aminoacyl-tRNA biosynthesis; citrate (TCA) cycle; oxidative phosphorylation; and valine, leucine, and isoleucine biosynthesis. Other pathways overrepresented by transcripts emphasize the importance of carbohydrates in cotton fiber, as well as pointing to more specific processes such as flavonoid biosynthesis (also potentially related to ROS scavenging), glycerolipid metabolism, and phosphatidylinositol signaling.
Overview of changes in the transcriptome in 10 to 28 DPA cotton fiber
We analyzed the changes in transcripts across the time-course of fiber development within one genotype and at the same DPA across genotypes (Fig. 1). Notably, about 88 % of the transcripts were expressed at 10 DPA in Gh or Gb fiber (33,310 or 33,837 transcripts, respectively), with similar expression levels maintained through 21 DPA. Some of these are alternative transcripts from the same locus, and the 10 DPA unique transcripts are homologous to 14,006 or 14,344 (37–38 %) of the 37,505 protein-coding loci in Gr D-genome diploid cotton. The largest change in gene expression for the fiber of both genotypes occurred between 21 to 28 DPA, representing the shift to predominantly SCW cellulose synthesis. By reference to 21 DPA, more transcripts were downregulated at 28 DPA (11,395 or 11,588 for Gh or Gb) than were upregulated (2971 or 3335 for Gh or Gb). This resulted in 23,834 or 24,297 transcripts expressed in 28 DPA Gb or Gh fiber (Additional files 1 and 2). Comparing 10 and 28 DPA, representing the dominance of primary vs secondary wall synthesis, similarly showed 15,074–15,881 downregulated transcripts at 28 DPA as compared to 4,125–4,149 upregulated transcripts. The GO terms that were overrepresented by the transcripts of both genotypes at 28 DPA compared to 21 DPA include: SCW biogenesis, cellulose microfibril organization, and numerous carbohydrate terms (sucrose, fructose, xylose, mannose, galactose, starch, trehalose). In addition, the overrepresented GO terms reflected the regulation of a major developmental shift: activation of MAPKK activity, negative regulation of ethylene-mediated signaling pathway, and transmembrane receptor protein serine/threonine kinase signaling pathway (Table 2). The homologous Gr gene IDs in each GO category can be found in Additional file 8.
Fig. 1.
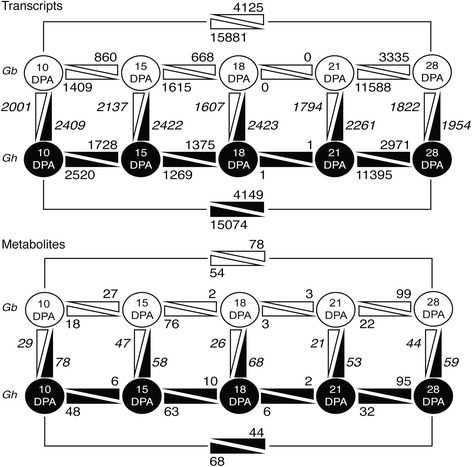
Quantitative changes in the transcriptome and metabolome within and between Gb and Gh fibers. Upper panel: the numbers of transcripts with at least 2 fold up- or downregulation (q ≤ 0.001). Lower panel: the numbers of metabolites with different concentrations (p ≤ 0.05). Each panel shows data for Gb fiber (top, white) and Gh fiber (bottom, black). On the horizontal axis, numbers are placed nearest the DPA when that number of transcripts or metabolites was higher. On the vertical axis, italicized numbers are placed nearest the genotype in which the transcripts or metabolites were higher on each DPA. The comparisons at 10 vs 28 DPA are also shown to highlight the contrast between primary and secondary wall synthesis, a comparison that is most distinct for Gh fiber that is no longer elongating at 28 DPA
Table 2.
Overrepresented Gene Ontology biological process terms in 28 DPA vs 21 DPA fiber. Transcripts that had higher expression (fold change ≥2) at 28 vs 21 DPA in both Gb and Gh fiber were analyzed for over-representation of GO biological process terms (p < 0.007, q < 0.05). False discovery rate q-values (FDR) were determined as in Table 1 (see p-values in Additional file 8)
| GO ID | GO Name | Gb-FDR | Gh-FDR |
|---|---|---|---|
| GO:0000186 | activation of MAPKK activity | 1.43E-04 | 8.60E-05 |
| GO:0010215 | cellulose microfibril organization | 4.17E-02 | 3.43E-02 |
| GO:0015996 | chlorophyll catabolic process | 2.05E-04 | 5.54E-05 |
| GO:0030206 | chondroitin sulfate biosynthetic process | 1.06E-05 | 6.00E-05 |
| GO:0035434 | copper ion transmembrane transport | 4.01E-05 | 6.06E-03 |
| GO:0010143 | cutin biosynthetic process | 8.11E-04 | 2.30E-05 |
| GO:0006535 | cysteine biosynthetic process from serine | 2.68E-04 | 7.06E-05 |
| GO:0006014 | D-ribose metabolic process | 1.43E-04 | 1.25E-04 |
| GO:0042732 | D-xylose metabolic process | 2.25E-03 | 2.61E-02 |
| GO:0006000 | fructose metabolic process | 2.82E-03 | 7.44E-07 |
| GO:0006012 | galactose metabolic process | 2.49E-05 | 3.06E-03 |
| GO:0009298 | GDP-mannose biosynthetic process | 2.23E-02 | 4.82E-02 |
| GO:0010417 | glucuronoxylan biosynthetic process | 5.97E-03 | 2.07E-02 |
| GO:0006424 | glutamyl-tRNA aminoacylation | 1.67E-03 | 1.60E-03 |
| GO:0019310 | inositol catabolic process | 4.01E-05 | 7.59E-03 |
| GO:0006559 | L-phenylalanine catabolic process | 2.25E-03 | 2.02E-03 |
| GO:0007142 | male meiosis II | 2.07E-02 | 2.01E-02 |
| GO:0019307 | mannose biosynthetic process | 5.97E-03 | 5.70E-03 |
| GO:0009556 | microsporogenesis | 2.07E-02 | 4.66E-04 |
| GO:0010105 | negative regulation of ethylene mediated signaling pathway | 2.06E-02 | 1.86E-02 |
| GO:0009878 | nodule morphogenesis | 1.67E-03 | 1.60E-03 |
| GO:0018106 | peptidyl-histidine phosphorylation | 8.84E-07 | 9.75E-05 |
| GO:0009915 | phloem sucrose loading | 2.07E-02 | 2.01E-02 |
| GO:0010205 | photoinhibition | 2.07E-02 | 2.01E-02 |
| GO:0010043 | response to zinc ion | 6.40E-05 | 6.51E-06 |
| GO:0009834 | secondary cell wall biogenesis | 7.21E-08 | 5.51E-07 |
| GO:0005982 | starch metabolic process | 6.95E-07 | 1.07E-05 |
| GO:0010345 | suberin biosynthetic process | 2.07E-02 | 5.70E-03 |
| GO:0005985 | sucrose metabolic process | 4.88E-10 | 6.86E-08 |
| GO:0007178 | transmembrane receptor protein serine/threonine kinase signaling pathway | 8.44E-05 | 2.37E-05 |
| GO:0005992 | trehalose biosynthetic process | 4.57E-04 | 2.16E-04 |
| GO:0006572 | tyrosine catabolic process | 1.43E-04 | 1.25E-04 |
Of ~38,000 non-redundant transcripts expressed across all DPA in each fiber genotype, approximately 94 % showed significantly different expression levels on at least one day between 10 to 28 DPA (q ≤0.001), and of these approximately 14 % showed fold change ≥ 2 (Additional files 9 and 10). Six major differential expression patterns were identified by use of Fuzzy C-means clustering [49] (Fig. 2). The color scale indicates the confidence level for cluster membership, with red to magenta indicating ≥ 70 % confidence in cluster assignment. The similarity of clusters for Gh and Gb fiber corresponds to many similarities in their fiber developmental programs [10]. The number of genes in each cluster ranged from 2,463 (Gb cluster e) to 9,370 (Gb cluster b), with only 3–9.6 % difference in the number of Gh or Gb genes in the paired clusters. The gene expression clusters correspond to known aspects of fiber development as interpreted below by reference to selected GO biological processes or genes overrepresented in both fiber genotypes (Additional file 11).
Fig. 2.
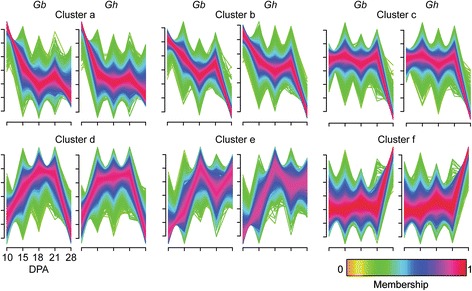
Gb and Gh fiber showed six similar clusters of gene expression patterns. All transcripts differentially expressed during fiber development (q ≤ 0.001) were clustered. Each x-axis tick corresponds to expression level at 10, 15, 18, 21, or 28 DPA, with RPKM expression values (on the y-axis) standardized to mean = 0 and standard deviation = 1. Transcript patterns in each cluster are color coded by membership value, with higher membership value indicating a higher likelihood that a given transcript belongs in a cluster
Cluster ‘a-transcripts’ includes genes upregulated at 10 DPA near the beginning of high-rate fiber elongation. Reflecting rapid cell growth and related biosynthetic demands, the overrepresented GO terms include: positive regulation of cell size, reductive tricarboxylic acid cycle, pentose phosphate shunt, fatty acid biosynthesis, and numerous terms related to amino acids. Cluster ‘b-transcripts’ includes genes upregulated at 10 DPA with another lower level of upregulation at 21 DPA. Genes in this cluster may be needed to initiate high-rate primary and secondary wall synthesis, and the overrepresented GO terms include: cellulose microfibril organization; carbon utilization; xylem development; ribosome biogenesis, chaperone mediated protein folding, and regulation of translational initiation. Cluster ‘c-transcripts’ includes genes upregulated at 10 to 21 during fiber elongation before the onset of SCW synthesis. The overrepresented GO terms in both genotypes are less informative for this cluster, but shared individual genes reflect the control of fiber elongation by turgor pressure (cotton genes related to Arabidopsis PIP2 and Gorai.002G002500.1) and expansin proteins (cotton genes related to AtEXP8 and Gorai.011G128500.2). Cluster ‘d-transcripts’ includes genes upregulated at 15 to 21 DPA during the initiation and maintenance of transitional cell wall remodeling. The overrepresented GO terms include: detection of ethylene-mediated signaling; cell wall macromolecule catabolic process; (1-3)-beta-D-glucan biosynthetic process; and ROS-related terms. Cluster ‘e-transcripts’ includes genes upregulated at 18 to 28 DPA with slightly lower expression at 21 DPA, during synthesis of the winding layer and the cellulose-rich SCW. The overrepresented GO terms include: early endosome to late endosome transport and protein stabilization, both of which may relate to the transport and function of cell wall biosynthetic enzymes. Cluster ‘f-transcripts’ includes genes upregulated at 28 DPA, and the overrepresented GO terms include: SCW biogenesis, cellulose microfibril organization, and terms related to sucrose, glucan biosynthesis, and vesicle trafficking.
Differences in the transcriptome between Gh and Gb fibers
A total of 20,830 transcripts were differentially expressed (fold change ≥ 2) between Gh and Gb fiber, with approximately 2,000 transcripts showing changed expression at any one DPA as compared to the other genotype (Fig. 1, Additional files 12 and 13). Among these, there were 4,551 or 5,252 non-redundant differentially expressed transcripts in Gb or Gh fiber, and most of these were uniquely found in one fiber genotype. The transcripts with higher expression in Gb fiber mapped to 25 overrepresented GO terms (q ≤ 0.01), including 6 terms associated with protein biosynthesis. In contrast, the transcripts with higher expression in Gh fiber mapped to 30 overrepresented GO terms, with 7 terms associated with biotic and abiotic stress responses (Table 3). The homologous Gr gene IDs in each GO category can be found in Additional file 14. The biological context of selected differences between genotypes will be discussed further below.
Table 3.
Overrepresented Gene Ontology biological process terms in Gb or Gh fiber. Transcripts that had higher expression (fold change ≥2) on at least one DPA in the cross genotype comparison were analyzed for over-representation of GO biological process terms (p < 0.006, q < 0.05). FDR q-values were determined as in Table 1 (see p-values in Additional file 14)
| Up in Gb fiber | Up in Gh fiber | ||||
|---|---|---|---|---|---|
| GO ID | GO Name | FDR | GO ID | GO Name | FDR |
| GO:0042254 | ribosome biogenesis | 1.15E-09 | GO:0015996 | chlorophyll catabolic process | 3.92E-06 |
| GO:0042026 | protein refolding | 3.28E-05 | GO:0010150 | leaf senescence | 3.92E-06 |
| GO:0010143 | cutin biosynthetic process | 2.11E-04 | GO:0019745 | pentacyclic triterpenoid biosynthetic process | 3.92E-06 |
| GO:0006730 | one-carbon metabolic process | 2.01E-03 | GO:0006090 | pyruvate metabolic process | 1.02E-05 |
| GO:0006821 | chloride transport | 2.81E-03 | GO:0009414 | response to water deprivation | 2.89E-05 |
| GO:0006448 | regulation of translational elongation | 9.92E-03 | GO:0006013 | mannose metabolic process | 1.23E-04 |
| GO:0006536 | glutamate metabolic process | 1.00E-02 | GO:0006094 | gluconeogenesis | 1.78E-04 |
| GO:0009637 | response to blue light | 1.60E-02 | GO:0006096 | glycolysis | 2.07E-04 |
| GO:0015808 | L-alanine transport | 1.63E-02 | GO:0006000 | fructose metabolic process | 3.34E-04 |
| GO:0050732 | negative regulation of peptidyl-tyrosine phosphorylation | 1.63E-02 | GO:0009651 | response to salt stress | 4.62E-04 |
| GO:0051085 | chaperone mediated protein folding requiring cofactor | 1.63E-02 | GO:0010193 | response to ozone | 1.11E-03 |
| GO:0048640 | negative regulation of developmental growth | 1.63E-02 | GO:0016556 | mRNA modification | 1.27E-03 |
| GO:0072521 | purine-containing compound metabolic process | 2.76E-02 | GO:0009965 | leaf morphogenesis | 1.34E-03 |
| GO:0006639 | acylglycerol metabolic process | 2.76E-02 | GO:0010363 | regulation of plant-type hypersensitive response | 1.97E-03 |
| GO:0051302 | regulation of cell division | 2.76E-02 | GO:0052542 | defense response by callose deposition | 2.47E-03 |
| GO:0006909 | phagocytosis | 2.76E-02 | GO:0030048 | actin filament-based movement | 2.47E-03 |
| GO:0046653 | tetrahydrofolate metabolic process | 2.76E-02 | GO:0009834 | secondary cell wall biogenesis | 2.47E-03 |
| GO:0006123 | mitochondrial electron transport, cytochrome c to oxygen | 2.76E-02 | GO:0019643 | reductive tricarboxylic acid cycle | 3.29E-03 |
| GO:0006497 | protein lipidation | 4.45E-02 | GO:0010167 | response to nitrate | 4.00E-03 |
| GO:0009220 | pyrimidine ribonucleotide biosynthetic process | 4.50E-02 | GO:0000165 | MAPK cascade | 5.62E-03 |
| GO:0015865 | purine nucleotide transport | 4.50E-02 | GO:0045492 | xylan biosynthetic process | 6.01E-03 |
| GO:0015721 | bile acid and bile salt transport | 4.50E-02 | GO:0010413 | glucuronoxylan metabolic process | 6.01E-03 |
| GO:0015701 | bicarbonate transport | 4.50E-02 | GO:0050832 | defense response to fungus | 6.46E-03 |
| GO:0009116 | nucleoside metabolic process | 4.59E-02 | GO:0051567 | histone H3-K9 methylation | 6.99E-03 |
| GO:0008033 | tRNA processing | 4.74E-02 | GO:0071805 | potassium ion transmembrane transport | 6.99E-03 |
| GO:0043068 | positive regulation of programmed cell death | 6.99E-03 | |||
| GO:0010200 | response to chitin | 8.09E-03 | |||
| GO:0000910 | cytokinesis | 9.02E-03 | |||
| GO:0019344 | cysteine biosynthetic process | 9.46E-03 | |||
| GO:0007030 | Golgi organization | 1.03E-02 | |||
Overview of changes in the metabolome of 10 to 28 DPA cotton fiber
The metabolites are definitive indicators of cellular activity, whereas transcripts may not be translated. Therefore, we compared metabolite concentrations across the time-course of fiber development within one genotype and at the same DPA across genotypes (Fig. 1). Of 206 total metabolites, 194 or 201 were present in 10 DPA Gb or Gh fiber, with similar numbers detected at all other DPA. The 20 occurrences of the highest concentration metabolites in Gb or Gh fiber are shown in Additional file 15, and these will be discussed below in their biological context.
In Gh or Gb fiber, 44 or 45 metabolites did not show a significant change between 10 to 28 DPA. Of these, 18 were unchanged in both genotypes, including several sugars and sugar derivatives (such as xylitol, sucrose, mannose-6-phosphate, and UDP-glucose) that relate to the importance of turgor pressure and polysaccharide synthesis for cotton fiber morphogenesis.
A total of 161 or 162 metabolites (approximately 78 % of total metabolites) had significantly different concentrations across DPA in Gb or Gh fiber (p ≤ 0.05); (Additional file 3). The greatest metabolic change occurred between 21 to 28 DPA in both genotypes, in contrast to the decrease in transcriptional complexity in the same interval. Compared to 21 DPA, 99 or 95 different metabolites were more concentrated in Gb or Gh fiber at 28 DPA (or 73 or 62 metabolites, excluding dipeptides) (Fig. 1). Metabolites with higher concentration at 28 DPA accounted for most of the >10-fold changes across DPA (Additional file 15). In Gb fiber, 21 different metabolites showed 10 to 126-fold concentration increases as fiber development progressed, and 20 of 21 cases reflected higher concentrations at 28 DPA as compared to 10 or 21 DPA. Similarly, Gh fiber had 15 different metabolites with 10 to 72-fold concentration increases across DPA, and 13 of 15 cases reflected the same developmental contrasts. Nine metabolites with ≥10-fold concentration differences across DPA were held in common between Gh and Gb fiber: adenosine-, cytosine-, guanosine-, or uridine-2’,3’-cyclic monophosphate; linoleamide; methyl-beta-glucopyranoside; fucosterol; phytosphingosine; and raffinose, and these are discussed in the context of metabolite clusters below.
Fuzzy C-means clustering revealed four major patterns of change in metabolite concentrations, with only three clusters being similar between genotypes (Fig. 3). Below we interpret the metabolite clusters by reference to metabolites with >10-fold concentration changes across DPA in both Gh and Gb fiber. Cluster ‘a-metabolites’ have higher concentration at 10 to 15 DPA with similar lower concentrations observed in both genotypes at 18 to 28 DPA. This cluster contains fucosterol (3β-Hydroxy-5,24(28)-stigmastadiene, Additional file 16). Phytosterols help to structure plasma membrane lipid domains that contain sphingolipids, which are derived from precursor phytosphingosine (4-D-Hydroxysphinganine) [50]. Phytosphingosine was in the ‘a’ or ‘c’ metabolite clusters in Gb or Gh fiber, but, like fucosterol, it occurred in high concentration at 10 DPA in both genotypes. Possibly, fucosterol and phytosphingosine are related to the organization of the plasma membrane to support high-rate elongation. The overall dissimilar Cluster ‘b-metabolites’ (Gb) and Cluster ‘c-metabolites’ (Gh) each contained linoleamide, with high concentration at 15 DPA in both genotypes. Linoleamide is derived from linoleic (18:2) fatty acid and modulates calcium levels in animal cells [51]. These two clusters (with 30 or 35 metabolites in Gb or Gh fiber) share the feature of lower metabolite concentrations at 28 DPA. About half of the molecules are held in common, including many fatty acids that could relate to building plasma membranes to support elongation. However one-third of the metabolites in Gb Cluster ‘b-metabolites’ have peak concentrations at 15 DPA, with potential functional consequences still to be determined. Cluster ‘d-metabolites’ are low at 18 to 21 DPA during transitional cell wall remodeling, but none showed >10-fold change across DPA in both genotypes. The biological context of this cluster will be discussed below as part of defining the transition stage. Cluster ‘e-metabolites’ have highest concentrations at 28 DPA in both genotypes and include: methyl-beta-glucopyranoside, raffinose, and the four nucleotide-2’,3’-cyclic monophosphates. The synthesis of methyl-beta-glucopyranoside may serve to detoxify methanol arising from pectin methylesterase activity during SCW synthesis [25, 52–54]. Raffinose is a sucrose-derived oligosaccharide that can act as an antioxidant to protect against oxidative stress, as well as exerting other protective effects [55]; see further discussion on ROS management below. The nucleotide-2’,3’-cyclic monophosphates are intermediate products of RNase activity that may signify increased nuclease activity in the initial stages of fiber cell death [56].
Fig. 3.
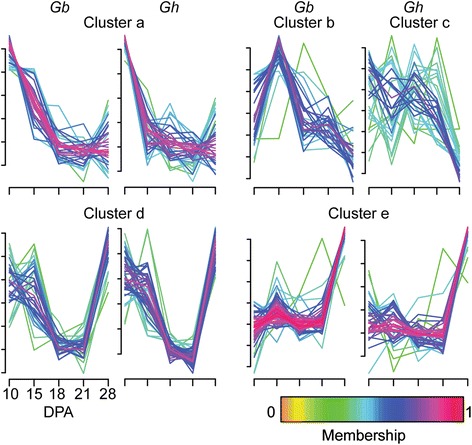
Four clusters of Gb and Gh fiber metabolites included three that were similar between genotypes. Metabolites with changes in concentration during fiber development (p ≤ 0.05) were clustered. The x-axis is as defined for Fig. 2. The y-axis is the scaled imputed mean value of metabolite concentration standardized to mean = 0 and standard deviation = 1. Color coding is as described in Fig. 2
Differences in the metabolome between Gh and Gb fibers
The difference in metabolite concentrations between Gh and Gb fiber was also determined at each DPA analyzed. Gh fiber had 105 metabolites with higher concentration than in Gb fiber, whereas 70 metabolites were more concentrated in Gb fiber (Additional file 3). Both genotypes differentially accumulated carbohydrates and amino acids, including ones associated with the metabolism of glutathione and the cellular control of redox homeostasis [46]. Gh fiber had 32 more dipeptides than Gb (see further discussion below). Gb fibers had higher levels of lipids, including three additional oxylipins that derive from the oxidation of linoleic or linolenic acid and participate in plant stress signaling [42, 57, 58]. Gb also had higher levels of six flavonoids (leucocyanidin, naringenin-7-O-glucoside, catechin, epicatechin, gallocatechin and epigallocatechin), whereas Gh fiber had higher levels of three other flavonoids (dihydromyricetin, dihydrokaempferol, and kaempferol 3-O-betaglucoside). These flavonoids may be acting as antioxidants or playing a role in modulating auxin signaling [59].
On each DPA, Gh fiber had 53–78 metabolites with higher concentration than Gb fiber, whereas Gb fiber had 21–47 higher concentration metabolites as compared to Gh fiber (Fig. 1). There were 23–33 dipeptides that were more highly concentrated on each DPA in Gh fiber, with only glycyltryptophan having higher concentration in Gb fiber (Additional file 3). The list of metabolites with greatest concentration difference (≥8-fold) in Gh as compared to Gb fiber showed 12 of 25 cases occurring at 10 DPA, with the list dominated by dipeptides (Table 4). Here, the ≥8-fold threshold was chosen to focus the discussion, and one metabolite is sometimes listed on multiple DPA. More dipeptides in Gh fiber contrasted with higher total protein content in Gb fibers throughout fiber development (Fig. 4). Overall the data support more active protein turnover in Gh fiber. The list of metabolites with ≥8-fold difference in Gb as compared to Gh fiber was dominated (13 of 20 cases) by antioxidants (including flavonoids, glutathione and ascorbate) at diverse DPA, although 7 of 20 cases occur at 28 DPA (Table 4). The section on ROS management contains additional related discussion.
Table 4.
Occurrences of ≥ 8-fold metabolite concentration differences between Gb and Gh fiber. Concentration differences were calculated from data in Additional file 3, which contains the p-values. FDR q-values were determined as in Table 1
| Higher concentration in Gb fiber | Higher concentration in Gh fiber | |||||||
|---|---|---|---|---|---|---|---|---|
| DPAa | Biochemical | Fold- change | FDR | DPAa | Biochemical | Fold-change | FDR | |
| 28 DPA | ascorbate | 138.27 | 0.00 | 10 DPA | serylleucine | 38.82 | 0.00 | |
| 15 DPA | leucocyanidin | 79.89 | 0.00 | 10 DPA | dihydrokaempferol | 17.02 | 0.00 | |
| 21 DPA | ascorbate | 50.36 | 0.04 | 10 DPA | N-acetylmethionine | 16.11 | 0.00 | |
| 18 DPA | ascorbate | 46.12 | 0.02 | 10 DPA | adenosine | 12.53 | 0.00 | |
| 15 DPA | catechin | 30.18 | 0.00 | 10 DPA | valylisoleucine | 11.81 | 0.00 | |
| 10 DPA | gallocatechin | 23.37 | 0.00 | 10 DPA | histidylleucine | 11.53 | 0.00 | |
| 10 DPA | glutamate | 23.27 | 0.00 | 10 DPA | leucylglutamate | 11.44 | 0.00 | |
| 28 DPA | adenosine 5’-diphosphate (ADP) | 22.51 | 0.00 | 10 DPA | lysylleucine | 11.12 | 0.00 | |
| 28 DPA | leucocyanidin | 17.02 | 0.00 | 10 DPA | threonylleucine | 11.10 | 0.00 | |
| 10 DPA | leucocyanidin | 13.84 | 0.01 | 18 DPA | phenethylamineb | 11.07 | 0.00 | |
| 28 DPA | catechin | 12.80 | 0.00 | 21 DPA | phenethylamineb | 10.86 | 0.00 | |
| 18 DPA | glutathione, reduced (GSH) | 12.61 | 0.00 | 10 DPA | alanylleucine | 10.59 | 0.00 | |
| 28 DPA | gallocatechin | 12.49 | 0.00 | 10 DPA | sucrose | 9.27 | 0.00 | |
| 15 DPA | gallocatechin | 11.40 | 0.00 | 10 DPA | serylphenyalanine | 9.25 | 0.00 | |
| 15 DPA | epicatechin | 11.26 | 0.00 | 10 DPA | alanylvaline | 9.23 | 0.00 | |
| 28 DPA | stachyose | 10.83 | 0.00 | 10 DPA | alanylphenylalanine | 8.86 | 0.00 | |
| 10 DPA | caffeate | 9.31 | 0.00 | 15 DPA | histidylleucine | 8.85 | 0.00 | |
| 21 DPA | glutamate | 9.07 | 0.03 | 18 DPA | thymidine | 8.69 | 0.00 | |
| 18 DPA | glutamate | 8.83 | 0.00 | 10 DPA | glycylleucine | 8.61 | 0.00 | |
| 28 DPA | glucarate | 8.51 | 0.00 | 10 DPA | valylleucine | 8.60 | 0.00 | |
| 15 DPA | alanylisoleucine | 8.38 | 0.00 | |||||
| 15 DPA | alanylvaline | 8.20 | 0.00 | |||||
| 18 DPA | adenosine | 8.19 | 0.00 | |||||
| 10 DPA | inosine | 8.15 | 0.00 | |||||
| 10 DPA | aspartylleucine | 8.00 | 0.00 | |||||
aDPA at which the high fold change relative to the other fiber genotype was observed; bisobar with 1-phenylethanamine
Fig. 4.
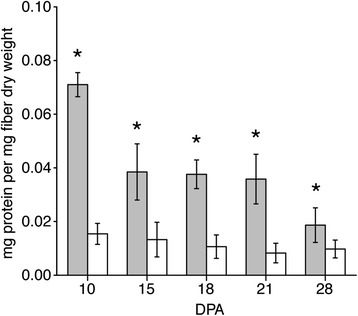
Total protein content in 10 to 28 DPA Gh and Gb fiber. Total protein was determined by Bradford assay and normalized to the dry weight of Gb (grey bars) and Gh (white bars) fiber. Mean values ± SD derive from 5 biological replicates at each DPA. Asterisks indicate a significant difference (p ≤ 0.017) between genotypes at each DPA as determined by t-test
Transitional cell wall remodeling is a distinct developmental stage
The data show that transitional cell wall remodeling is a distinct phase of fiber development that remains relatively stable at both the transcriptional and biochemical level for four days (between 18 to 21 DPA) under these experimental conditions. Only 0 or 2 transcripts and 6 or 8 metabolites changed concentrations between 18 and 21 DPA in Gb or Gh fiber, in contrast to substantial differences between 15 and 18 DPA and even greater differences between 21 and 28 DPA (Fig. 1). The expression pattern of the gene encoding a small GTPase-family protein (GhRAC13) [GenBank S79308.1], which is homologous to Gorai.011G031400.2 and AtRAC2 (At5g45970), confirmed the consistency of these results with known aspects of the transition stage in cotton fiber [60]. In the RNA-Seq data, GhRAC13 or its ortholog in Gb fiber were upregulated between 15 to 21 DPA (Fig. 5a). They were also the most highly expressed RAC homologs at 18 DPA (Additional file 17), consistent with the prior data on Gh fiber [34].
Fig. 5.
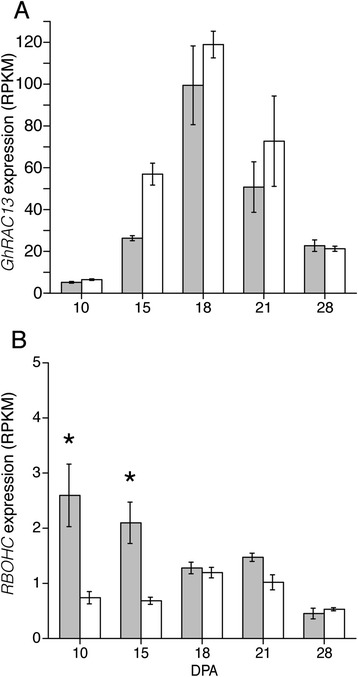
Expression of GhRAC13 peaks during transitional cell wall remodeling. (a) The level of expression of GhRAC13 (GenBank: S79308.1) is shown for Gb (grey bars) and Gh (white bars) fiber at 10 to 28 DPA, as derived from the RNA-Seq data. In both fibers, the similar timing of upregulated expression corresponds to the similar timing of the transition stage. (b) Expression of an RBOHC homolog (related to Gorai.001G053300.1) that may contribute to the oxidative burst by producing superoxide. Asterisks indicate a significant difference in the cross-genotype comparison (p ≤ 1E-4). Error bars are standard deviation
The GhRAC13 protein is thought to regulate the activity of NADPH oxidase, a producer of the H2O2 signal required to initiate SCW synthesis [60, 61]. Plant Ras/Rho GTPases activate NADPH oxidases, which generate superoxide by transferring electrons from NADPH inside the cell across the membrane and coupling them to molecular oxygen to produce superoxide anion. The superoxide anion is then converted to H2O2 either spontaneously or by superoxide dismutase [29]. Only one NADPH oxidase transcript had RPKM > 2 (related to Gorai.001G053300 .1). It was more highly expressed in Gb at 10 and 15 DPA, but similarly expressed in both genotypes at 18–28 DPA including lower expression after the transition stage (Fig. 5b). This transcript is homologous to RBOHC/RHD2 (E-value = 0, Additional file 17) with a role in Arabidopsis root hair elongation [62]. The activity of NADPH oxidase is influenced by post-translational modifications and calcium [63], which may help to explain its somewhat divergent expression pattern between the genotypes as contrasted with the highly similar RAC13 expression pattern. The expression of other transcripts potentially involved in the oxidative burst was generally similar between genotypes, except for one additional Arabidopsis RAC2 homolog that was expressed in Gh but not Gb fiber (related to Gorai.010G242900.1) (Additional file 18).
Curiously Cluster ‘d-transcripts’, which were upregulated during transitional cell wall remodeling, were mirrored by Cluster ‘d-metabolites’ with lower concentrations at 18 and 21 DPA. As reported before for Gh fiber [24], the concentrations of glucose and fructose decreased beginning at 18 DPA with a continued decline until 28 DPA in both genotypes (Additional file 3). There were 33 or 46 metabolites in Cluster ‘d-metabolites’ for Gb or Gh fiber (30 or 25, excluding dipeptides). Nine of these metabolites were held in common between the two genotypes and help to illustrate special features of the transition stage in both Gh and Gb fiber (Additional file 16). In addition to one amino acid (valine) and a related dipeptide (valylvaline), the seven other common metabolites with lower concentrations were adenine, cytidine, dihydromyricetin, guanosine, histidine, tryptophan, and uridine. Adenine is used to synthesize ATP, NAD, and FAD during respiration, which occurs at a lower rate during transitional cell wall remodeling in Gh fiber [24]. Similarly, histidine is a precursor of alpha-keto-glutarate in the TCA cycle, which would be required at lower levels when the respiration rate is reduced. Adenine, cytidine, guanosine, and uridine are precursors of RNA, and their lower concentrations may indicate reduced RNA synthesis during a relatively stable developmental phase with almost no transcriptional change in either fiber genotype. Tryptophan is an amino acid used in the synthesis of auxin and serotonin, which may inhibit auxin activity in plants [64]. Similarly, dihydromyricetin is a flavonoid that mediates ethanol effects on the brain in interaction with γ-aminobutyric acid (GABA) and glutamate [65], both of which are present in Cluster ‘a-metabolites’ with high concentration during elongation of Gh and Gb cotton fiber. In plants, glutamate is a newly emerging signaling molecule [66] as well as a precursor to ROS scavenging glutathione (see below), whereas GABA modulates the activity of Ca++ channels [67]. Therefore, the lower concentrations of tryptophan and dihydromyricetin at 18 to 21 DPA likely signify shifts in intracellular signaling at this distinctive developmental stage.
Gh and Gb fiber show similar expression profiles for cellulose synthase (CESA), and cellulose-synthase-like (CSL) genes
Glycosyltransferases within the cellulose synthase superfamily, including cellulose synthases (CESAs) and cellulose-synthase-like transcripts (CSLs), are required to produce para-crystalline cellulose fibrils and the polysaccharides in the cell wall matrix [17, 18]. The expression pattern of this gene family is often similar between Gh and Gb fiber, serving to highlight the logical correlation of gene expression with the similar early fiber elongation rates and timing of the onset of SCW formation in the two genotypes.
CESA genes: Arabidopsis thaliana provides a reference point for groups of CESA genes in seed plants [17]. The ten Arabidopsis CESA genes are grouped into six clades typified by: AtCESA1 (and including AtCESA10); AtCESA3; AtCESA4; AtCESA6 (and including AtCESA2,5,9); AtCESA7; and AtCESA8. The synthesis of expanding primary walls (as in growing roots and hypocotyls) requires the activity of AtCESA1, 3, and 6, whereas SCW synthesis (as in xylem tracheary elements and interfascicular fibers) requires the activity of AtCESA4, 7, and 8. For both primary and secondary wall synthesis, a distinct heteromeric cellulose synthesis complex contains at least 18 total CESAs [68]. The fifteen CESA loci in the Gr genome [47], as compared to 10 CESA loci in Arabidopsis, is generally consistent with the comparative gene richness in these two species [69].
Both Gh and Gb fiber express genes mapping to each of the 15 Gr CESA loci (Additional file 17), with remarkable similarity in pattern and magnitude of expression of each isoform. There were 10–15 CESA loci expressed on the days analyzed here (Fig. 6). As expected, loci homologous to AtCESA1, 3, and 6 were expressed during cotton fiber elongation, although the expression of transcripts in the AtCESA2, 5, 6, 9 clade was low overall. Expression of these ‘primary wall CESAs’ persists through 28 DPA even in Gh fiber where elongation had stopped five days earlier. Transcripts homologous to one of the AtCESA8-like loci (and Gorai.009G161200.1) are downregulated at 28 DPA in both genotypes, suggesting that an AtCESA8-like CESA may have adopted a novel role in early elongating cotton fiber (Additional file 17).
Fig. 6.
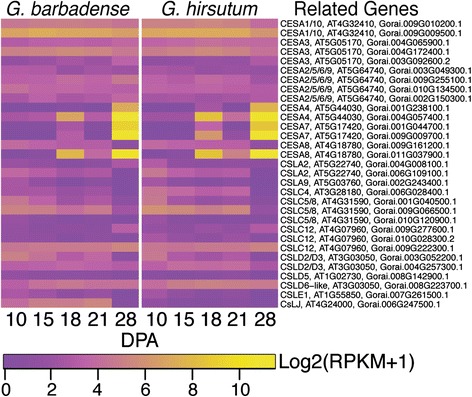
Expression patterns of CESA and CSL genes in Gb and Gh fibers. Heat map colors correspond to the LOG2 transformed RPKM value plus one for each transcript analyzed. Higher and lower expression levels are shown in yellow or purple, respectively. The data from cotton transcripts are referenced to ‘Related genes’ including the gene name and locus identifier for the most homologous Arabidopsis gene and the transcript identifier for the most homologous Gr gene
The homologs of AtCESA4, 7, and 8 were upregulated at 18 and 28 DPA during early and late cell wall thickening (Fig. 6). Interestingly, these show less expression at 21 DPA, consistent with the relatively static stage of transitional cell wall remodeling and winding layer synthesis between 18 and 21 DPA. All 15 CESA loci were expressed at 18 DPA during transitional cell wall remodeling, showing the potential for cellulose synthesis complexes of diverse types to be formed within cotton fiber if all of the transcripts are translated. Expression of many CESA loci in multiple clades continued at 28 DPA, even in Gh fiber that was no longer elongating.
CSL genes: The CESA superfamily includes 9 cellulose-synthase-like (CSL) families in addition to CESAs, inclusive of clades CSLA to CSLH and CSLJ. The CSLs have motifs indicative of ß-glycosyltransferase activity, and some have been proven to participate in synthesis of the hemi-cellulosic cell wall matrix polysaccharides including xyloglucan and mannan [18, 70]. The Gr genome has at least 35 CSL loci within the CSLA to CSLE, CLSG, and CSLJ families, compared to 31 CSLs in Arabidopsis [47]. Both Gh and Gb fiber express 16 CSL genes within four families (CSLA, CSLC, CSLD, CSLE), although the expression of the only CSLE homolog is weak (Additional file 17). At least one gene in the CSLA, CSLC, and CSLD, families is expressed relatively strongly from 10 to 21 DPA in elongating fiber of both genotypes. In addition, representatives of the CSLC and CSLD families are expressed strongly at 28 DPA when only Gb fiber is continuing to elongate. Gb fiber at 10 to 21 DPA expresses a homolog of a fifth CSL family, CSLJ, which exists in many angiosperms (but not Arabidopsis) and has unknown function [70] (Fig. 6).
Arabidopsis CSLC4 (with one cotton fiber homolog) supports the synthesis of the ß-1,4-glucan backbone of xyloglucan [71], which is found within the primary wall of Gh and Gb fiber [10]. Cotton fiber shows relatively weak expression of a homolog of AtCSLA2 and even lower expression of an AtCSLA9 homolog: both of the encoded proteins have ß-1,4-mannan and/or glucomannan synthase activity in vitro [72]. Mannan has been detected in extracts of tightly bound cell wall matrix polymers of 6 to 30 DPA Gh fiber and mature Gh and Gb fibers that are still surrounded by a primary wall [25, 73]. Tentatively, it may contribute positively to the ‘elongation’ parameter, or the extent of fiber stretching before breaking [73]. Members of the CSLD family can support the synthesis of cellulose, cellulose-like polymers, and/or mannan. One cotton gene homologous to AtCSLD2/3 (two closely related isoforms in Arabidopsis) has strongest expression at 10 DPA in both genotypes, and another is expressed at a low level throughout 10 to 28 DPA. Proteins in the CSL2/3 clade are required for normal root hair growth in diverse plant species [74, 75], and their roles in cotton fiber can be explored in further research.
Cotton fiber modifies an ancient gene regulatory network to repress lignification
The genetic program for cotton fiber SCW synthesis resembles the ancient hierarchical gene network that regulates the differentiation of sclerenchyma cells [6, 15, 76, 77]. However, unlike those cell types in the plant body, cotton fiber represses hemicellulose, protein, and lignin biosynthesis during SCW synthesis so that cellulose becomes predominant. Our data show that cotton fiber expresses 92 primary loci that are homologous to 45 transcriptional regulators of Arabidopsis sclerenchyma cell differentiation [19, 20] (Additional file 17). Of these, 39 cotton homologs have fold-change ≥2 between 10 to 28 DPA in either Gh or Gb fiber, and the expression patterns are highly similar in both genotypes (Fig. 7).
Fig. 7.

Expression patterns of genes encoding secondary wall related transcription factors in Gb and Gh fibers. The heat map parameters are explained in the legend of Fig. 6
Our data showed that cotton fiber expresses genes that are highly homologous to Arabidopsis SCW transcriptional regulators named NST1, SND2, SND, MYB4, MYB42, MYB46, MYB52, MYB55, MYB61, MYB83, MYB85, MYB103, KNAT7, VNI2, myb-like (At3g11280), BLH6, and RDUF1 (Additional file 17). We will draw analogies below to gene/protein functions in Arabidopsis sclerenchyma cells [20, 22, 77], although sequence changes in Gossypium may also affect the precise regulatory roles in cotton. The transcriptional network controlling sclerenchyma SCW synthesis is described in hierarchical tiers with higher levels having broader control of SCW differentiation processes [20]. Cotton fiber expresses only one of five ‘Tier 3’ master regulators of SCW synthesis: homologues of NST1 (NAC043) that is active in Arabidopsis stem fibers (but not vessels). In both cotton genotypes, two of four NST1 homologues were upregulated at 15 DPA, just before transitional cell wall remodeling, and remained highly expressed through 28 DPA (Fig. 7). The other two loci were upregulated more strongly in 28 DPA Gh fiber than in Gb, pointing to potential isoform-specific effects on the extent of SCW thickening. Other genes, such as a homolog of BLH6 that appears to activate SCW thickening in Arabidopsis stem fibers, may also help to regulate cotton fiber SCW synthesis.
Cotton fiber expresses five of seven ‘Tier 2’ transcription factor targets of NST1, each of which is independently capable of inducing SCW synthesis in Arabidopsis (Fig. 7). In cotton fiber, MYB61 was upregulated at 15 DPA like NST1, whereas the expression of SND3 (NAC010), MYB83, and MYB103 were strongly upregulated by 18 DPA. The expression of a single locus homologous to MYB46, which is redundant in Arabidopsis with MYB83, was weakly and transiently upregulated at 18 DPA. At least some homologs of SND3, MYB83, and MYB103 showed a high-low-high expression pattern at 18-21-28 DPA, which correlates with the relatively stable phase of transitional cell wall remodeling at 18 to 21 DPA followed by the distinct phase of mainly cellulose synthesis. These transcription factors may interact with different partners to modulate the synthesis of different layers as the cotton fiber thickens, i.e. the thin transitional “winding” layer followed by the thick SCW composed primarily of cellulose.
In correlation with dominance of cellulose synthesis during SCW thickening, the cotton fiber genotypes analyzed here expressed only five homologs of the 19 ‘Tier 1’ transcription factors (Fig. 7). In Arabidopsis, the ‘Tier 1’ transcription factors control particular SCW processes such as cellulose, hemicellulose, and lignin synthesis [20]. The expression of three KNAT7 (IXR11) homologs at variable levels over time in cotton fiber is consistent with the modulation of the amount of secondary wall cellulose, which is important because overly thick walls prevent cotton fibers from drying with the ‘kidney bean’ shape required for yarn spinning [78]. In other plants, KNAT7 can activate or repress SCW synthesis in different cell types, depending on its protein partners. A Gh ortholog of AtKNAT7 (KC200250, related to Gorai.004G206600.1) can modulate the SCW thickness of interfasicular fibers in transgenic Arabidopsis, although the mechanism behind the effect was unclear [79], and this gene was upregulated between 18 to 28 DPA in our data. Other ‘Tier 1’ regulators expressed in cotton fiber include: 2 loci homologous to SND2 (NAC073), both with upregulated expression only at 18 and 28 DPA; and 2–4 loci of MYB52 with weak expression at 28 DPA. The roles of SND2 and MYB52 in sclerenchyma cells are not completely clear, although MYB52 may repress lignification or the whole SCW differentiation program [19, 20]. In addition to recognized ‘Tier 1’ regulators, cotton fiber also shows strongly upregulated expression between 15 to 28 DPA of one of four loci homologous to Arabidopsis LIM1 (WLIM1; related to Gorai.012G147400). Over-expression in cotton of another LIM1 homolog [GenBank: JX648310], which is related to Gorai.010G026100.1, resulted in ~20 % thinner fibers with 1.4 % lignin/lignin-like-phenolics compared to 1 % in wild type [80]. Therefore, this gene family may help to control the balance between cellulose and lignin content in cotton fibers.
Corresponding to little or no lignin in the fiber of advanced commercial cotton cultivars [81], the cotton homologs of nine lignin-regulating transcription factors (MYB20, MYB58, MYB63, MYB69, MYB75, MYB79, ATHB18, BP, At3g46080) [20] were not expressed at RPKM ≥ 2 (Additional file 17). In contrast, cotton fiber expresses 5 loci homologous to MYB4, which is a widely conserved repressor of lignification in angiosperms [22] (Fig. 7). One of the MYB4 loci was strongly upregulated at 28 DPA during the synthesis of SCW with little or no lignin, perhaps indicating a specific role for this isoform in repressing SCW lignification in cotton fiber. In addition, cotton fiber expresses homologs of MYB-like At3g11280 and RDUF1, which repress lignification and/or the whole SCW differentiation program in Arabidopsis [19]. In cotton fiber, one locus homologous to At3g11280 was upregulated at 28 DPA, implying that the encoded protein may indeed repress lignification specifically. Conversely, the cotton gene encoding RDUF1-like protein was downregulated at 28 DPA, suggesting that it may act as repressor of SCW cellulose synthesis. Similarly, the transcription of three loci encoding homologs of VNI2 (NAC083), a SCW repressor in Arabidopsis, shows a general pattern of downregulation at 28 DPA when cotton fiber is undergoing massive SCW thickening.
The results show that lignin synthesis in commercial cotton fiber is repressed at the transcriptional level. Consistently, no lignin monomers (p-coumaroyl-, coniferyl-, or sinapyl-alcohols) were found in the fiber metabolome, although monolignols were readily detected even in young tree shoots by similar methods [82] (Additional file 3). Nonetheless, 74 loci homologous to lignification-related structural genes [20, 21] (Additional file 17) were expressed in at least one cotton fiber sample, often at low levels. Transcripts encoding three enzymes needed to convert phenylalanine to p-coumaryl CoA were detected (phenylalanine ammonia lyase, PAL; C4-hydroxylase, C4H; and 4-coumaroyl-CoA-ligase, 4CL, or 4CL-like). However, these enzymes support the synthesis of both lignin monomers and flavanoids [83], which were abundant in the cotton fiber metabolome. The flavonoid branch of the phenylpropanoid pathway is modulated by chalcone synthase (CHS, with 3-5 loci expressed in cotton fiber). Alternatively hydroxycinnamoyl-CoA shikimate/quinate hydroxycinnamoyl transferase (HCT) controls flux toward lignin. Gb fiber had stronger expression of more HCT loci as compared to Gh fiber, but both genotypes had low expression of only one HCT locus at 28 DPA during SCW synthesis (Fig. 8). Therefore, flux toward lignin may be limited during cotton fiber SCW synthesis despite the (overall low) expression of down-stream genes on the lignin branch pathway. The expression patterns of lignin-related structural genes are displayed in Fig. 8 including homologues of: C3-hydroxylase (C3H); cinnamoyl-CoA reductase-like (CCRL); cinnamyl alcohol dehydrogenase (CAD or CAD-like); ferulate 5-hydroxylase (F5H); caffeoyl-CoA O-methyltransferase (CCOMT of CCOMT-like); and caffeic acid O-methyltransferase (COMT or COMT-like). These could be related to the presence of minor amounts of lignin-like phenolics in cotton fiber [28, 80, 81].
Fig. 8.

Expression patterns of genes related to lignin biosynthesis in Gb and Gh fibers. The heat map parameters are explained in the legend of Fig. 6
Differences in ascorbate synthesis and recycling may enhance ROS management and elongation in Gb cotton fiber
Previous indications of the importance of ROS management in cotton fiber development and quality improvement were provided in the introduction. Here, we provide new insights into the genetic and biochemical basis of this effect. The fibers from Gb and Gh accumulate and manage antioxidants differently, especially at the transition stage and afterwards when increasing ROS levels help to regulate SCW synthesis while elongation in Gb fiber also continues. As detailed below, Gb fiber has greater capacity to manage ROS, which is consistent with its higher protein levels (as detected in Bradford assay) as contrasted with the higher levels of dipeptides (derived from protein degradation) in Gh fiber, as summarized previously.
The heat map of expression levels of genes related to ROS management via the glutathione-ascorbate cycle [84] is shown in Fig. 9, and the combined transcriptomic and metabolomics data are shown in Fig. 10. The relevant specialized cotton fiber metabolites that were detected in this study included gulono-1,4-lactone, ascorbate, dehydroascorbate, nicotinamide adenine dinucleotide (NAD+), and reduced and oxidized glutathione (GSH and GSSG). Relevant cotton fiber transcripts were homologous to genes encoding gulono-1,4-lactone oxidase (GLOase, or GulLO6), APX, monodehydroascorbate reductase (MDHAR or MDAR), DHAR, glutathione disulfide-reductase (GSR, or GR), and glutathione-dependent peroxidase (GPX) (Additional file 17). At neutral pH most ascorbate is in the form of ascorbate(H−), a reducing agent/anti-oxidant that readily donates a hydrogen atom/electron to H2O2, converting it to harmless H2O in a reaction modulated by APX. The (often short-lived) monodehydroascorbate product can donate a second electron to yield dehydroascorbate in a non-enzymatic process. Reduced ascorbate can be regenerated by: (a) MDHAR using monodehydroascorbate and NADH cofactor; or (b) DHAR using dehydroascorbate and reduced glutathione cofactor. In addition, reduced glutathione acts as an anti-oxidant through donating an electron to H2O2 or oxidized biomolecules, followed by formation of dimeric oxidized glutathione, as catalyzed by GPX enzyme activity. A low GSH:GSSG ratio has been used as a metabolic indicator of oxidative stress in plants [85], and GSR enzyme activity maintains the pool of GSH that is available for ROS scavenging and use as a co-factor during replenishment of the ascorbate pool via DHAR activity.
Fig. 9.
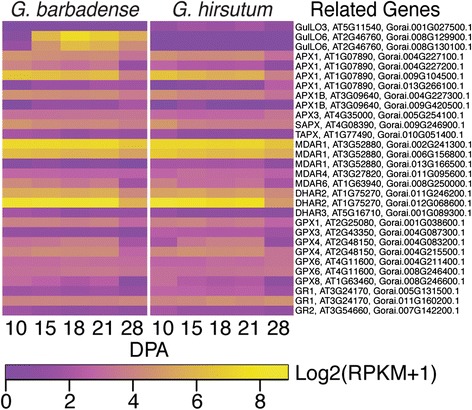
Expression patterns of genes related to ROS management via the glutathione-ascorbate cycle and the glutathione-perioxidase cycle in Gb and Gh fibers. The heat map parameters are explained in the legend of Fig. 6
Fig. 10.
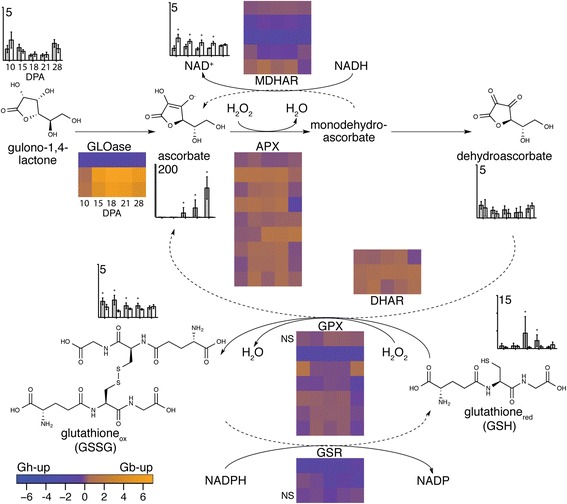
Ratio of transcripts and metabolite levels in the glutathione-ascorbate cycle and the glutathione peroxidase cycle in Gb and Gh fiber. The pathway is adapted from [93]. Metabolites: Graphs show the level of detected metabolites across DPA in Gb (grey bars) and Gh (white bars) fiber, with each X-axis tick representing 10, 15, 18, 21, or 28 DPA. The Y-axis shows the scaled imputed means for metabolite concentrations, with the maximum value shown numerically at the top. The detected metabolites are indicated by chemical structures, except for NAD+ that was omitted for clarity Oxidized or reduced glutathione are indicated with the subscripts ‘ox’ or ‘red’. Asterisks indicate a significant difference in the cross-genotype comparison (p ≤ 0.05). Ratios of transcripts: The heat maps show the ratio of expression of different loci in Gb vs Gh fiber, with each DPA represented on the horizontal. Each row represents a different gene, portrayed in the same order for each gene type as in Fig. 9. Enzyme names putatively corresponding to the transcripts are shown adjacent to reaction arrows. Forward or reverse reactions are indicated by solid or dashed lines. All of the transcripts shown had a significantly different expression level between Gh and Gb fiber on at least one DPA, except where NS is shown in two cases
In contrast to the heat map in Fig. 9 that shows the levels of gene expression in both genotypes, the heat maps in Fig. 10 are ratios of gene expression between Gb and Gh fiber at each DPA. Only five genes have consistently higher expression in Gh fiber (dark purple rows), but these have very low expression in both fiber genotypes. These are homologs of GLOase (Gorai.001G027500), MHDAR (Gorai.013G166500 and Gorai.006G156800), GPX (Gorai.004G087300), and GSR (Gorai.005G131500). In following, we emphasize the many other genes with moderate to high expression levels that also have a higher Gb:Gh expression ratios.
Abundance and synthesis of antioxidants
The metabolomics analysis revealed a much higher level of ascorbate in Gb fiber at 18 to 28 DPA, with a 138-fold increase at 28 DPA as compared to Gh fiber. Gb fiber also expressed two GLOase genes at 365-5,385-fold higher level at 15 to 28 DPA (related to Gorai.008G129900 and Gorai.008G130100; both homologs of At2g46760, AtGUlLO6). This may be a general attribute of Gb accessions, a hypothesis that is supported by our analysis of published microarray data [7]; Additional file 19. In total, our data showed that only three GLOase homologs were expressed at RPKM ≥ 2 in cotton fiber. Interestingly, At2g46760 is in a co-expression network (http://atted.jp/cgi-bin/locus.cgi?loc=At2g46760) with the VND7 transcription factor gene (At1g71930) that initiates SCW synthesis in water-conducting vessels [86] rather than supportive xylem fibers. The GLOase enzyme converts gulono-1,4-lactone into ascorbate, and gulono-1,4-lactone was detected at low similar levels in fibers of both genotypes. At all DPA tested both Gh and Gb fiber had similar levels of precursors of gulono-1,4-lactone biosynthesis, myo-inositol-1-P and myo-inositol. Collectively, the data support the possibility that a major difference in GLOase gene expression flows into increased GLOase protein and enzyme activity to produce more ascorbate in Gb fiber.
Gb fiber also had higher levels of one or both forms of glutathione at 10 to 21 DPA as compared to Gh fiber. Reduced glutathione was most concentrated at 18 DPA near the onset of cell wall thickening. Correspondingly, the level of glutamate, the precursor for glutathione, was also significantly higher in Gb fiber at 10, 18, and 28 DPA. The levels of transcripts homologous to glutathione synthetase or gamma-glutamylcysteine synthetase genes, which encode the enzymes needed for conversion of glutamate to glutathione, were not significantly different between genotypes.
Effects of ascorbate on cell elongation
In addition to acting as a general cellular reductant, ascorbate may also contribute to the production of highly reactive hydroxyl radicals in the apoplast. In the presence of copper, ascorbate reduces molecular oxygen to peroxide and Cu++ to Cu+ resulting in the formation of hydroxyl radicals through a Fenton reaction. The hydroxyl radicals can cleave cell wall polysaccharides such as xyloglucan and pectins, potentially increasing the fluidity of cell wall polymers and enabling cell expansion [87, 88]. This mechanism could operate during the prolonged elongation period of Gb fiber when ascorbate levels are high. Proving that ascorbate serves one or both roles during late elongation in Gb fiber will be aided by determining the sub-cellular localization of ascorbate and the extent of apoplastic hydroxyl radical production during and after the transition stage.
Use and replenishment of ROS scavengers
Both Gh and Gb fiber expressed numerous genes encoding APX, which mediates the use of ascorbate as a ROS scavenger. The levels of oxidized ascorbate, or dehydroascorbate, were low and similar in 10 to 28 DPA Gh and Gb fiber, which is consistent with H2O2 scavenging via ascorbate in both fiber types. Nine APX loci expressed in cotton fiber were homologous to five Arabidopsis APX genes, with most of them being more highly expressed in Gb fiber. The cellular ascorbate pool can potentially be diminished through the degradation of dehydroascorbate into 2,3-diketogulonate, or the reduced ascorbate pool can be replenished through DHAR and/or MDHAR enzyme activity. DHAR was expressed more strongly in Gb fiber, which may indicate its preferential use of the glutathione-dependent DHAR-mediated dehydroascorbate recycling pathway to promote fiber elongation. Consistent with this possibility, transgenic plant experiments showed that DHAR enhanced ascorbate content more than MDHAR [89], improved antioxidant capacity, reduced oxidation of lipids, and improved root growth [90].
Both genotypes showed generally similar GPX transcript levels, but two of three GSR transcripts were higher in Gh fiber. This difference may partly account for the 1.4–2.8-fold higher levels of oxidized glutathione in Gb from 10 to 21 DPA. Reduced glutathione was 4.5–12.6-fold higher in Gb at 18 and 21 DPA during transitional cell wall remodeling, and the GSH:GSSG ratio was higher in Gb fiber (Fig. 11a). Given that a high GSH:GSSG ratio indicates less oxidative stress in plant cells [85], this result is consistent with lower oxidative stress in Gb fiber as compared to Gh fiber. Gb fiber also had significantly lower levels of alternative oxidase transcript (related to Gorai.005G220500.1 and At5g64210/AOX2) at 15 and 18 DPA (with the same trend observed at 21 DPA) (Fig. 11b). AOX is induced during stress to attenuate ROS accumulation and energy production by bypassing the electron transport chain [91], and higher AOX levels are correlated with low fiber quality in the im cotton fiber mutant [92]. Higher oxidative stress in the fiber of Gh cv Deltapine 90 may be one explanation for its lower quality as compared to the fiber of Gb cv Phytogen 800.
Fig. 11.
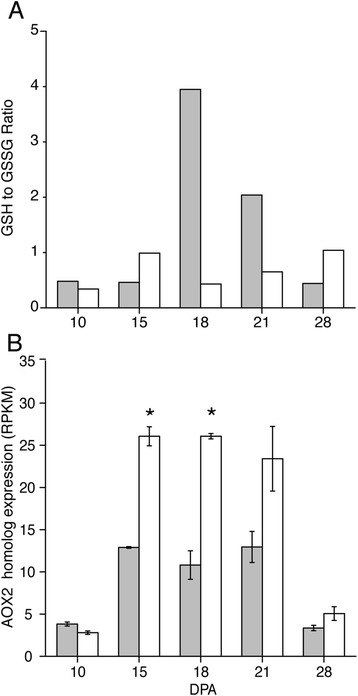
Metabolomic and transcriptomic evidence that transition-stage Gh fiber experiences more oxidative stress than Gb fiber. a) The ratio of reduced glutathione (GSH) to oxidized glutathione (GSSG) in Gb (grey bars) and Gh (white bars) fiber. The ratio is based on the scaled imputed mean for both metabolites. b) The expression level (RPKM) of a putative alternative oxidase (AOX) gene in Gh (white bars) and Gb (grey bars) fiber as derived from RNA-Seq data. The homologous protein in Arabidopsis (AOX2, At5G64210) attenuates ROS production during respiration [91], and the related Gr transcript is Gorai.005G220500.1. Asterisks indicate significant differences between genotypes at a given DPA. Error bars are standard deviation
Molecular and biochemical supports for increasing cotton fiber length
Our transcriptomic and metabolomic data support the prediction that it will be possible to generate longer fibers in Gh and other short fibered cotton (such as G. arboreum grown in Asia) through use of breeding or biotechnology strategies to enhance the levels of ascorbate in fiber, to use ascorbate for ROS scavenging via APX, and to recycle dehydroascorbate via DHAR. Over-expression of chloroplast-targeted APX provided preliminary indications of an approximately 10 % increase in Gh fiber length, while over-expression of GR had no effect compared to wild type [33]. Consistently, Gb fiber did not show any upregulation of GR gene expression in the RNA-Seq data. Our data reveal genes encoding particular isoforms of GLOase, MDHAR, APX, DHAR, and GPX that could be over-expressed to increase the quality of Gh fiber (indicated by orange colors assigned to higher Gb/Gh gene expression ratios in Fig. 10). In particular, we predict that over-expression of the GLOase genes that are uniquely and strongly expressed in Gb fiber (related to Gorai.008G129900 and Gorai.008G130100) will improve the length of Gh fiber. Higher expression of rat GLOase increased ascorbate levels in plants [93]. It may also be beneficial to increase the metabolic flux to gulono-1,4-lactone at the same time so that GLOase does not become substrate limited. Although we did not detect the alternative ascorbate precursor, L-Galactono-1,4-lactone, or any of its upstream metabolites [93] in 10 to 28 DPA cotton fiber, generating flux through this pathway might confer advantages in cotton fiber as well.
To identify other transcripts that might contribute to the sustained elongation of Gb fibers, we compared the intersection of three gene sets. The first gene set included Gb transcripts with ≥ 2-fold higher or similar expression level in the 28/21 DPA comparison, denoting continued expression during late-stage elongation in the fiber of Gb cv Phytogen 800. (Similar expression level was defined as < 2-fold change, or Gb 28/21 DPA ratio ≥ 0.5.) The second gene set included Gh transcripts with 21/28 DPA expression ratio of ≥ 2-fold, denoting a substantial decline in 28 DPA Gh fibers that were no longer elongating. The third gene set included transcripts that were expressed in either genotype at 10 DPA during early high-rate elongation, implying an assumption in this analysis that late-stage elongation in Gb fibers was mechanistically similar to early elongation. This assumption was supported by prior data showing that Gb fiber had a longer period of plasmodesmatal closure at the fiber foot as compared to Gh fiber. The resulting symplastic isolation is thought to increase the turgor pressure that contributes to fast elongation [94].
These three data sets overlapped by 1,288 transcripts (Fig. 12). Of this subset, the transcripts most highly expressed in 28 DPA Gb fibers relative to 28 DPA Gh fibers include ones that encode proteins that can logically support continued elongation: a tonoplast intrinsic protein/aquaporin (AtTIP1;3-like); a pectin (pectate) lyase (At1G04680-like); and a UDP-glycosyltransferase (AtUGT73B4-like) (Additional file 20). For each of these highlighted cases (and others not discussed here), there is no other primary transcript mapping to the same Arabidopsis gene identifier in this Venn intersection set, supporting the possibility that the implicated functions and/or protein isoforms may support prolonged fiber elongation.
Fig. 12.
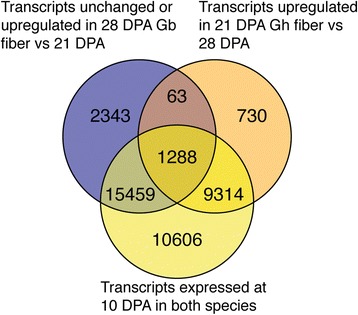
Venn diagram implicating transcripts that may contribute to the extended elongation period of Gb fibers. The three transcript sets compared were: (1) upregulated or unchanged from 21 to 28 DPA in Gb fibers; (2) downregulated from 21 to 28 DPA in Gh fibers; and (3) expressed in both genotypes at 10 DPA during early high-rate elongation. The 1,288 transcripts held in common may support the continued elongation of Gb fiber
The aquaporin gene is homologous to water-transporting AtTIP1;3 (in the Tonoplast Intrinsic Sub-family of aquaporins). This protein localizes to the vacuole and, together with AtTIP5;1, is required for normal Arabidopsis pollen tube growth [95, 96]. The aquaporin gene family in cotton is predicted to be important for the generation of turgor pressure to drive fiber elongation [97]. The pectin lyase is homologous to polygalacturonanase At3g48950, within a complex 67-member Arabidopsis gene family [98]. Pectin degradation may facilitate cell wall loosening and continuing fiber elongation [99]. For example, a pectin lyase with peak expression in 10 DPA Gh fiber positively regulates fiber elongation [100]. Arabidopsis UGT73B4 can glycosylate the flavanol anti-oxidant quercetin, which is expected to increase its solubility and stability [101]. A cotton UGT73B4-like protein may function similarly to support the role of flavonoids in ROS management. Related GO biological processes overrepresented in this dataset include: water transport, hydrogen peroxide catabolic process, and cell redox homeostasis (Additional file 21).
The metabolite data also revealed differences in 28 DPA Gb fiber that could support prolonged elongation. Of metabolites with ≥10-fold concentration difference across DPA, 11 of 29 were more concentrated in 28 DPA Gb fiber, as compared to 21 DPA, whereas 5 of 23 were more concentrated in 28 DPA Gh fiber (Additional file 15). The upregulated metabolites at 28 DPA in both genotypes, including raffinose, were discussed previously. The five unique metabolites that were upregulated between 21 to 28 DPA in Gb fiber were adenosine, guanosine 3’-monophosphate (3’-GMP), galactinol, stachyose, and phosphoethanolamine. Metabolites that were strongly upregulated (8–138 fold) in 28 DPA Gb vs Gh fiber included adenosine 5’-diphosphate (ADP), three flavonoids (leucocyanidin, catechin, gallocatechin), stachyose, glucarate, and ascorbate (Table 4). Phosphoethanolamine is a precursor for synthesis of the phospholipids required to support continuing fiber elongation. In addition to ascorbate and flavonoids, galactinol and raffinose can scavenge free radicals [102]. Stachyose is also within the family of ‘raffinose oligosaccharides’ and may play a similar role in ROS remediation in cotton fiber. These oligosaccharides may also act as osmolytes to increase turgor pressure during a prolonged elongation period [55]. Glucarate (or D-glucaro-1,4-lactone) is able to protect biomolecules, such as lipids and proteins, against oxidative damage by ROS, including H2O2 [103]. In summary, several ways of looking at the data implicate enhanced ROS management as a key feature supporting the prolonged elongation and greater final length of the fiber of Gb cv Phytogen 800 as compared to Gh cv Deltapine 90.
Conclusions
Deep transcriptomic and metabolomic analyses provided unparalleled insights into similarities and differences of fiber development in two genotypes of commercial allotetraploid cotton with well-characterized fiber development profiles. The implications of this rich data set can be experimentally tested in numerous ways. As examples, the expression levels of putatively important transcripts can be manipulated, including analysis of the presence and activity of the encoded proteins. Attempts can be made to change the metabolome of short-fibered cotton to be more like this cultivar of Gb fiber. Cotton diversity panels can be analyzed to look for repetitive correlations between useful fiber traits and particular genes and metabolites implicated by our data.
The specific stages analyzed in this study included primary wall synthesis to support high-rate elongation, transitional cell wall remodeling, and secondary wall thickening via mainly cellulose synthesis. During high rate elongation at 10 DPA, the transcriptomes and metabolomes of the fibers of both genotypes were complex with about 88 % of the transcripts and 95 % of the metabolites analyzed in this study being detected. Transitional cell wall remodeling was a discrete, relatively stable developmental stage lasting at least four days (18 to 21 DPA). Transcription factors associated with secondary wall synthesis in sclerenchyma cells began to be expressed then and persisted until 28 DPA when cellulose synthesis dominates. However, numerous transcripts encoding lignification activators were missing, whereas transcripts encoding a lignin repressor were present at 28 DPA. Correspondingly, the monolignol precursors of lignin were not detected in the cotton fiber metabolome. Therefore, commercial cotton fiber is white, soft, and absorbent due to the repression of lignification at the transcriptional level.
The data consistently supported a greater capacity for managing ROS in long cotton fiber, which correlated with higher protein levels and less protein degradation in the Gb cultivar as compared to the Gh cultivar we analyzed. The data showed a greatly increased level of ascorbate during the late elongation phase of the fiber in Gb cv Phytogen 800, and transcriptional clues suggested this is likely due to the activity of particular GLOase isoforms. In ongoing experiments, we are testing our prediction that enhanced ascorbate biosynthesis and recycling contributes to the extended elongation period and longer final length of Gb fiber either through general ROS management, by indirect modification of cell wall polysaccharides, or both. Further mining of the data will point to many fruitful avenues for future experimentation on the genetic and biochemical regulators of cotton fiber morphogenesis and quality.
Methods
Plant growth
The cotton cultivars used in this study were Gossypium hirsutum cv Deltapine 90 and Gossypium barbadense cv Phytogen 800. Plants were grown in parallel at the North Carolina State University Phytotron in a controlled greenhouse with a 26 °C/22 °C day/night temperature cycle. Under these well-controlled conditions, the plants developed vigorously with similar vegetative growth. The temporal progression of fiber development was repeatable between growing cycles. As needed, 1000 W metal halide lights were used to supplement day length to approximately 12 h a day. Plants (one per 6-L pot) were potted in a 2:1 (v/v) mixture of washed and sterilized gravel (# 16, construction grade) and Redi-earth Plug and Seedling Mix (#F1153; Sun Gro Horticulture Canada; based on the original “Cornell Mix”, [104]. Plants were watered twice daily with a solution containing macro- and micronutrients [105]. Fourteen plants of each cotton genotype were arranged in 4 rows of 7 plants each on wheeled carts holding one plant from each genotype. The carts were rotated once per week to equalize exposure of each plant to light on the edges of the plant block. Samples combined into one RNA pool were selected from positions distributed throughout the plant block.
Fiber sampling
Flowers were tagged upon opening, and fiber for each DPA sampled (10, 15, 18, 21, and 28 DPA) was collected from a 1st position boll between nodes 10 and 16 (low to middle on the plant) between September and January. Bolls were primarily collected from 12:00-4:00 pm to minimize potential expression variability associated with time of day. All bolls included in the samples had diameter in a range previously established as typical for that DPA (data not shown), indicating normal growth of the fruits containing the assayed fibers. Within 5 min after the boll was harvested, bolls were cut open with a razor blade and cotton seed and fiber were frozen in liquid nitrogen. The samples were gently ground under nitrogen in a mortar and pestle until the developing seeds and fibers had separated, then seeds were removed with forceps. The remaining cotton fiber was then ground to a fine powder and stored at −80 °C until RNA and metabolite extraction.
Overview of the statistical and gene identifier framework
Methods for statistical analysis are described in pertinent sections below. In the data interpretations, we generally highlighted significant differences defined for: (a) transcriptomics, q-values ≤ 0.001 and fold changes ≥ 2; and (b) metabolomics, p-values ≤ 0.05 and q-values ≤ 0.1, which indicates that ≤10 % of the discoveries called significant are possibly false positives. The q-values associated with p-values ≤ 0.05 in the metabolomics data are often considerably lower than 0.1 and both values are available in the supporting data. Arabidopsis gene identifiers, names and associated information are available at TAIR (http://www.arabidopsis.org/), and Gr gene identifiers and associated information are available at (www.phytozome.net). The heat maps and discussion generally relate only to primary transcripts (designated as such by read abundance in Gr, [47]), but data for all transcripts from one locus are available in the additional files.
Transcriptomics
As detailed below, the transcriptome from each of 10 samples (5 DPA × 2 genotypes) was derived from three cDNA sequencing libraries, and each library contained RNA from the fiber of three bolls collected from different plants. Therefore, the transcriptome of each sample cumulatively represents fiber from three biological replicates inclusive of nine bolls from nine different plants. As described in detail below, > 2 billion reads were compiled from 37–97 M reads from each of 30 libraries (3 libraries × 5 DPA × 2 genotypes).
RNA extraction and quality control
RNA was extracted from 100–250 μl volumes of ground cotton fiber using a kit (Sigma Spectrum Total Plant RNA Kit; Sigma-Aldrich, www.sigmaaldrich.com) per the manufacturer’s instructions for protocol A and including an on-column DNAse digestion (Sigma DNase70 Kit; Sigma-Aldrich, www.sigmaaldrich.com). RNA quality was first assessed by spectrophotometry (Nanodrop; Thermo Scientific, www.thermoscientific.com) to ensure that all samples had a 260/280 absorbance between 2.0–2.2. Prior to library preparation RNA pool quality was assessed on the Agilent bioanalyzer (www.agilent.com).
Library preparation
For each sample, three cDNA libraries were prepared fee-for-service by the Genomic Sciences Laboratory at North Carolina State University (research.ncsu.edu/gsl/) using the Illumina TruSeq RNA sample prep kit v2 (www.illumina.com). Total RNA (1 μg) was purified using poly-T oligo-attached magnetic beads to isolate poly-A containing mRNA. The mRNA was chemically fragmented using divalent cations and fragments were reverse transcribed into first strand cDNA using random primers. Second strand synthesis was carried out using DNA Polymerase I and RNaseH. Ends were repaired on the double-stranded cDNA to convert overhangs into blunt ends, 3’ ends were adenylated, and adapters with indexes for multiplexing were ligated. The library was amplified with a PCR reaction and then small fragments were removed (AmpureXP beads; www.beckmancoulter.com). The final library was quantified on the Agilent Bioanalyzer High Sensitivity DNA Chip; (www.agilent.com) prior to pooling.
Sequencing
Six libraries per timepoint (3 per genotype) were pooled at equimolar concentration, denatured, diluted with hybridization buffer, and clustered at 9.5pM concentration via cBot in one lane of an Illumina TruSeq PE v3 flowcell. A PhiX v3 control library spike-in was included in the lane to monitor clustering and error rate during sequencing. The flowcell was sequenced on a HiSeq2000 instrument with TruSeq SBS v3-HS chemistry (Illumina; www.illumina.com) following the manufacturer’s instructions for 2 ×100 bp reads with single indexing.
Mapping reference and annotation
To generate a mapping reference, we combined information from Gr (D genome) and Ga (A genome) cotton, representative of the diploid ancestral genomes of modern tetraploid cotton. Gr transcripts were obtained from the D genome resource at PHYTOZOME (http://www.phytozome.net/) [47], and the assembled Ga ESTs were described in [48]. The Gr genome contains 37,505 protein coding genes and 77,267 annotated protein coding transcripts. Blastn was used to identify redundant sequences between the Gr transcripts and the Ga ESTs, and all sequences with ≥ E-10 match to a Gr transcript were omitted from the Ga contig assemblies in order to give priority to Gr genome information. A total of 98,807 cotton fiber transcripts generated in the current study were mapped to the reference, with 98 % of them matching Gr sequences. Note that this does not indicate an allelic expression bias. Instead it indicates our use of the Gr genome resources for further analysis whenever possible. This is a valid approach as indicated by only 1.8 % average amino acid difference between A and D protein orthologs [106].
Annotation information, including gene IDs for Arabidopsis homologs, was adopted from the cotton D genome resource at www.phytozome.net. These annotations were validated using Blastp against the TAIR peptide database (updated 12/14/2010). Of the RNA-Seq reads that we analyzed further (RPKM ≥ 2), only 2,106 of 41,566 reads (5 %) mapped uniquely to the Ga EST assemblies (NCBI Transcriptome Shotgun Archive (TSA) accession number GBYK00000000), and these were annotated if possible using Blastx against TAIR with 1E-5 cut-off. Additional sequence annotation and KEGG (www.genome.jp) reference pathway mapping was performed using Blast2GO version 2.7.2 (www.blast2go.com) [107]. Blastx was used to compare sequences against the NCBI non-redundant database with an expectation value cut-off of 1E-10 for sequences mapping to the Gr reference and 1E-4 for A genome contig assemblies. GO annotation was performed using Blast2GO default settings (1E-5) and was refined using the Blast2GO ANNEX tool.
Quantification of raw reads
Raw reads from the two genotypes of cotton fiber were mapped to the reference set using Burrows-Wheeler Aligner (BWA version 0.6.2; http://bio-bwa.sourceforge.net/) allowing for a maximum of 4 % mismatches. Sequence reads mapping to the reference were counted through BEDTOOLS (version 2.16.2; https://github.com/arq5x/bedtools2) after file format conversion using SAMTOOLS (version 0.1.18; http://samtools.sourceforge.net/). For normalization of read counts, RPKM values were calculated on the three replicates of each sample as: number of mapped reads per gene divided by total mapped reads and transcript size, followed by multiplication by 1 million. RPKM values for each gene were averaged from replicates for further analysis.
Differential gene expression
Differential gene expression was compared in a pair-wise manner within and between fiber genotypes. For Gh and Gb fiber, successive DPA were compared and the 10 vs 28 DPA comparison was included to emphasize the contrast between primary and secondary wall synthesis. For the between genotype comparisons, the data from the same DPA were compared in Gh vs Gb fiber. For differential gene expression, three biological replicates per sample were then subject to statistical tests, using the samWrapper package (DEGseq version 2.6; http://bioconductor.org/packages/release/bioc/html/DEGseq.html) in R (www.r-project.org) followed by multiple correction (FDR ≤ 0.001). The computational analysis of differential gene expression was performed in a Linux environment at TACC (Texas Advanced Computation Center) at University of Texas at Austin.
Metabolomics
Five biological samples of 100 mg ground fiber were processed for each DPA. For 44 of the 50 samples, the same material used for sequencing was also used for metabolomic analysis (Metabolon Inc., Durham NC; www.metabolon.com). In a few cases, additional fiber was collected for metabolomics: 2 Gb bolls at 10 DPA and 1 boll each for Gb and Gh fiber at 18 and 21 DPA. Normalization was based on fiber dry weight, with a ratio of 15 μl extraction solvent per 1 mg. Protein was not used for data normalization due to the decline in total protein content (analyzed by Bradford assay) during fiber development and the higher protein level in Gb fiber (Fig. 4). Due to the approximately two-fold increase in the fiber weight per unit length ratio in both Gh and Gb fiber between 21 and 28 DPA [10], the 28 DPA samples were presumed to have less cytoplasmic contribution to the metabolite pool. Therefore, in the 28 DPA samples and within the 2-fold range, metabolite increases may be under-estimated and metabolite decreases may be over-estimated in terms of actual cytoplasmic concentration.
An automated sample preparation process was used (MicroLab STAR® system; hamiltonrobotics.com). For quality control, recovery standards were added prior to the first step in an aqueous and organic extraction process designed to remove proteins while maximizing recovery of small molecules. The resulting extract of each sample was divided into two aliquots for analysis by Liquid and Gas chromatography/mass spectrometry (GC/MS; LC/MS; LC/MS2). Residual organic solvent was evaporated (TurboVap®/Zymark; www.perkinelmer.com), and then each sample was frozen and dried under vacuum. The LC/MS and GC/MS methods were described previously [108, 109]. For GC/MS the column was 5 % phenyl and the temperature was ramped from 40° to 300 °C over 16 min. LC/MS and GC/MS data were automatically matched to a library of chemical standards as described in [110]. Compounds were identified as described previously [111, 112], but for our analysis a chemical library based on approximately 3000 standards was used. Normalization for instrument inter-day tuning differences, data scaling, and imputation of missing values were performed as described in [113].
Analysis using R ver 3.1.1
Overrepresentation of KEGG pathways was calculated using the R function phyper and the family-wise error rate and false discovery rates were calculated using the R function p.adjust. Heat maps were produced on transcripts with RPKM ≥ 2 in either fiber genotype using the R function heat map.2 in the gplots package version 2.14.2 [114]. Due to display size limitations, alternative transcripts (as referenced to the Gr genome; www.phytozome.net; [47] were omitted from the heat map datasets, with the exception of the respiratory oxidative burst genes. This omission was done after checking that the correlation between primary and secondary transcripts from the same locus was ≥0.73 and that the annotations at Phytozome were identical. The only exception to this high correlation rule was for an alternative transcript related to chalcone synthase that was expressed in Gh fiber (R = 0.99 compared to the primary transcript), but the corresponding locus had essentially no expression in Gb fiber (RPKM < 0.14). Plots were generated using the R function barplot in the gplots package. Cluster analysis was done using the R package Mfuzz [49]. The optimal fuzzifier values were calculated for each set of transcripts and metabolites using the function mestimate. Potential optimal cluster numbers were estimated using the minimum centroid distance with the function dmin. Venn diagrams were drawn using the package VennDiagram [115].
Availability of supporting data
The raw sequencing data supporting the results of this article have been deposited to the NCBI sequence read archive (http://www.ncbi.nlm.nih.gov/sra/) under accession SRP049330 and bioproject archive (http://www.ncbi.nlm.nih.gov/bioproject/) PRJNA263926. The contig assemblies from G. arboreum were deposited in the NCBI Transcriptome Shotgun Archive (TSA) (http://www.ncbi.nlm.nih.gov/genbank/tsa) under accession GBYK00000000. The processed data sets supporting the results of this article are included within the article and its additional files.
Acknowledgements
We thank: Cory Dashiell for producing the sequencing libraries; Dr. Joshua Udall for contributing A genome ESTs used in our RNAseq mapping reference; and Dr. Michael R. Stiff for helpful discussions. This work was supported by a National Science Foundation grant to ZJC, BES, and CHH (award #1025947). The funding agency had no role in the collection, analysis, and interpretation of data; in the writing of the manuscript; and in the decision to submit the manuscript for publication.
Additional files
Gb Transcriptome data. Transcript levels (RPKM) for Gb fiber at 10, 15, 18, 21, and 28 DPA.
Gh Transcriptome data. Transcript levels (RPKM) for Gh fiber at 10, 15, 18, 21, and 28 DPA.
Metabolome data. Metabolite levels and the results of pair-wise statistical comparisons for Gb and Gh fiber at 10, 15, 18, 21, and 28 DPA.
KEGG pathway enrichment_Metabolites. Overrepresented KEGG pathways in the fiber metabolome.
Blast2GO sequence annotation. Top NCBI BLASTX hits for reference sequences with RPKM ≥ 2.
Blast2GO KEGG pathway mapping. Predicted KEGG pathways and enzyme codes for reference sequences with RPKM ≥ 2.
KEGG pathway enrichment_Transcripts. Overrepresented KEGG pathways as inferred from the combined fiber transcriptome from Gb and Gh fiber.
GO Biological Processes 28 vs 21 DPA. Gene Ontology biological processes that are overrepresented in 28 DPA transcripts compared to 21 DPA in Gb or Gh fiber.
Gb differentially expressed genes across DPA. Transcripts differentially expressed for pairwise comparisons of Gb fiber at 10, 15, 18, 21, and 28 DPA.
Gh differentially expressed genes across DPA. Transcripts differentially expressed for pairwise comparisons of Gh fiber at 10, 15, 18, 21, and 28 DPA.
Transcript clusters. The six transcript clusters in Gb or Gh fiber and the overrepresented GO biological processes associated with each cluster.
Gb differentially expressed genes across genotypes. Transcripts more highly expressed in Gb fiber compared to Gh fiber at 10, 15, 18, 21, and 28 DPA.
Gh differentially expressed genes across genotypes. Transcripts more highly expressed in Gh fiber compared to Gb fiber at 10, 15, 18, 21, and 28 DPA.
Cross-genotype GO Biological Processes enrichment. Gene Ontology biological processes overrepresented in transcripts differentially expressed between Gb and Gh fiber.
High concentration and high change metabolites. Tables of the most concentrated metabolites in Gb or Gh fiber and those with a 10 fold or greater change between 10 and 28 DPA.
Metabolite clusters. Lists of metabolites in each of the four metabolite clusters found in Gb or Gh fiber.
TAIR blastp. Homology and annotation information derived from TAIR Blastp for transcripts displayed in the heat maps and graphs.
Transcripts associated with respiratory oxidative burst. Heat map of transcripts encoding Rho-like GTPases (RAC), respiratory burst oxidase homologs (RBOH), and copper and manganese superoxide dismutase (CSD, MSD).
Comparing RNA-Seq and Microarray Data.
Transcripts for extended elongation. A list of 1288 transcripts that potentially contribute to extended elongation in Gb fiber at 28 DPA.
Extended elongation GO Biological Processes. Gene Ontology biological processes overrepresented in the 1288 transcripts correlated with extended elongation in Gb fiber at 28 DPA.
Footnotes
Competing interests
DCA is an employee of Metabolon Inc., which was paid fee-for-service for the metabolomics analyses reported here. The other authors declare that they have no competing interests.
Authors’ contributions
CHH, JRT, and BES designed the study. JRT collected samples and extracted RNA. BES and MVD performed sequencing. GN, QS, JRT performed bioinformatics analyses. DCA performed metabolomics and associated statistical analysis. CHH and JRT wrote the manuscript. All authors approved the final manuscript.
Contributor Information
John R. Tuttle, Email: jrtuttle@ncsu.edu
Gyoungju Nah, Email: gyoungju.nah@gmail.com.
Mary V. Duke, Email: Mary.Duke@ars.usda.gov
Danny C. Alexander, Email: dalexander@metabolon.com
Xueying Guan, Email: xueying107@gmail.com.
Qingxin Song, Email: qingxinsong@gmail.com.
Z. Jeffrey Chen, Email: zjchen@mail.utexas.edu.
Brian E. Scheffler, Email: brian.scheffler@ars.usda.gov
Candace H. Haigler, Email: candace_haigler@ncsu.edu
References
- 1.Haigler CH, Betancur L, Stiff MR, Tuttle JR. Cotton fiber: a powerful single-cell model for cell wall and cellulose research. Front Plant Sci. 2012;3:104. doi: 10.3389/fpls.2012.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wendel JF, Flagel LE, Adams KL. Jeans, genes, and genomes: cotton as a model for studying polyploidy. In: Soltis PS, Soltis DE, editors. Polyploidy and genome evolution. Heidelberg: Springer Berlin Heidelberg; 2012. pp. 181–207. [Google Scholar]
- 3.Wendel JF. New world tetraploid cottons contain old world cytoplasm. Proc Natl Acad Sci U S A. 1989;86(11):4132–6. doi: 10.1073/pnas.86.11.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grover C, Grupp K, Wanzek R, Wendel J. Assessing the monophyly of polyploid Gossypium species. Plant Syst Evol. 2012;298(6):1177–83. [Google Scholar]
- 5.Wakelyn PJ, Bertoniere NR, French AD, Thibodeaux DP, Triplett BA, Rousselle M, Goynes Jr WR, Edwards JV, Hunter L, McAlister DD. Cotton fiber chemistry and technology. Boca Raton: CRC Press; 2010. [Google Scholar]
- 6.Al-Ghazi Y, Bourot S, Arioli T, Dennis ES, Llewellyn DJ. Transcript profiling during fiber development identifies pathways in secondary metabolism and cell wall structure that may contribute to cotton fiber quality. Plant Cell Physiol. 2009;50(7):1364–81. doi: 10.1093/pcp/pcp084. [DOI] [PubMed] [Google Scholar]
- 7.Chen X, Guo W, Liu B, Zhang Y, Song X, Cheng Y, Zhang L, Zhang T. Molecular mechanisms of fiber differential development between G. barbadense and G. hirsutum revealed by genetical genomics. PLoS ONE. 2012;7(1):e30056. doi: 10.1371/journal.pone.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lacape J, Claverie M, Vidal RO, Carazzolle MF, Pereira GAG, Ruiz M, Pré M, Llewellyn D, Al-Ghazi Y, Jacobs J. Deep sequencing reveals differences in the transcriptional landscapes of fibers from two cultivated species of cotton. PLoS ONE. 2012;7(11):e48855. doi: 10.1371/journal.pone.0048855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan J, Tu L, Deng F, Hu H, Nie Y, Zhang X. A genetic and metabolic analysis revealed that cotton fiber cell development was retarded by flavonoid naringenin. Plant Physiol. 2013;162(1):86–95. doi: 10.1104/pp.112.212142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Avci U, Pattathil S, Singh B, Brown VL, Hahn MG, Haigler CH. Cotton fiber cell walls of Gossypium hirsutum and Gossypium barbadense have differences related to loosely-bound xyloglucan. PLoS ONE. 2013;8(2):e56315. doi: 10.1371/journal.pone.0056315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meinert MC, Delmer DP. Changes in biochemical composition of the cell wall of the cotton fiber during development. Plant Physiol. 1977;59(6):1088–97. doi: 10.1104/pp.59.6.1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seagull RW. Cytoskeletal involvement in cotton fiber growth and development. Micron. 1993;24(6):643–60. [Google Scholar]
- 13.Hsieh Y, Honik E, Hartzell M. A developmental study of single fiber strength: greenhouse grown SJ-2 Acala cotton. Text Res J. 1995;65(2):101–12. [Google Scholar]
- 14.Hinchliffe DJ, Meredith WR, Delhom CD, Thibodeaux DP, Fang DD. Elevated growing degree days influence transition stage timing during cotton fiber development resulting in increased fiber-bundle strength. Crop Sci. 2011;51(4):1683–92. [Google Scholar]
- 15.Betancur L, Singh B, Rapp RA, Wendel JF, Marks MD, Roberts AW, Haigler CH. Phylogenetically distinct cellulose synthase genes support secondary wall thickening in Arabidopsis shoot trichomes and cotton fiber. J Int Plant Biol. 2010;52(2):205–20. doi: 10.1111/j.1744-7909.2010.00934.x. [DOI] [PubMed] [Google Scholar]
- 16.Arpat AB, Waugh M, Sullivan JP, Gonzales M, Frisch D, Main D, Wood T, Leslie A, Wing RA, Wilkins TA. Functional genomics of cell elongation in developing cotton fibers. Plant Mol Biol. 2004;54(6):911–29. doi: 10.1007/s11103-004-0392-y. [DOI] [PubMed] [Google Scholar]
- 17.Somerville C. Cellulose synthesis in higher plants. Annu Rev Cell Dev Biol. 2006;22:53–78. doi: 10.1146/annurev.cellbio.22.022206.160206. [DOI] [PubMed] [Google Scholar]
- 18.Scheller HV, Ulvskov P. Hemicelluloses. Annu Rev Plant Biol. 2010;61(1):263–89. doi: 10.1146/annurev-arplant-042809-112315. [DOI] [PubMed] [Google Scholar]
- 19.Cassan-Wang H, Goue N, Saidi MN, Legay S, Sivadon P, Goffner D, Grima-Pettenati J. Identification of novel transcription factors regulating secondary cell wall formation in Arabidopsis. Front Plant Sci. 2013;4:189. doi: 10.3389/fpls.2013.00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hussey SG, Mizrachi E, Creux NM, Myburg AA. Navigating the transcriptional roadmap regulating plant secondary cell wall deposition. Front Plant Sci. 2013;4:325. doi: 10.3389/fpls.2013.00325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ehlting J, Mattheus N, Aeschliman D, Li E, Hamberger B, Cullis I, Zhuang J, Kaneda M, Mansfield S, Samuels L, Ritland K, Ellis B, Bohlmann J, Douglas C. Global transcript profiling of primary stems from Arabidopsis thaliana identifies candidate genes for missing links in lignin biosynthesis and transcriptional regulators of fiber differentiation. Plant J. 2005;42(5):618–40. doi: 10.1111/j.1365-313X.2005.02403.x. [DOI] [PubMed] [Google Scholar]
- 22.Zhao Q, Dixon RA. Transcriptional networks for lignin biosynthesis: more complex than we thought? Trends Plant Sci. 2011;16(4):227–33. doi: 10.1016/j.tplants.2010.12.005. [DOI] [PubMed] [Google Scholar]
- 23.Wang N, Liu W, Peng Y. Gradual transition zone between cell wall layers and its influence on wood elastic modulus. J Mater Sci. 2013;48(14):5071–84. [Google Scholar]
- 24.Martin LK, Haigler CH. Cool temperature hinders flux from glucose to sucrose during cellulose synthesis in secondary wall stage cotton fibers. Cellulose. 2004;11(3–4):339–49. [Google Scholar]
- 25.Singh B, Avci U, Eichler Inwood SE, Grimson MJ, Landgraf J, Mohnen D, Sorensen I, Wilkerson CG, Willats WG, Haigler CH. A specialized outer layer of the primary cell wall joins elongating cotton fibers into tissue-like bundles. Plant Physiol. 2009;150(2):684–99. doi: 10.1104/pp.109.135459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tokumoto H, Wakabayashi K, Kamisaka S, Hoson T. Changes in the sugar composition and molecular mass distribution of matrix polysaccharides during cotton fiber development. Plant Cell Physiol. 2002;43(4):411–8. doi: 10.1093/pcp/pcf048. [DOI] [PubMed] [Google Scholar]
- 27.Tokumoto H, Wakabayashi K, Kamisaka S, Hoson T. Xyloglucan breakdown during cotton fiber development. J Plant Physiol. 2003;160(11):1411–4. doi: 10.1078/0176-1617-01066. [DOI] [PubMed] [Google Scholar]
- 28.Gou JY, Wang LJ, Chen SP, Hu WL, Chen XY. Gene expression and metabolite profiles of cotton fiber during cell elongation and secondary cell wall synthesis. Cell Res. 2007;17(5):422–34. doi: 10.1038/sj.cr.7310150. [DOI] [PubMed] [Google Scholar]
- 29.Sharma P, Jha AB, Dubey RS, Pessarakli M. Reactive oxygen species, oxidative damage, and antioxidative defense mechanism in plants under stressful conditions. J Botany. 2012;2012. ArticleID 217037. doi.org/10.1155/2012/217037.
- 30.Zhang D, Zhang T, Guo W. Effect of H2O2 on fiber initiation using fiber retardation initiation mutants in cotton (Gossypium hirsutum). J Plant Physiol. 2010;167(5):393–9. [DOI] [PubMed]
- 31.Mei W, Qin Y, Song W, Li J, Zhu Y. Cotton GhPOX1 encoding plant class III peroxidase may be responsible for the high level of reactive oxygen species production that is related to cotton fiber elongation. J Genet Genomics. 2009;36(3):141–50. [DOI] [PubMed]
- 32.Li H, Qin Y, Pang Y, Song W, Mei W, Zhu Y. A cotton ascorbate peroxidase is involved in hydrogen peroxide homeostasis during fibre cell development. New Phytol. 2007;175(3):462–71. doi: 10.1111/j.1469-8137.2007.02120.x. [DOI] [PubMed] [Google Scholar]
- 33.Gottula J, Price K, Allen RD, Mullinix B, Wright RJ. Efficacy of antioxidant overproduction on fiber growth and maturation in cotton. Crop Sci. 2009;49(5):1733–41. [Google Scholar]
- 34.Potikha TS, Collins CC, Johnson DI, Delmer DP, Levine A. The involvement of hydrogen peroxide in the differentiation of secondary walls in cotton fibers. Plant Physiol. 1999;119(3):849–58. doi: 10.1104/pp.119.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Y, Bian S, Yao Y, Liu J. Comparative proteomic analysis provides new insights into the fiber elongating process in cotton. J Proteome Res. 2008;7(11):4623–37. doi: 10.1021/pr800550q. [DOI] [PubMed] [Google Scholar]
- 36.Kurek I, Kawagoe Y, Jacob-Wilk D, Doblin M, Delmer D. Dimerization of cotton fiber cellulose synthase catalytic subunits occurs via oxidation of the zinc-binding domains. Proc Natl Acad Sci U S A. 2002;99(17):11109–14. doi: 10.1073/pnas.162077099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hovav R, Udall JA, Chaudhary B, Hovav E, Flagel L, Hu G, Wendel JF. The evolution of spinnable cotton fiber entailed prolonged development and a novel metabolism. PLOS GENET. 2008;4(2):e25. doi: 10.1371/journal.pgen.0040025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chaudhary B, Hovav R, Rapp R, Verma N, Udall JA, Wendel JF. Global analysis of gene expression in cotton fibers from wild and domesticated Gossypium barbadense. Evol Dev. 2008;10(5):567–82. doi: 10.1111/j.1525-142X.2008.00272.x. [DOI] [PubMed] [Google Scholar]
- 39.Rapp RA, Haigler CH, Flagel L, Hovav RH, Udall JA, Wendel JF. Gene expression in developing fibres of Upland cotton (Gossypium hirsutum L.) was massively altered by domestication. BMC Biol. 2010;8:139. doi: 10.1186/1741-7007-8-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou M, Sun G, Sun Z, Tang Y, Wu Y. Cotton proteomics for deciphering the mechanism of environment stress response and fiber development. J Proteomics. 2014;105:74–84. doi: 10.1016/j.jprot.2014.03.017. [DOI] [PubMed] [Google Scholar]
- 41.Hu G, Koh J, Yoo M, Pathak D, Chen S, Wendel JF. Proteomics profiling of fiber development and domestication in upland cotton (Gossypium hirsutum L.) Planta. 2014;240(6):1237–51. doi: 10.1007/s00425-014-2146-7. [DOI] [PubMed] [Google Scholar]
- 42.Moller IM, Jensen PE, Hansson A. Oxidative modifications to cellular components in plants. Annu Rev Plant Biol. 2007;58:459–81. doi: 10.1146/annurev.arplant.58.032806.103946. [DOI] [PubMed] [Google Scholar]
- 43.Jacques S, Ghesquière B, Van Breusegem F, Gevaert K. Plant proteins under oxidative attack. Proteomics. 2013;13(6):932–40. doi: 10.1002/pmic.201200237. [DOI] [PubMed] [Google Scholar]
- 44.Xiong Y, Contento AL, Nguyen PQ, Bassham DC. Degradation of oxidized proteins by autophagy during oxidative stress in Arabidopsis. Plant Physiol. 2007;143(1):291–9. doi: 10.1104/pp.106.092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fini A, Brunetti C, Di Ferdinando M, Ferrini F, Tattini M. Stress-induced flavonoid biosynthesis and the antioxidant machinery of plants. Plant Signal Behav. 2011;6(5):709–11. doi: 10.4161/psb.6.5.15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meyer AJ. The integration of glutathione homeostasis and redox signaling. J Plant Physiol. 2008;165(13):1390–403. doi: 10.1016/j.jplph.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 47.Paterson AH, Wendel JF, Gundlach H, Guo H, Jenkins J, Jin D, Llewellyn D, Showmaker KC, Shu S, Udall J, Yoo M, Byers R, Chen W, Doron-Faigenboim A, Duke MV, Gong L, Grimwood J, Grover C, Grupp K, Hu G, Lee T, Li J, Lin L, Liu T, Marler BS, Page JT, Roberts AW, Romanel E, Sanders WS, Szadkowski E, et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature. 2012;492(7429):423–7. doi: 10.1038/nature11798. [DOI] [PubMed] [Google Scholar]
- 48.Guan X, Nah G, Song Q, Udall JA, Stelly DM, Chen ZJ. Transcriptome analysis of extant cotton progenitors revealed tetraploidization and identified genome-specific single nucleotide polymorphism in diploid and allotetraploid cotton. BMC Res Notes. 2014;7(1):493. doi: 10.1186/1756-0500-7-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar L, Futschik ME. Mfuzz: a software package for soft clustering of microarray data. Bioinformation. 2007;2(1):5–7. doi: 10.6026/97320630002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cacas J, Furt F, Le Guédard M, Schmitter J, Buré C, Gerbeau-Pissot P, Moreau P, Bessoule J, Simon-Plas F, Mongrand S. Lipids of plant membrane rafts. Prog Lipid Res. 2012;51(3):272–99. doi: 10.1016/j.plipres.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 51.Huang J, Jan C. Linoleamide, a brain lipid that induces sleep, increases cytosolic Ca2+ levels in MDCK renal tubular cells. Life Sci. 2001;68(9):997–1004. doi: 10.1016/s0024-3205(00)01002-x. [DOI] [PubMed] [Google Scholar]
- 52.Aubert S, Choler P, Pratt J, Douzet R, Gout E, Bligny R. Methyl-beta-D-glucopyranoside in higher plants: accumulation and intracellular localization in Geum montanum L. leaves and in model systems studied by 13C nuclear magnetic resonance. J Exp Bot. 2004;55(406):2179–89. doi: 10.1093/jxb/erh235. [DOI] [PubMed] [Google Scholar]
- 53.Liu Q, Talbot M, Llewellyn DJ. Pectin methylesterase and pectin remodelling differ in the fibre walls of two gossypium species with very different fibre properties. PLoS ONE. 2013;8(6):e65131. doi: 10.1371/journal.pone.0065131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Komarova TV, Sheshukova EV, Dorokhov YL. Cell wall methanol as a signal in plant immunity. Front Plant Sci. 2014;5:101. doi: 10.3389/fpls.2014.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.ElSayed A, Rafudeen M, Golldack D. Physiological aspects of raffinose family oligosaccharides in plants: protection against abiotic stress. Plant Biol. 2014;16(1):1–8. doi: 10.1111/plb.12053. [DOI] [PubMed] [Google Scholar]
- 56.Fukuda H. Programmed cell death of tracheary elements as a paradigm in plants. Plant Mol Biol. 2000;44(3):245–53. doi: 10.1023/a:1026532223173. [DOI] [PubMed] [Google Scholar]
- 57.Wanjie SW, Welti R, Moreau RA, Chapman KD. Identification and quantification of glycerolipids in cotton fibers: reconciliation with metabolic pathway predictions from DNA databases. Lipids. 2005;40(8):773–85. doi: 10.1007/s11745-005-1439-4. [DOI] [PubMed] [Google Scholar]
- 58.Eckardt NA. Oxylipin signaling in plant stress responses. Plant Cell. 2008;20(3):495–7. doi: 10.1105/tpc.108.059485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Besseau S, Hoffmann L, Geoffroy P, Lapierre C, Pollet B, Legrand M. Flavonoid accumulation in Arabidopsis repressed in lignin synthesis affects auxin transport and plant growth. Plant Cell. 2007;19(1):148–62. doi: 10.1105/tpc.106.044495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Delmer DP, Pear JR, Andrawis A, Stalker DM. Genes encoding small GTP-binding proteins analogous to mammalian rac are preferentially expressed in developing cotton fibers. Mol Gen Genetics. 1995;248(1):43–51. doi: 10.1007/BF02456612. [DOI] [PubMed] [Google Scholar]
- 61.Brembu T, Winge P, Bones AM. The small GTPase AtRAC2/ROP7 is specifically expressed during late stages of xylem differentiation in Arabidopsis. J Exp Bot. 2005;56(419):2465–76. doi: 10.1093/jxb/eri239. [DOI] [PubMed] [Google Scholar]
- 62.Foreman J, Demidchik V, Bothwell JH, Mylona P, Miedema H, Torres MA, Linstead P, Costa S, Brownlee C, Jones JD. Reactive oxygen species produced by NADPH oxidase regulate plant cell growth. Nature. 2003;422(6930):442–6. doi: 10.1038/nature01485. [DOI] [PubMed] [Google Scholar]
- 63.Takeda S, Gapper C, Kaya H, Bell E, Kuchitsu K, Dolan L. Local positive feedback regulation determines cell shape in root hair cells. Science. 2008;319(5867):1241–4. doi: 10.1126/science.1152505. [DOI] [PubMed] [Google Scholar]
- 64.Pelagio-Flores R, Ortiz-Castro R, Mendez-Bravo A, Macias-Rodriguez L, Lopez-Bucio J. Serotonin, a tryptophan-derived signal conserved in plants and animals, regulates root system architecture probably acting as a natural auxin inhibitor in Arabidopsis thaliana. Plant Cell Physiol. 2011;52(3):490–508. doi: 10.1093/pcp/pcr006. [DOI] [PubMed] [Google Scholar]
- 65.Shen Y, Lindemeyer AK, Gonzalez C, Shao XM, Spigelman I, Olsen RW, Liang J. Dihydromyricetin as a novel anti-alcohol intoxication medication. J Neurosci. 2012;32(1):390–401. doi: 10.1523/JNEUROSCI.4639-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Forde BG. Glutamate signalling in roots. J Exp Bot. 2014;65(3):779–87. doi: 10.1093/jxb/ert335. [DOI] [PubMed] [Google Scholar]
- 67.Yu GH, Zou J, Feng J, Peng XB, Wu JY, Wu YL, et al. Exogenous gamma-aminobutyric acid (GABA) affects pollen tube growth via modulating putative Ca2 + -permeable membrane channels and is coupled to negative regulation on glutamate decarboxylase. J Exp Bot. 2014;65(12):3235–48. doi: 10.1093/jxb/eru171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jarvis MC. Cellulose biosynthesis: counting the chains. Plant Physiol. 2013;163(4):1485–6. doi: 10.1104/pp.113.231092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin L, Tang H, Compton RO, Lemke C, Rainville LK, Wang X, Rong J, Rana MK, Paterson A. Comparison analysis of Gossypium and Vitis genomes indicates genome duplication specific to the Gossypium lineage. Genomics. 2011;97:313–20. doi: 10.1016/j.ygeno.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 70.Yin Y, Johns MA, Cao H, Rupani M. A survey of plant and algal genomes and transcriptomes reveals new insights into the evolution and function of the cellulose synthase superfamily. BMC Genomics. 2014;15(1):260. doi: 10.1186/1471-2164-15-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cocuron JC, Lerouxel O, Drakakaki G, Alonso AP, Liepman AH, Keegstra K, Raikhel N, Wilkerson CG. A gene from the cellulose synthase-like C family encodes a beta-1,4 glucan synthase. Proc Natl Acad Sci U S A. 2007;104(20):8550–5. doi: 10.1073/pnas.0703133104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liepman A, Wilkerson C, Keegstra K. Expression of cellulose synthase-like (Csl) genes in insect cells reveals that CslA family members encode mannan synthases. Proc Natl Acad Sci U S A. 2005;102(6):2221–6. doi: 10.1073/pnas.0409179102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rajasundaram D, Runavot J, Guo X, Willats WG, Meulewaeter F, Selbig J. Understanding the relationship between cotton fiber properties and non-cellulosic cell wall polysaccharides. PLoS ONE. 2014;9(11):e112168. doi: 10.1371/journal.pone.0112168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Park S, Szumlanski AL, Gu F, Guo F, Nielsen E. A role for CSLD3 during cell-wall synthesis in apical plasma membranes of tip-growing root-hair cells. Nat Cell Biol. 2011;13(8):973–80. doi: 10.1038/ncb2294. [DOI] [PubMed] [Google Scholar]
- 75.Yoshikawa T, Eiguchi M, Hibara K, Ito J, Nagato Y. Rice slender leaf 1 gene encodes cellulose synthase-like D4 and is specifically expressed in M-phase cells to regulate cell proliferation. J Exp Bot. 2013;64(7):2049–61. doi: 10.1093/jxb/ert060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Haigler CH, Singh B, Wang G, Zhang D. Genomics of cotton fiber secondary wall deposition and cellulose biogenesis. In: Paterson AH, editor. Genetics and Genomics of Cotton, Plant Genetics and Genomics: Crops and Models 3. New York: Springer Science + Business Media, LLC; 2009. pp. 385–417. [Google Scholar]
- 77.Zhong R, Lee C, Ye Z. Evolutionary conservation of the transcriptional network regulating secondary cell wall biosynthesis. Trends Plant Sci. 2010;15(11):625–32. doi: 10.1016/j.tplants.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 78.Haigler C, Zhang D, Wilkerson C. Biotechnological improvement of cotton fibre maturity. Physiol Plantarum. 2005;124(3):285–94. [Google Scholar]
- 79.Gong S, Huang G, Sun X, Qin L, Li Y, Zhou L, Li X. Cotton KNL1, encoding a class II KNOX transcription factor, is involved in regulation of fibre development. J Expt Bot. 2014;65(15):4133–47. doi: 10.1093/jxb/eru182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Han L, Li Y, Wang H, Wu X, Li C, Luo M, Wu S, Kong Z, Pei Y, Jiao G, Xia G. The dual functions of WLIM1a in cell elongation and secondary wall formation in developing cotton fibers. Plant Cell. 2013;25(11):4421–38. doi: 10.1105/tpc.113.116970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fan L, Shi W, Hu W, Hao X, Wang D, Yuan H, Yan H. Molecular and biochemical evidence for phenylpropanoid synthesis and presence of wall-linked phenolics in cotton fibers. J Int Plant Biol. 2009;51(7):626–37. doi: 10.1111/j.1744-7909.2009.00840.x. [DOI] [PubMed] [Google Scholar]
- 82.Lee Y, Alexander D, Wulff J, Olsen J. Changes in metabolite profiles in Norway spruce shoot tips during short-day induced winter bud development and long-day induced bud flush. Metabolomics. 2014;10(5):842–58. [Google Scholar]
- 83.Fraser CM, Chapple C. The phenylpropanoid pathway in Arabidopsis. Arabidopsis Book. 2011;9:e0152. doi: 10.1199/tab.0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Noctor G, Foyer CH. Ascorbate and glutathione: keeping active oxygen under control. Annu Rev Plant Biol. 1998;49(1):249–79. doi: 10.1146/annurev.arplant.49.1.249. [DOI] [PubMed] [Google Scholar]
- 85.Kranner I, Birtić S, Anderson KM, Pritchard HW. Glutathione half-cell reduction potential: a universal stress marker and modulator of programmed cell death? Free Radical Bio Med. 2006;40(12):2155–65. doi: 10.1016/j.freeradbiomed.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 86.Kubo M, Udagawa M, Nishikubo N, Horiguchi G, Yamaguchi M, Ito J, Mimura T, Fukuda H, Demura T. Transcription switches for protoxylem and metaxylem vessel formation. Genes Dev. 2005;19(16):1855–60. doi: 10.1101/gad.1331305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fry S. Oxidative scission of plant cell wall polysaccharides by ascorbate-induced hydroxyl radicals. Biochem J. 1998;332:507–15. doi: 10.1042/bj3320507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dumville JC, Fry SC. Solubilisation of tomato fruit pectins by ascorbate: a possible non-enzymic mechanism of fruit softening. Planta. 2003;217(6):951–61. doi: 10.1007/s00425-003-1061-0. [DOI] [PubMed] [Google Scholar]
- 89.Haroldsen VM, Chi-Ham CL, Kulkarni S, Lorence A, Bennett AB. Constitutively expressed DHAR and MDHAR influence fruit, but not foliar ascorbate levels in tomato. Plant Physiol Biochem. 2011;49(10):1244–9. doi: 10.1016/j.plaphy.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yin L, Wang S, Eltayeb AE, Uddin MI, Yamamoto Y, Tsuji W, Takeuchi Y, Tanaka K. Overexpression of dehydroascorbate reductase, but not monodehydroascorbate reductase, confers tolerance to aluminum stress in transgenic tobacco. Planta. 2010;231(3):609–21. doi: 10.1007/s00425-009-1075-3. [DOI] [PubMed] [Google Scholar]
- 91.Van Aken O, Giraud E, Clifton R, Whelan J. Alternative oxidase: a target and regulator of stress responses. Physiol Plantarum. 2009;137(4):354–61. doi: 10.1111/j.1399-3054.2009.01240.x. [DOI] [PubMed] [Google Scholar]
- 92.Kim HJ, Tang Y, Moon HS, Delhom CD, Fang DD. Functional analyses of cotton (Gossypium hirsutum L.) immature fiber (im) mutant infer that fiber cell wall development is associated with stress responses. BMC Genomics. 2013;14(1):889. doi: 10.1186/1471-2164-14-889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lisko KA, Aboobucker SI, Torres R, Lorence A. Engineering elevated Vitamin C in plants to improve their nutritional content, growth, and tolerance to abiotic stress. In: Jetter R, editor. Phytochemicals—biosynthesis, function, and application. New York: Springer Science + Business Media; 2014. pp. 109–28. [Google Scholar]
- 94.Ruan YL, Xu SM, White R, Furbank RT. Genotypic and developmental evidence for the role of plasmodesmatal regulation in cotton fiber elongation mediated by callose turnover. Plant Physiol. 2004;136(4):4104–13. doi: 10.1104/pp.104.051540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Soto G, Alleva K, Mazzella MA, Amodeo G, Muschietti JP. AtTIP1; 3 and AtTIP5; 1, the only highly expressed Arabidopsis pollen-specific aquaporins, transport water and urea. FEBS Lett. 2008;582(29):4077–82. doi: 10.1016/j.febslet.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 96.Wudick MM, Luu DT, Tournaire-Roux C, Sakamoto W, Maurel C. Vegetative and sperm cell-specific aquaporins of Arabidopsis highlight the vacuolar equipment of pollen and contribute to plant reproduction. Plant Physiol. 2014;164(4):1697–706. doi: 10.1104/pp.113.228700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Park W, Scheffler BE, Bauer PJ, Campbell BT. Identification of the family of aquaporin genes and their expression in upland cotton (Gossypium hirsutum L.) BMC Plant Biol. 2010;10:142. doi: 10.1186/1471-2229-10-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cao J. The pectin lyases in Arabidopsis thaliana: evolution, selection, and expression profiles. PLoS ONE. 2012;7(10):e46944. doi: 10.1371/journal.pone.0046944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Senechal F, Wattier C, Rusterucci C, Pelloux J. Homogalacturonan-modifying enzymes: structure, expression, and roles in plants. J Exp Bot. 2014;65(18):5125–60. doi: 10.1093/jxb/eru272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang H, Guo Y, Lv F, Zhu H, Wu S, Jiang Y, Li F, Zhou B, Guo W, Zhang T. The essential role of GhPEL gene, encoding a pectate lyase, in cell wall loosening by depolymerization of the de-esterified pectin during fiber elongation in cotton. Plant Mol Biol. 2010;72(4–5):397–406. doi: 10.1007/s11103-009-9578-7. [DOI] [PubMed] [Google Scholar]
- 101.Lim E, Ashford DA, Hou B, Jackson RG, Bowles DJ. Arabidopsis glycosyltransferases as biocatalysts in fermentation for regioselective synthesis of diverse quercetin glucosides. Biotechnol Bioeng. 2004;87(5):623–31. doi: 10.1002/bit.20154. [DOI] [PubMed] [Google Scholar]
- 102.Nishizawa A, Yabuta Y, Shigeoka S. Galactinol and raffinose constitute a novel function to protect plants from oxidative damage. Plant Physiol. 2008;147(3):1251–63. doi: 10.1104/pp.108.122465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Olas B, Saluk-Juszczak J, Nowak P, Glowacki R, Bald E, Wachowicz B. Protective effects of D-glucaro 1, 4-lactone against oxidative/nitrative modifications of plasma proteins. Nutrition. 2007;23(2):164–71. doi: 10.1016/j.nut.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 104.Sheldrake R, Boodley J. Cornell peat-like mixes for commercial plant growing. Coop Ext Info Bull. 1973;43. [http://www.greenhouse.cornell.edu/crops/factsheets/peatlite.pdf]
- 105.Saravitz CH, Downs RJ, Thomas JF. North Carolina State University Phytotron Procedural Manual. [http:// http://www.ncsu.edu/phytotron/manual.pdf]
- 106.Hu G, Koh J, Yoo M, Grupp K, Chen S, Wendel JF. Proteomic profiling of developing cotton fibers from wild and domesticated Gossypium barbadense. New Phytol. 2013;200(2):570–82. doi: 10.1111/nph.12381. [DOI] [PubMed] [Google Scholar]
- 107.Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 108.Evans AM, DeHaven CD, Barrett T, Mitchell M, Milgram E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem. 2009;81(16):6656–67. doi: 10.1021/ac901536h. [DOI] [PubMed] [Google Scholar]
- 109.Yobi A, Wone BW, Xu W, Alexander DC, Guo L, Ryals JA, Oliver MJ, Cushman JC. Metabolomic profiling in Selaginella lepidophylla at various hydration states provides new insights into the mechanistic basis of desiccation tolerance. Mol Plant. 2013;6(2):369–85. doi: 10.1093/mp/sss155. [DOI] [PubMed] [Google Scholar]
- 110.DeHaven CD, Evans AM, Dai H, Lawton KA. Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J Cheminformatics. 2010;2(1):9. doi: 10.1186/1758-2946-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lawton KA, Berger A, Mitchell M, Milgram KE, Evans AM, Guo L, Hanson RW, Kalhan SC, Ryals JA, Milburn MV. Analysis of the adult human plasma metabolome. Pharmacogenomics. 2008;9(4):383–97. doi: 10.2217/14622416.9.4.383. [DOI] [PubMed] [Google Scholar]
- 112.Evans A, Mitchell M, Dai H, DeHaven C. Categorizing ion-features in liquid chromatography/mass spectrometry metabolomics data. Metabolomics. 2012;2:110. [Google Scholar]
- 113.Oliver MJ, Guo L, Alexander DC, Ryals JA, Wone BW, Cushman JC. A sister group contrast using untargeted global metabolomic analysis delineates the biochemical regulation underlying desiccation tolerance in Sporobolus stapfianus. Plant Cell. 2011;23(4):1231–48. doi: 10.1105/tpc.110.082800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, Lumley T, 1345 Maechler M, Magnusson A, Moeller S. gplots: various R programming tools for plotting data. [http://cran.r-project.org/web/packages/gplots/index.html]
- 115.Chen H, Boutros PC. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinf. 2011;12:35. doi: 10.1186/1471-2105-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]