

Abstract
Tetrodotoxin-sensitive persistent sodium currents, INaP, that activate at subthreshold voltages, have been detected in numerous vertebrate and invertebrate neurons. These currents are believed to be critical for regulating neuronal excitability. However, the molecular mechanism underlying INaP is controversial. In this study, we identified an INaP with a broad range of voltage dependence, from −60 mV to 20 mV, in a Drosophila sodium channel variant expressed in Xenopus oocytes. Mutational analysis revealed that two variant-specific amino acid changes, I260T in the S4–S5 linker of domain I (ILS4–S5) and A1731V in the voltage sensor S4 of domain IV (IVS4), contribute to the INaP. I260T is critical for the portion of INaP at hyperpolarized potentials. The T260-mediated INaP is likely the result of window currents flowing in the voltage range where the activation and inactivation curves overlap. A1731V is responsible for impaired inactivation and contributes to the portion of INaP at depolarized potentials. Furthermore, A1731V causes enhanced activity of two site-3 toxins which induce persistent currents by inhibiting the outward movement of IVS4, suggesting that A1731V inhibits the outward movement of IVS4. These results provided molecular evidence for the involvement of distinct mechanisms in the generation of INaP: T260 contributes to INaP via enhancement of the window current, whereas V1731 impairs fast inactivation probably by inhibiting the outward movement of IVS4.
Keywords: persistent current, Drosophila sodium channel, window current
Graphical Abstract
Voltage-gated sodium channels are critical for the initiation and propagation of action potentials in neurons and other excitable cells (Catterall, 2000). They consist of a large pore-forming α-subunit, which is associated with a variable number of smaller β-subunits in different excitable tissues. The α-subunit comprises four homologous domains (I–IV), each having six membrane spanning segments (S1–6) (Catterall, 2000). The S5 and S6 segments form the inner pore, whereas the reentrant loops (called the P-region) connecting S5 and S6 segments form the outer pore, which serves as the ion selectivity filter. The S1–S4 segments function as the voltage sensor; each S4 segment has five to eight basic residues, either arginine or lysine, separated from one another by two neutral residues. In response to membrane depolarization, S4s move outward, initiating conformational changes, which results in channel opening (i.e., activation) and an inward sodium current. This current is transient because a few milliseconds after opening, the channels are inactivated (i.e., closed). The short intracellular loop connecting domains III and IV, known as the fast inactivation gate, serves as an intracellular blocking particle that occludes the pore during inactivation (Catterall, 2012).
Besides the transient (i.e., fast inactivating) sodium currents (INaT), which are responsible for the upstroke of action potentials, there are also a tetrodotoxin (TTX)-sensitive non-inactivating or persistent currents (INaP), which activate at subthreshold voltages and cannot inactivate completely even with prolonged depolarization (Taylor, 1993, Crill, 1996, Stafstrom, 2007, Kiss, 2008). INaP have been detected in numerous types of vertebrate neurons in the brain, such as the suprachiasmatic nucleus (Pennartz et al., 1997, Jackson et al., 2004); cerebellar nuclei (Raman et al., 2000); and tuberomammillary neurons (Llinas and Alonso, 1992, Uteshev et al., 1995, Taddese and Bean, 2002). INaP are also found in invertebrate neurons, such as squid giant axons (Rakowski et al., 2002, Clay, 2003), squid olfactory receptor neurons (Chen and Lucero, 1999), leech spontaneously active heart interneurons (Opdyke and Calabrese, 1994), Drosophila aCC/RP2 motor neurons (Mee et al., 2004), and cockroach terminal abdominal efferent dorsal unpaired median (DUM) octopaminergic neurons (Lapied et al., 1990). It is generally believed that they are critical for acceleration of firing rates, boosting synaptic inputs, and promotion of oscillatory neural activities (Taylor, 1993, Crill, 1996, Stafstrom, 2007). Whether INaP arise from distinct sodium channels or rather from different gating modes of a common sodium channel is an important yet unresolved issue (Kiss et al., 2009).
Mammals have nine genes that encode nine sodium channel α-subunit isoforms, with different gating properties and different expression patterns in various cell types, tissues, and developmental stages, presumably to accommodate unique physiological functions in specific neuronal and non-neuronal cells (Catterall, 2000, Goldin et al., 2000, Frank and Catterall, 2003). In contrast to mammals, most insects including Drosophila melanogaster appear to have only a single sodium channel gene that encodes an equivalent of the α-subunit of mammalian sodium channels (Loughney et al., 1989, Feng et al., 1995, Warmke et al., 1997, Dong, 2010). Despite having only a single gene, insects generate many sodium channel variants with different gating and pharmacological properties by alternative splicing and RNA editing (Dong, 2007, 2010). For example, an RNA editing event leading to an F to I substitution at the C-terminal end of a cockroach sodium channel resulted in the generation of INaP (Liu et al., 2004). Also, two mutually exclusive exons l and k of the Drosophila sodium channel, DmNav, regulate the size of INaP current (Lin et al., 2009).
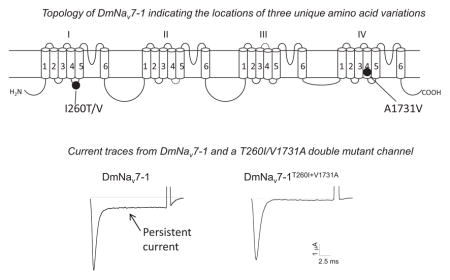
In the course of functional analysis of DmNav variants from D. melanogaster, we found that DmNav7-1 exhibits a prominent INaP (10–15% of the size of INaT) with extremely hyperpolarizing voltage dependence of both activation and inactivation compared with other DmNav variants (Olson et al., 2008). Mutational and functional analyses of this variant in Xenopus oocytes revealed two amino acid substitutions, which are unique to DmNav7-1, that contribute to the generation of the INaP. They are I260T in the linker connecting S4 and S5 in domain I (IL45), and A1731V in S4 of domain IV (IVS4). I260T is responsible for the hyperpolarizing portion of INaP;, whereas A1731V is responsible for the depolarizing part of INaP. The T260-mediated INaP is the result of window currents flowing in the voltage range where the activation and inactivation curves overlap. In contrast, the V1731-mediated INaP is the result of impaired fast inactivation. In addition, we found that the V1731-mediated INaP enhanced the action of two site 3 peptide toxins, which are known to induce INaP by inhibiting the outward movement of IVS4. These results suggest that the V1731-mediated INaP originates from an impaired movement of IVS4. The findings from this study link the generation of INaP in Drosophila sodium channels to both window currents and possible perturbations in the movement of the voltage sensor in domain IV.
EXPERIMENTAL PROCEDURES
Sodium channel variants of Drosophila melanogaster
Sodium channel variants from D. melanogaster (DmNav) were previously cloned and characterized in Xenopus oocytes (Olson et al., 2008).
Site-directed mutagenesis
Site-directed mutagenesis was performed by Polymerase Chain Reaction (PCR) using specific primers and Pfu Turbo DNA polymerase (Stratagene, La Jolla, CA). All mutagenesis results were verified by DNA sequencing.
Expression of DmNav channels in Xenopus oocytes
The procedures for oocyte preparation and cRNA injection were identical to those described previously (Olson et al., 2008). For robust expression of sodium currents, cRNA was co-injected into oocytes with Drosophila melanogaster tipE cRNA (1:1 ratio), which enhances sodium channel expression (Feng et al., 1995, Warmke et al., 1997).
Electrophysiological recording and analysis
The gating properties of DmNav channels were determined using the two electrode voltage clamp technique. Protocols for electrophysiological recording and data analysis were similar to those described previously (Tan et al., 2005). The voltage dependence of sodium channel conductance (G) was calculated by measuring the peak current at test potentials (20 ms) ranging from −80 mV to +65 mV in 5-mV increments and divided by the driving force (V−Vrev), where V is the test potential and Vrev is the reversal potential for sodium. Peak conductance values were normalized to the maximal peak conductance (Gmax) and fitted with a two-state Boltzmann equation of the form G/Gmax=[1 + exp (V−V1/2)/k]−1, in which V is the potential of the voltage pulse, V1/2 is the voltage for half-maximal activation, and k is the slope factor. The voltage dependence of fast inactivation was determined using 100-ms prepulses ranging from −120 to 40 mV in 5-mV increments and then a −10-mV test pulse for 20 ms. The normalized peak current was plotted as a function of the prepulse potential. The data were fitted with a Boltzmann equation to generate V0.5, the midpoint of the inactivation curve, and k, the slope factor.
A double pulse protocol was used to determine the onset of fast inactivation. Prepulse potentials ranging from −80 to 20 mV in 10-mV increments of varying durations were applied from a holding potential of −120 mV followed by a test pulse to 10 mV for 20 ms. To determine recovery from fast inactivation, sodium channels were inactivated by a 100 ms depolarizing pulse to −10 mV and then repolarized from −120 mV to −50 mV for an interval of variable durations followed by a 20 ms test pulse to −10 mV.
A slow depolarizing ramp protocol from a holding potential of −100 to 20 mV at a ramp rate of 0.2mV/ms, which promotes inactivation of the rapidly inactivating component of sodium current, was used to examine the properties of INaP.
The development of fast inactivation was determined by applying a prepulse of −60 mV of varying durations from a holding potential of −120 mV followed by a −10 mV test pulse for 20 ms to measure the fraction of sodium channels that were inactivated during the prepulse. Data for the development of inactivation were fitted to a single exponential function and plotted as a function of the development pulse voltage (from −70 mV to −30 mV in 10-mV increments).
The recovery from fast inactivation was measured using a two-pulse protocol, in which channels were fast-inactivated by a 100 ms pulse to −10 mV, and then were allowed to recover at −80 mV for an increasing time, and finally a 20 ms of −10 mV test pulse to measure the fraction of recovered channels. Data for recovery from fast inactivation were fitted to a single exponential function and plotted as a function of the recovery voltage (from −120 mV to −50 mV in 10-mV increments).
Measurement of toxin effects
Two site 3 toxins in their recombinant forms were used: Av3, a 27 amino-acid-peptide toxin from the sea anemone Anemonia viridis (previously named Anemonia sulcata) (Moran et al., 2007) and LqhαIT, a 64 residue long polypeptide from the scorpion Leiurus quinquestriatus (Zilberberg et al., 1996). Stock solutions of Av3 (50 μM) and LqhαIT (10 μM) were prepared in distilled water containing 10% bovine serum albumin to prevent toxin adherence to vials. The extent of fast inactivation was assayed by measuring the peak current as well as the current 20 ms after depolarization onset. The ratio I20ms/Ipeak gives an estimate of the probability for channels not to be inactivated after 20 ms; a value of zero represents complete inactivation in 20 ms, whereas a value of one indicates no inactivation (Chen et al., 2000).
Statistical analysis
Data are presented as the mean ± SEM. Statistical analysis was performed using a one-way ANOVA test and Scheffe’s post hoc analysis. Significance values were set at P < 0.05 or as indicated in the table and figure legends.
RESULTS
Substitutions T260I and V1731A eliminate INaP and the hyperpolarized shifts in gating
The DmNav7-1 variant was co-expressed in Xenopus oocytes with TipE, which is known to enhance the expression of insect sodium channels (Feng et al., 1995, Warmke et al., 1997). Representative sodium currents from oocytes expressing DmNav7-1 and TipE are shown in Fig. 1A. Besides the major component of transient sodium current (INaT), there is a fraction of sodium current that failed to inactivate, i.e., persistent sodium current (INaP). In addition, DmNav7-1 channels activate and inactivate at more hyperpolarized membrane potentials compared with other DmNav variants (Olson et al., 2008) (Fig. 1B). INaP was normalized to INaT and the voltage dependence of normalized INaP was plotted in Fig. 1B along with the voltage-dependence of INaT. INaP currents activated over a broad voltage range, starting at ca. −70 mV and peaking at ca. −20 mV. The amplitude of the maximal INaP was 13% of INaT (Fig. 1B). Both INaT and INaP were completely abolished in the presence of 10 nM tetrodotoxin (TTX), which specifically blocks voltage-gated sodium channels.
Fig. 1. Large persistent current with broad voltage dependence of activation.
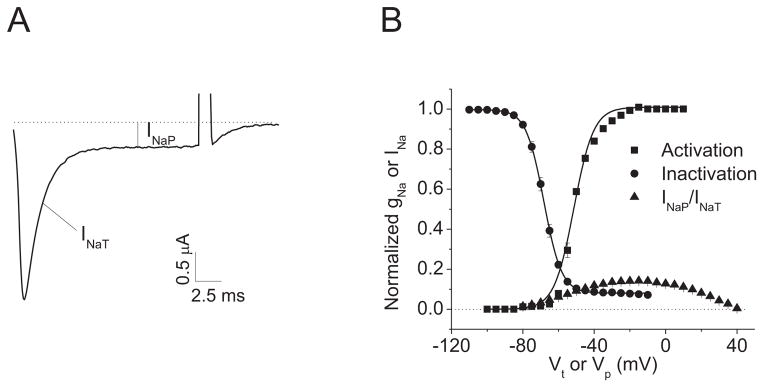
A. A current trace indicating INaP. The sodium current trace was elicited by a 20 ms test pulse to −10 mV from a holding potential of −120 mV. B. Voltage dependences of channel activation and inactivation. The percentage of INaP was normalized to the INaT peak.
A total of 65 DmNav variants were previously isolated from the adult D. melanogaster and characterized in Xenopus oocytes (one variant reported in Liu et al., 2004; and 64 variants described in Olson et al., 2008). The 64 variants were grouped into 29 types based on the alternative exon usage (Olson et al., 2008). Compared to a DmNav cDNA sequence previously deposited in GenBank (Accession number: M32078), 12 amino acid changes are found in DmNav7-1 (Olson et al., 2008). Six of them (I260T, K303R, W319R, E771G, H1676R and A1731V) are unique to DmNav7-1 (Fig. 2A), whereas other variants possess I260, K303, W319, E771, H1676 and A1731 like the deposited cDNA sequence. To determine which amino acid change(s) is responsible for the INaP and hyperpolarized gating of DmNav7-1, we individually replaced the six unique residues in DmNav7-1 with residues found in the deposited cDNA and other DmNav variants. The resultant six mutant channels, T260I, R303K, R319W, G771E, R1676H and V1731A, were functionally examined in Xenopus oocytes.
Fig. 2. Substitutions T260I and V1731A abolished the INaP in DmNav7-1.

A. Schematic diagram of DmNav7-1 indicating the six amino acid changes unique to this DmNav variant. B. Representative peak current traces showing INaT and INaP of DmNav7-1 and three mutant channels. Sodium currents were elicited by a 20-ms test pulse to −20 mV (DmNav7-1), −40 mV (V1731A) or −10 mV (T260I and T260I+V1731A) from a holding potential of −120 mV. C. INaP of DmNav7-1 and six mutant channels. The percentage of INaP was normalized to the peak of INaT. D and E. Voltage dependence of activation (D) and inactivation (E) of DmNav7-1 and three mutants. The half-maximal voltage for activation and inactivation (V1/2) and slope factor (k) of DmNav7-1 and mutant channels are shown in Table 1.
The amplitude of INaP was decreased in two mutant channels, T260I and V1731A, and was essentially abolished in the double mutant, T260I + V1731A (Fig. 2B and 2C). However, the other four substitutions had no effect on INaP (Fig. 2C). Moreover, T260I shifted the voltage dependence of channel activation in the depolarizing direction by approximately 22 mV (Fig. 2D, Table 1), but had no significant effect on the voltage dependence of inactivation (Fig. 2E, Table 1). In contrast, V1731A shifted the voltage dependence of inactivation in the depolarizing direction by 14 mV (Fig. 2E, Table 1), but had no significant effect on the voltage dependence of activation (Fig. 2D and Table 1). The other four substitutions had no significant effect on the voltage dependence of activation and inactivation (Table 1). Furthermore, the T260I + V1731A double mutant exhibited depolarizing shifts in the voltage dependence of both activation and inactivation (Fig. 2D and 2E, Table 1).
Table 1.
Voltage-dependences of activation and inactivation of DmNav7-1 and mutant channels
| Na+ Channel Type | Activation | Inactivation | n | ||
|---|---|---|---|---|---|
|
| |||||
| V1/2 (mV) | k (mV) | V1/2 (mV) | k (mV) | ||
| DmNav7-1 | −45.8 ± 1.4 | 6.7 ± 0.7 | −66.9 ± 0.6 | 4.7 ± 0.2 | 6 |
| V1731A | −47.7 ± 1.0 | 3.4 ± 0.3 | −52.9 ± 0.5* | 5.0 ± 0.7 | 13 |
| T260I | −26.1 ± 1.7* | 9.2 ± 1.0 | −69.8 ± 0.7 | 6.6 ± 0.3 | 10 |
| T260I+ V1731A | −23.5 ± 1.4* | 8.4 ± 0.8 | −49.4 ± 0.5* | 6.2 ± 0.3 | 10 |
| R303K | −45.5 ± 1.1 | 4.7 ± 0.4 | −66.0 ± 1.1 | 4.9 ± 0.2 | 7 |
| R319W | −47.0 ± 0.7 | 4.9 ± 0.2 | −67.4 ± 0.9 | 5.1 ± 0.3 | 7 |
| G771E | −50.6 ± 1.1 | 6.3 ± 0.8 | −69.1 ± 1.1 | 4.6 ± 0.2 | 6 |
| R1676H | −47.1 ± 1.0 | 3.9 ± 0.6 | −65.0 ± 0.8 | 4.5 ± 0.3 | 6 |
The voltage dependences of conductance and inactivation were fitted with a two-state Boltzmann equation to determine V1/2, the voltage for half-maximal conductance or inactivation, and K, the slope factor for conductance or inactivation. The values in the table represent the mean ± SD. and n is the number of oocytes used. The asterisks indicate significant differences from the wild-type DmNav7-1 channel as determined by using one-way analysis of variance with Scheffé’s post hoc analysis (p<0.05).
Identification of two components of INaP in DmNav7-1
We plotted the voltage dependence of INaP with the lower part of the conductance and inactivation curves of INaT in Fig. 3 for the wild-type DmNav7-1, T260I, V1731A and T260I+V1731A mutant channels. For the DmNav7-1 channel, the voltage dependence of channel activation and inactivation of INaT overlapped in the potential range from −60 mV to −40 mV, resulting in a window current (Fig. 3A). The hyperpolarizing portion of INaP (normalized to INaT) partially overlapped with the window current (Fig. 3A). In contrast, the more depolarizing portion of INaP was in a voltage range in which channel activation was saturated, but channel inactivation was incomplete. Intriguingly, substitution T260I eliminated the window current by shifting the voltage dependence of activation in the depolarizing direction;, and also eliminated the portion of INaP at hyperpolarizing potentials (Fig. 3B), suggesting a link between the window current and the portion of INaP at hyperpolarizing potentials. In contrast, the V1731A substitution eliminated the more depolarizing portion of INaP, but enlarged the window current due to a depolarizing shift of inactivation (Fig. 3C). The double substitution eliminated most of the INaP (Fig. 3D). The voltage dependence of INaP in the wild-type and three mutant channels are summarized in Fig. 3E.
Fig. 3. Voltage dependence of INaP, window currents and ramp currents.

A-D. The lower sections of the conductance and inactivation curves of INaT and the voltage dependence of INaP for DmNav7-1 (A) T260I (B), V1731A (C) and T260I+V1731A (D). E. The percentage of INaP was normalized to the INaT peak for DmNav7-1 and the three mutant channels. F. Ramp currents evoked during a voltage ramp from −100 mV to +20 mV over 600 ms (0.2 mV/ms). The ramp data were normalized to the transient peak current (INaT). Each curve was the average of measurements of at least 10 oocytes.
We then used a slow voltage ramp from −100 mV to 20 mV at 0.2 mV/ms to further characterize the gating properties of INaP. This ramp protocol inactivates INaT, leaving just the window and persistent currents. The voltage-dependence of the persistent current resulting from the ramp protocol (Fig. 3F) was extremely similar to that of INaP in Fig. 3E. These results further indicate that I260T in DmNav7-1 is responsible for both the window current and the hyperpolarizing portion of INaP. However, the fact that the A1731V change enlarged the window current but did not affect the ramp or INaP currents demonstrates that the depolarizing portion of INaP does not result from the window current.
Substitution I260T is due to a T to C change in the transcript encoding DmNav7-1 (atg to acc), possibly by a U-to-C editing event, which remains to be confirmed. However, an A-to-I editing event at the same codon but at a different nucleotide (underlined in atg to gcc) was identified in two other variants, DmNav4-2 and DmNav5-1, resulting in substitution I260V (Olson et al., 2008). While DmNav4-2 did not produce sufficient currents in oocytes for functional analysis, DmNav5-1 exhibited INaP, but with a smaller amplitude compared to DmNav7-1 (Fig. 4A). When V260 was replaced with I (i.e., V260I) in DmNav5-1, the INaP decreased (Fig. 4B). Furthermore, consistent with the earlier report (Olson et al., 2008), the V260I substitution also shifted the voltage-dependence of activation in the depolarizing direction. However, the shift by V260I was much smaller than that by T260I, 10 mV vs. 22 mV (Fig. 4C; Table 2). Like T260I, the V260I substitution had no effect on the voltage-dependence of channel inactivation (Fig. 4D; Table 2). The voltage-dependence of INaP appears to coincide with the voltage dependence of the window current (Fig. 4E), suggesting that substitution I260V in DmNav5-1 contributes to both the window current and INaP.
Fig. 4. Substitution I260V contributed to INaP in another DmNav variant.

A. Current trace of I260V mutant channel showing INaP. B. Voltage dependence of INaP. The percentage of INaP was normalized to the peak of INaT. C and D. Voltage dependence of activation (C) and inactivation (D) of DmNav5-1 and V260I mutant channel. E. The lower parts of the conductance and inactivation curves of INaT and the voltage dependence of INaP for DmNav5-1. The half-maximal voltage for activation and inactivation (V1/2) and slope factor (k) of DmNav5-1 and mutant channels are shown in Table 2.
Table 2.
Voltage-dependences of activation and inactivation of DmNav5-1 and mutant channel
| Na+ Channel Type | Activation | Inactivation | n | ||
|---|---|---|---|---|---|
|
| |||||
| V1/2 (mV) | k (mV) | V1/2 (mV) | k (mV) | ||
| DmNav5-1 | −32.6 ± 0.9 | 4.0 ± 0.2 | −43.4 ± 0.9 | 4.6 ± 0.1 | 10 |
| V260I | −24.4 ± 0.8* | 6.9 ± 0.4 | −44.3 ± 0.9 | 5.2 ± 0.1 | 9 |
The voltage dependences of conductance and inactivation were fitted with a two-state Boltzmann equation to determine V1/2, the voltage for half-maximal conductance or inactivation, and K, the slope factor for conductance or inactivation. The values in the table represent the mean ± SED. and n is the number of oocytes used. The asterisks indicate significant differences from the wild-type DmNav5-1 channel as determined by using one-way analysis of variance with Scheffé’s post hoc analysis (p<0.05).
Substitution V1731A slows entry into and accelerates recovery from fast inactivation
To determine whether the V1731-mediated depolarized portion of INaP results from impaired fast inactivation, we investigated the development of fast inactivation and recovery from fast inactivation of DmNav7-1 and mutant channels. Compared with DmNav7-1 channels that exhibit fast entry into and recovery from inactivation, the V1731A substitution and the double substitution slowed entry into fast inactivation (Fig. 5A) and accelerated recovery from fast inactivation (Fig. 5B). In contrast, substitution T260I did not alter the kinetics of entry into inactivation, but slowed recovery from fast inactivation (Fig. 5A and 5B). Furthermore, the voltage dependence of the kinetics for both entry into and recovery from fast inactivation was shifted in the depolarizing direction for V1731A and V1731A + T260I (Fig. 5C and 5D). T260I caused a hyperpolarizing shift in the voltage dependence for recovery from fast inactivation (Fig. 5D), but not in the voltage dependence of entry into fast inactivation (Fig. 5C).
Fig. 5. Effects of substitutions T260I and V1731A on inactivation kinetics.

A. The development of fast inactivation for DmNav7-1, V1731A, T260I and double mutant channels. B. Recovery from fast inactivation. C. Voltage dependence of the time constant for entry into fast inactivation. D. Voltage dependence of recovery from fast inactivation. The recording protocols are shown above the plots. Values are mean ± SEM for measurements from five to eight oocytes.
Substitution A1731V increases INaP likely by interfering with the outward movement of IVS4
A1731V is located between the 6th and 7th of eight positively charged residues in IVS4 of DmNav7-1. The outward movement of positively charged IVS4 facilitates the docking of the inactivation particle to its receptor site and therefore this outward movement is critical for fast inactivation (Kuhn and Greeff, 1999, Yang and Kuo, 2003). We predicted that A1731V could cause incomplete inactivation by interfering with the outward movement of IVS4, thereby impairing the docking of the inactivation particle to its receptor. To test this hypothesis, we used two sodium channel site 3 toxins which are known to induce persistent current by inhibiting the outward movement of IVS4 (Hanck and Sheets, 2007; Moran et al., 2007). If A1731V in DmNav7-1 channels induces persistent current by inhibiting the outward movement of IVS4, we reasoned that site 3 toxins would be more effective on DmNav7-1 channels over V1731A mutant channels. Therefore, we examined the sensitivity of DmNav7-1 and V1731A and T260I mutant channels to Av3 and LqhαIT, two insect-selective site 3 venom toxins.
Av3 and LqhαIT inhibited fast inactivation and induced persistent currents, as expected. In the presence of 250 nM Av3, the percentage of INaP dramatically increased, from 13% to 90% (Fig. 6A and 6B). Av3 also increased the peak current (Fig. 6A). Av3 had similar effects on V1731A and T260I mutant channels (Fig. 6A). However, V1731A channels were less sensitive to Av3 than DmNav7-1 and T260I channels, consistent with our prediction (Fig. 6A and 6B). The V1731A substitution also reduced channel sensitivity to LqhαIT, whereas the T260I substitution did not (Fig. 6C and 6D).
Fig. 6. Effects of substitutions T260I and V1731A on channel sensitivity to Av3 and LqhαIT toxins.

A and C. Representative sodium current traces in the absence and presence of Av3 (250 nM) or LqhαIT (10 nM) for DmNav7-1, T260I and V1731A mutant channels. Sodium currents were elicited by a 20-ms step depolarization to −10 mV from a holding potential of −120 mV. B and D. The percentage of toxin modification of fast inactivation by Av3 (B) and LqhαIT (D). The inhibitory effect on inactivation by toxins was calculated by measuring the current remaining at 20 ms (I20ms) divided by the peak current (Ipeak) after toxin application.
DISCUSSION
Substantial research has been focused on the origin of INaP and its contribution to the regulation of neuronal activities in various regions of mammalian brains. However, the molecular mechanism underlying INaP currents is largely elusive. Previous studies have indicated that persistent currents can result from either alternative gating modes or splice/RNA editing variants, with possibly multiple mechanisms (Liu et al., 2004, Stafstrom, 2007, Lin et al., 2009, Chatelier et al., 2010). To gain insight into the molecular basis of INaP, we conducted mutational analysis of a Drosophila sodium channel variant, DmNav7-1, that exhibits a large INaP when expressed in Xenopus oocytes. Our data show that INaP originates from two unique amino acid substitutions: I260T in the S4–S5 linker in domain I and A1731V in the voltage sensor S4 in domain IV. Substitution I260T is responsible for a hyperpolarizing shift in activation, resulting in a large window current which contributes to the INaP at hyperpolarized potentials. The mutant channel bearing substitution A1731V generates INaP at more depolarized potentials, due to the impairment of fast inactivation probably by inhibiting the outward movement of IVS4. Our findings reveal that a combination of two sequence features equipped with two distinct mechanisms underlies the unique gating of DmNav7-1 channels.
The involvement of window currents in the generation of INaP has been controversial (Kiss et al., 2009). Our findings indicate that I260T-mediated INaP at hyperpolarized potentials originates from window currents. First, the T260-mediated 22 mV hyperpolarizing shift in the voltage-dependence of activation increases the overlap between activation and inactivation curves, resulting in both a larger window current and a larger INaP. Second, the voltage range for the window current completely coincides with the voltage range for the activation of the T260-mediated INaP (Fig. 3). Third, substitution T260I abolishes both the window current and the portion of INaP at more negative potentials. Fourth, substitution I260V, due to an A-to-I editing event (Olson et al., 2008), causes a smaller hyperpolarizing shift of only 10 mV, which results in smaller window and INaP currents.
Among the 65 available DmNav variants, DmNav7-1 is the only variant with I260T and A1731V substitutions, indicating that the frequency of these putative RNA editing events is extremely low (~1.5%). However, an A-to-I editing at I260 resulting in substitution I260V was previously observed in two DmNav that belong to different splicing types (Olson et al. 2008). We recently conducted bulk Solexa sequencing and RNAseq of DmNav cDNAs from fly heads and brains in an attempt to independently detect these putative editing sites in additional DmNav variants. We realized that these sequencing techniques cannot accurately detect transcripts that are below 3–5% and hence, were not effective in detecting I260T and A1731V substitutions in bulk cDNA populations. Future experiments are needed to detect I260T and A1731V substitutions in other DmNav variants by analyzing a much greater number of cDNA clones from specific nerve tissues. In a previous study (Liu et al., 2004), we identified an INaP generated by substitution F1950S in DmNav (i.e., F1919S in BgNav) as a result of a U-to-C editing event at the C-terminal domain (Liu et al. 2004). The frequency of this U-to-C editing was also extremely low. Initially, this substitution was identified in one of 69 BgNav variants; and one of 65 DmNav variants (Liu et al., 2004). Subsequent targeted analysis of 561 clones isolated from a specific tissue (cockroach terminal ganglia), where an INaP was previously reported in cockroach, showed that 3.2% of BgNav transcripts were edited (Liu et al., 2004). Sodium channel opening at these subthreshold voltages (such as −60 mV) is significant because the resulting inward currents tend to depolarize the resting membrane potential and regulate the excitability of neurons in which these RNA editing events occur. Potentially, RNA editing at position 260 occurs at different nucleotides, resulting in different amino acid substitutions. The fact that different substitutions modify the gating differently suggests that various neurons may achieve precise levels of modulation of sodium channel gating by RNA editing to fulfill their unique functional roles in the nervous system. Identifying the localization or spatiotemporal functioning of such neurons in insects might greatly advance the understanding of how persistent currents regulate neuronal activities in vivo.
Sodium channel opening is initiated by the outward movement of the positively charged S4 voltage sensors through the electric field, as evident from the measurements of gating currents (Chanda and Bezanilla, 2002). Short intracellular linkers between the S4 and S5 segments transmit the movements of the voltage sensing modules to the S6 segments during channel opening and closing (Catterall, 2012). Furthermore, the contribution of voltage sensors to channel gating is domain-specific. Voltage sensors in domains I, II and III are primarily involved in channel activation, whereas the voltage sensor in domain IV is critical for inactivation (Chahine et al., 1994, Chen et al., 1996, Sheets et al., 1999, Chanda and Bezanilla, 2002). Domains I, II and III generate the fast component of the gating current that correlates with the opening of the channel, while domain IV generates the slow component of the gating current that is a prelude to the inactivation process (Chanda and Bezanilla, 2002, Goldschen-Ohm et al., 2013). Consistent with this general dogma, substitutions I260T/V in the S4–S5 linker of domain I affect activation, whereas substitution A1731V in S4 of domain IV affects inactivation.
The I260T substitution is situated in the linker connecting S4 and S5 of domain I. I260 is conserved among all sodium channels. In fact, the entire IL45 sequence is highly conserved among insect and mammalian sodium channels (Fig. 7), suggesting an important role of this linker in modulating sodium channel function. Indeed, the same mutation (I234T) in hNav1.7, which is associated with inherited erythromelalgia (IEM), causes similar gating modifications (Ahn et al., 2010). Like I260T, the I234T mutation induces a large (18 mV) hyperpolarizing shift in the voltage dependence of activation and also increases the window currents (Ahn et al., 2010). These altered gating properties associated with I234T could contribute to the increased excitability of nociceptive dorsal root ganglion neurons where hNav1.7 channels are located. It is possible that the I260T substitution would result in similar physiological effects in Drosophila neurons where the I260T editing occurs.
Fig. 7. Alignments of amino acid sequences in ILS45 (A) and IVS4 (B) of DmNav7-1 and rat Nav1.1-1.9.

All other DmNav variants have I260 and A1731.
Neutralization of the positively charged residues in IVS4, particularly the outermost one, reduced the voltage sensitivity of fast inactivation (Chen et al., 1996, Kontis et al., 1997). Here we show that a neutral residue located between the 6th and 7th positively charged residues in IVS4 also has an important role in the inactivation process potentially by controlling the outward movement of IVS4. This alanine is extremely conserved among insect and mammalian sodium channels (Fig. 7B). The fact that a subtle change from A1371 to V1371 drastically alters the voltage dependence of inactivation indicates the importance of neutral residues in IVS4 in fast inactivation. Furthermore, the A1731V substitution in DmNav7-1 enhanced the action of two site 3 scorpion and sea anemone toxins on inactivation. Site 3 toxins inhibit sodium channel fast inactivation by binding at the extracellular loops near IVS4 (Tejedor and Catterall, 1988, Thomsen and Catterall, 1989, Rogers et al., 1996; Gur et al., 2011, Wang et al., 2011) and preventing the outward movement of IVS4 (Sheets and Hanck, 1995, 1999, 2005, Campos and Beirao, 2006, Sheets and Hanck, 2007). Residue 1731 is located toward the inner end of IVS4 and is probably not directly involved in toxin binding. Therefore, the A1731V substitution likely inhibits the outward movement of IVS4. Consequently, the binding of the inactivation particle to its docking receptor site would be hindered, resulting in the generation of an INaP, supporting the notion that the V1731-mediated INaP is the result of incomplete fast inactivation.
Several mammalian sodium channels, such as Nav1.3, Nav1.6 and Nav1.9, have been reported to generate persistent currents (Smith et al., 1998, Dib-Hajj et al., 2002, Lampert et al., 2006a, Lampert et al., 2006b, Sun et al., 2007). However, INaP from various neurons in the mammalian brain exhibit a wider range of gating properties than those of brain sodium channel isoforms, and the underlying causes of these INaP are largely unknown at the molecular level (Pennartz et al., 1997, Magistretti et al., 1999a, b, Taddese and Bean, 2002, Jackson et al., 2004, Khaliq and Bean, 2010, Yamada-Hanff and Bean, 2013). It is possible that the expression of INaP in neurons is different from that observed in Xenopus oocytes due to neuron-specific posttranslational regulation. Alternatively and/or in addition, alternative splicing and RNA editing also play an important role in the generation of INaP in mammals, a possibility that remains to be investigated. In contrast, the role of alternative splicing and RNA editing in regulating INaP in insects has been demonstrated. A previous study shows that two mutually exclusive exons l/k encoding IIIS3–S4 in DmNav significantly affected the magnitude of INaP, which ranges from 1.5 to 2.4% of transient current for variants including exon k, but increases to 4.1–9.5% in variants including exon l (Lin et al., 2009). Apparently, alternative splicing resulting in the inclusion of exon l in replacement of exon k is associated with increased neuronal excitability (Lin et al., 2012). Interestingly, Pasilla, an RNA-binding protein involved in alternative splicing of DmNav, regulates the magnitude of INaP by modulating the relative proportions of exons k/l (Park et al., 2004; Lin et al. 2009; 2012). Our previous and current studies showed that the magnitude of RNA editing-mediated INaP ranges from about 50% of INaT for F1950S editing site (Liu et al., 2004) to 2% of INaT for I260V editing site. Taken together, these studies suggest that alternative splicing and RNA editing are likely important mechanisms in modulating the excitability of insect neurons.
In conclusion, we identified two residues, T260 in the S4–S5 linker of domain I and V1731 in IVS4, that are responsible for INaP in a Drosophila sodium channel variant. Our findings provide molecular evidence for the contribution of window currents to the generation of persistent currents. Furthermore, analysis of the action of site 3 neurotoxins on DmNav7-1 and mutant channels revealed the role of a neutral residue in IVS4 in regulating the voltage sensor movement and channel inactivation.
Highlights.
A Drosophila sodium channel, DmNav7-1, exhibits a unique persistent current (INaP).
This INaP activates over a broad voltage range.
I260T contributes to INaP at hyperpolarizing potentials via enhancement of window currents
A1731V contributes to INaP at depolarizing potentials probably by inhibiting the outward movement of IVS4.
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM057440) to K.D.; the National Natural Science Foundation of China (81273113) to R.G. and K.D.; the Israeli Science Foundation (1085/12) to M.G.; and the Binational Agricultural Research and Development Fund (IS-4313-10C) to M.G. and D.G.. R.G. was supported by a scholarship from the government of Jiangsu Province, China.
Abbreviations
- INaP
persistent sodium current
- INaT
transient sodium current
- Av3
sea anemone toxin from Anemonia viridis
- LqhαIT
scorpion toxin from Leiurus quinquestriatus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn HS, Dib-Hajj SD, Cox JJ, Tyrrell L, Elmslie FV, Clarke AA, Drenth JP, Woods CG, Waxman SG. A new Nav1.7 sodium channel mutation I234T in a child with severe pain. Eur J Pain. 2010;14:944–950. doi: 10.1016/j.ejpain.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Campos FV, Beirao PS. Effects of bound Ts3 on voltage dependence of sodium channel transitions to and from inactivation and energetics of its unbinding. Cell Biochem Biophys. 2006;44:424–430. doi: 10.1385/CBB:44:3:424. [DOI] [PubMed] [Google Scholar]
- Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Voltage-gated sodium channels at 60: structure, function and pathophysiology. J Physiol. 2012;590:2577–2589. doi: 10.1113/jphysiol.2011.224204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahine M, George AL, Jr, Zhou M, Ji S, Sun W, Barchi RL, Horn R. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Chanda B, Bezanilla F. Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J Gen Physiol. 2002;120:629–645. doi: 10.1085/jgp.20028679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatelier Al, Zhao J, Bois P, Chahine M. Biophysical characterisation of the persistent sodium current of the Nav1. 6 neuronal sodium channel: a single-channel analysis. Pflugers Arch. 2010;460:77–86. doi: 10.1007/s00424-010-0801-9. [DOI] [PubMed] [Google Scholar]
- Chen H, Gordon D, Heinemann SH. Modulation of cloned skeletal muscle sodium channels by the scorpion toxins Lqh II, Lqh III, and LqhαIT. Pflugers Arch. 2000;439:423–432. doi: 10.1007/s004249900181. [DOI] [PubMed] [Google Scholar]
- Chen LQ, Santarelli V, Horn R, Kallen RG. A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. J Gen Physiol. 1996;108:549–556. doi: 10.1085/jgp.108.6.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Lucero MT. Transient and persistent tetrodotoxin-sensitive sodium currents in squid olfactory receptor neurons. J Comp Physiol [A] 1999;184:63–72. doi: 10.1007/s003590050306. [DOI] [PubMed] [Google Scholar]
- Clay JR. On the persistent sodium current in squid giant axons. J Neurophysiol. 2003;89:640–644. doi: 10.1152/jn.00652.2002. [DOI] [PubMed] [Google Scholar]
- Crill WE. Persistent sodium current in mammalian central neurons. Annu Rev Physiol. 1996;58:349–362. doi: 10.1146/annurev.ph.58.030196.002025. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj S, Black JA, Cummins TR, Waxman SG. NaN/Nav1.9: a sodium channel with unique properties. Trends Neurosci. 2002;25:253–259. doi: 10.1016/s0166-2236(02)02150-1. [DOI] [PubMed] [Google Scholar]
- Dong K. Insect sodium channels and insecticide resistance. Invert Neurosci. 2007;7:17–30. doi: 10.1007/s10158-006-0036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong K. Progress in insect sodium channel research. In: Gilbert LI, Gill SS, editors. Insect Pharmacology, Channels, Receptors, Toxins and Enzymes. Academic Press; 2010. pp. 25–27. [Google Scholar]
- Feng G, Deak Pt, Chopra M, Hall LM. Cloning and functional analysis of tipE, a novel membrane protein that enhances drosophila para sodium channel function. Cell. 1995;82:1001–1011. doi: 10.1016/0092-8674(95)90279-1. [DOI] [PubMed] [Google Scholar]
- Frank HY, Catterall WA. Overview of the voltage-gated sodium channel family. Genome Bio. 2003;4:207. doi: 10.1186/gb-2003-4-3-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Netter YB. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28:365. doi: 10.1016/s0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Goldschen-Ohm MP, Capes DL, Oelstrom KM, Chanda B. Multiple pore conformations driven by asynchronous movements of voltage sensors in a eukaryotic sodium channel. Nature Commun. 2013;4:1350. doi: 10.1038/ncomms2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur M, Kahn R, Regev N, Wang J, Catterall WA, Gordon D, Gurevitz M. Elucidation of the molecular basis of selective recognition uncovers the interaction site for the core-domain of scorpion alpha-toxins on sodium channels. J Biol Chem. 2011;286:35209–35217. doi: 10.1074/jbc.M111.259507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanck DA, Sheets MF. Site-3 toxins and cardiac sodium channels. Toxicon. 2007;49:181–193. doi: 10.1016/j.toxicon.2006.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AC, Yao GL, Bean BP. Mechanism of spontaneous firing in dorsomedial suprachiasmatic nucleus neurons. J Neurosci. 2004;24:7985–7998. doi: 10.1523/JNEUROSCI.2146-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq ZM, Bean BP. Pacemaking in dopaminergic ventral tegmental area neurons: depolarizing drive from background and voltage-dependent sodium conductances. J Neurosci. 2010;30:7401–7413. doi: 10.1523/JNEUROSCI.0143-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss T. Persistent Na-channels: Origin and function. Acta Biologica Hungarica. 2008;59:1–12. doi: 10.1556/ABiol.59.2008.Suppl.1. [DOI] [PubMed] [Google Scholar]
- Kiss T, Pirger Z, Kemenes Gr. Food-aversive classical conditioning increases a persistent sodium current in molluscan withdrawal interneurons in a transcription dependent manner. Neurobiol Learn Mem. 2009;92:114–119. doi: 10.1016/j.nlm.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Kontis KJ, Rounaghi A, Goldin AL. Sodium channel activation gating is affected by substitutions of voltage sensor positive charges in all four domains. J Gen Physiol. 1997;110:391–401. doi: 10.1085/jgp.110.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn FJP, Greeff NG. Movement of voltage sensor S4 in domain 4 is tightly coupled to sodium channel fast inactivation and gating charge immobilization. J Gen Physiol. 1999;114:167–184. doi: 10.1085/jgp.114.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampert A, Dib-Hajj SD, Tyrrell L, Waxman SG. Size matters: Erythromelalgia mutation S241T in Nav1.7 alters channel gating. J Biol Chem. 2006a;281:36029–36035. doi: 10.1074/jbc.M607637200. [DOI] [PubMed] [Google Scholar]
- Lampert A, Hains BC, Waxman SG. Upregulation of persistent and ramp sodium current in dorsal horn neurons after spinal cord injury. Exp Brain Res. 2006b;174:660–666. doi: 10.1007/s00221-006-0511-x. [DOI] [PubMed] [Google Scholar]
- Lapied B, Le Corronc H, Hue B. Sensitive nicotinic and mixed nicotinic-muscarinic receptors in insect neurosecretory cells. Brain Res. 1990;533:132–136. doi: 10.1016/0006-8993(90)91805-q. [DOI] [PubMed] [Google Scholar]
- Lin W-H, Wright DE, Muraro NI, Baines RA. Alternative splicing in the voltage-gated sodium channel DmNav regulates activation, inactivation, and persistent current. J Neurophysiol. 2009;102:1994–2006. doi: 10.1152/jn.00613.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WH, Gunay C, Marley R, Prinz AA, Baines RA. Activity-dependent alternative splicing increases persistent sodium current and promotes seizure. J Neurosci. 2012;32:7267–7277. doi: 10.1523/JNEUROSCI.6042-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Song W, Dong K. Persistent tetrodotoxin-sensitive sodium current resulting from U-to-C RNA editing of an insect sodium channel. Proc Natl Acad Sci USA. 2004;101:11862–11867. doi: 10.1073/pnas.0307695101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas RR, Alonso A. Electrophysiology of the mammillary complex in vitro. I. Tuberomammillary and lateral mammillary neurons. Neurophysiol. 1992;68:1307–1320. doi: 10.1152/jn.1992.68.4.1307. [DOI] [PubMed] [Google Scholar]
- Loughney K, Kreber R, Ganetzky B. Molecular analysis of the para locus, a sodium channel gene in Drosophila. Cell. 1989;58:1143–1154. doi: 10.1016/0092-8674(89)90512-6. [DOI] [PubMed] [Google Scholar]
- Magistretti J, Ragsdale DS, Alonso A. Direct demonstration of persistent Na+ channel activity in dendritic processes of mammalian cortical neurones. J Physiol. 1999a;521(Pt 3):629–636. doi: 10.1111/j.1469-7793.1999.00629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti J, Ragsdale DS, Alonso A. High conductance sustained single-channel activity responsible for the low-threshold persistent Na(+) current in entorhinal cortex neurons. J Neurosci. 1999b;19:7334–7341. doi: 10.1523/JNEUROSCI.19-17-07334.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mee CJ, Pym ECG, Moffat KG, Baines RA. Regulation of neuronal excitability through pumilio-dependent control of a sodium channel gene. J Neurosci. 2004;24:8695–8703. doi: 10.1523/JNEUROSCI.2282-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran Y, Kahn R, Cohen L, Gur M, Karbat I, Gordon D, Gurevitz M. Molecular analysis of the sea anemone toxin Av3 reveals selectivity to insects and demonstrates the heterogeneity of receptor site-3 on voltage-gated Na+ channels. Biochem J. 2007;406:41–48. doi: 10.1042/BJ20070233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson RO, Liu ZQ, Nomura Y, Song WZ, Dong K. Molecular and functional characterization of voltage-gated sodium channel variants from Drosophila melanogaster. Insect Biochem Mol Biol. 2008;38:604–610. doi: 10.1016/j.ibmb.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opdyke CA, Calabrese RL. A persistent sodium current contributes to oscillatory activity in heart interneurons of the medicinal leech. J Comp Physiol [A] 1994;175:781–789. doi: 10.1007/BF00191850. [DOI] [PubMed] [Google Scholar]
- Park JW, Parisky K, Celotto AM, Reenan RA, Graveley BR. Identification of alternative splicing regulators by RNA interference in Drosophila. PNAS. 2004;101:15974–15979. doi: 10.1073/pnas.0407004101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennartz CMA, Bierlaagh MA, Geurtsen AMS. Cellular mechanisms underlying spontaneous firing in rat suprachiasmatic nucleus: involvement of a slowly inactivating component of sodium current. J Neurophysiol. 1997;78:1811–1825. doi: 10.1152/jn.1997.78.4.1811. [DOI] [PubMed] [Google Scholar]
- Rakowski RF, Gadsby DC, De Weer P. Single ion occupancy and steady-state gating of Na channels in squid giant axon. J Gen Physiol. 2002;119:235–250. doi: 10.1085/jgp.20028500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Gustafson AE, Padgett D. Ionic currents and spontaneous firing in neurons isolated from the cerebellar nuclei. J Neurosci. 2000;20:9004–9016. doi: 10.1523/JNEUROSCI.20-24-09004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JC, Qu Y, Tanada TN, Scheuer T, Catterall WA. Molecular determinants of high affinity binding of β--scorpion toxin and sea anemone toxin in the S3–S4 extracellular loop in domain IV of the Na+ channel α-subunit. J Biol Chem. 1996;271:15950–15962. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- Sheets MF, Hanck DA. Voltage-dependent open-state inactivation of cardiac sodium channels: gating current studies with Anthopleurin-A toxin. J Gen Physiol. 1995;106:617–640. doi: 10.1085/jgp.106.4.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets MF, Hanck DA. Gating of skeletal and cardiac muscle sodium channels in mammalian cells. J Gen Physiol. 1999;514:425–436. doi: 10.1111/j.1469-7793.1999.425ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets MF, Hanck DA. Charge immobilization of the voltage sensor in domain IV is independent of sodium current inactivation. J Gen Physiol. 2005;563:83–93. doi: 10.1113/jphysiol.2004.077644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets MF, Hanck DA. Outward stabilization of the S4 segments in domains III and IV enhances lidocaine block of sodium channels. J Gen Physiol. 2007;582:317–334. doi: 10.1113/jphysiol.2007.134262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets MF, Kyle JW, Kallen RG, Hanck DA. The Na channel voltage sensor associated with inactivation is localized to the external charged residues of domain IV, S4. Biochem J. 1999;77:747–757. doi: 10.1016/S0006-3495(99)76929-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, Smith RD, Plummer NW, Meisler MH, Goldin AL. Functional analysis of the mouse Scn8a sodium channel. J Neurosci. 1998;18:6093–6102. doi: 10.1523/JNEUROSCI.18-16-06093.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafstrom CE. Persistent sodium current and its role in epilepsy. Epilepsy Curr. 2007;7:15–22. doi: 10.1111/j.1535-7511.2007.00156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun GC, Werkman TR, Battefeld A, Clare JJ, Wadman WJ. Carbamazepine and topiramate modulation of transient and persistent sodium currents studied in HEK293 cells expressing the Na(v)1.3 alpha-subunit. Epilepsia. 2007;48:774–782. doi: 10.1111/j.1528-1167.2007.01001.x. [DOI] [PubMed] [Google Scholar]
- Taddese A, Bean BP. Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron. 2002;33:587–600. doi: 10.1016/s0896-6273(02)00574-3. [DOI] [PubMed] [Google Scholar]
- Tan J, Liu Z, Wang R, Huang ZY, Chen AC, Gurevitz M, Dong K. Identification of amino acid residues in the insect sodium channel critical for pyrethroid binding. Mol Pharmacol. 2005;67:513–522. doi: 10.1124/mol.104.006205. [DOI] [PubMed] [Google Scholar]
- Taylor CP. Na+ currents that fail to inactivate. Trends Neurosci. 1993;16:455–460. doi: 10.1016/0166-2236(93)90077-y. [DOI] [PubMed] [Google Scholar]
- Tejedor FJ, Catterall WA. Site of covalent attachment of alpha-scorpion toxin derivatives in domain I of the sodium channel alpha subunit. Proc Natl Acad Sci USA. 1988;85:8742–8746. doi: 10.1073/pnas.85.22.8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen WJ, Catterall WA. Localization of the receptor site for alpha-scorpion toxins by antibody mapping: implications for sodium channel topology. Proc Natl Acad Sci USA. 1989;86:10161–10165. doi: 10.1073/pnas.86.24.10161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uteshev V, Stevens DR, Haas HL. A persistent sodium current in acutely isolated histaminergic neurons from rat hypothalamus. Neuroscience. 1995;66:143–149. doi: 10.1016/0306-4522(94)00593-t. [DOI] [PubMed] [Google Scholar]
- Wang J, Yarov-Yarovoy V, Kahn R, Gordon D, Gurevitz M, Scheuer T, Catterall WA. Mapping the receptor site for α-scorpion toxins on a sodium channel voltage sensor. Proc Natl Acad Sci USA. 2011;108:15426–15431. doi: 10.1073/pnas.1112320108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warmke JW, Reenan RAG, Wang PY, Qian S, Arena JP, Wang JX, Wunderler D, Liu K, Kaczorowski GJ, VanderPloeg LHT, Ganetzky B, Cohen CJ. Functional expression of Drosophila para sodium channels - Modulation by the membrane protein TipE and toxin pharmacology. J Gen Physiol. 1997;110:119–133. doi: 10.1085/jgp.110.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada-Hanff J, Bean BP. Persistent sodium current drives conditional pacemaking in CA1 pyramidal neurons under muscarinic stimulation. J Neurosci. 2013;33:15011–15021. doi: 10.1523/JNEUROSCI.0577-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y-C, Kuo C-C. The position of the fourth segment of domain 4 determines status of the inactivation gate in Na+ channels. J Neurosci. 2003;23:4922–4930. doi: 10.1523/JNEUROSCI.23-12-04922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberberg N, Gordon D, Pelhate M, Adams ME, Norris TM, Zlotkin E, Gurevitz M. Functional expression and genetic alteration of an alpha scorpion neurotoxin. Biochemistry. 1996;35:10215–10222. doi: 10.1021/bi9528309. [DOI] [PubMed] [Google Scholar]