

Abstract
Ravidomycin V and related compounds, e.g., FE35A-B, exhibit potent anticancer activities against various cancer cell lines in the presence of visible light. The amino sugar moieties (d-ravidosamine and its analogues, respectively) in these molecules contribute to the higher potencies of ravidomycin and analogues when compared to closely related compounds with neutral or branched sugars. Within the ravidomycin V biosynthetic gene cluster, five putative genes encoding NDP-d-ravidosamine biosynthetic enzymes were identified. Through the activities of the isolated enzymes in vitro, it is demonstrated that ravD, ravE, ravIM, ravAMT and ravNMT encode TDP-d-glucose synthase, TDP-4-keto-6-deoxy-d-glucose-4,6-dehydratase, TDP-4-keto-6-deoxy-d-glucose-3,4-ketoisomerase, TDP-3-keto-6-deoxy-d-galactose-3-aminotransferase, and TDP-3-amino-3,6-dideoxy-d-galactose-N,N-dimethyl-transferase, respectively. A protocol for a one-pot enzymatic synthesis of TDP-d-ravidosamine has been developed. The results presented here now set the stage to produce TDP-d-ravidosamine routinely for glycosylation studies.
Introduction
Glycosylated microbial natural products possess a wide range of pharmaceutical properties such as anticancer, antiviral, antifungal or antibiotic.1 The sugar moieties of these molecules are often deoxygenated to various degrees, and are appended to the backbone through O- or C-, and rarely through N- or S-glycosidic linkages.2–4 Structural and functional studies have demonstrated that the sugar moieties contribute significantly to the biological activities of the parent compounds.
Due to the large structural diversity of deoxysugars, and due to their huge importance for biological activities, much attention has been given to elucidate their biosynthetic pathways.5–13 Recent progress in understanding these pathways has proven that an array of glycosylated natural products can be generated through enzymatic glycodiversification.14–17 However, the successful application of this concept requires an in-depth knowledge of various deoxysugar biosynthetic pathway enzymes including their catalytic activities, their preferred substrates, and their flexibility towards unnatural substrates. Additionally, glycodiversification of natural products also requires glycosyltransferases (GTs) with sugar-donor substrate flexibility, allowing the attachment of various sugar residues to target aglycones. Although gene sequences for dozens of GTs are known, only a marginal number of these enzymes have been fully characterized. Unavailability of functional GTs, or their acceptor or sugar donor substrates continue to contribute to the slow progress of GT studies.
Ravidomycin V (1) and deacetylravidomycin V (2) are potent antitumor antibiotics produced by Streptomyces ravidus.18,19 Both antibiotics contain a common angucyclinone-derived tetracyclic aromatic benzo[d]naptho[1,2-b]pyran-6-one core moiety, which is connected to their respective sugar residues at C-4 position through a C–C bond (Fig. 1). Deacetylravidomycin V (2) contains d-ravidosamine (N,N-dimethyl-3-amino-3,6-dideoxy-d-galactose) and exhibits superior anticancer/antibacterial activities than ravidomycin V (1) itself. The 4′-hydroxyl group of d-ravidosamine is acetylated in ravidomycin V. Ravidomycin M (3) and deacetylravidomycin M (4) possess a methyl side chain at C-8 position instead of the vinyl side chain present in the major congeners 1 and 2. The enantiomeric sugar l-ravidosamine was found in the anthracycline antibiotic komodoquinone A (7).20,21 The substitution pattern specifically at the 3′-amino and 4′-hydroxyl groups of d-ravidosamine are different in the other reported ravidomycin analogues FE35A (5) and FE35B (6). Gilvocarcin V,22 chrysomycin V,25 Mer 1020 dA-D,24 BE 12406 A-B,25,26 and polycarcin V27 are other closely related analogues, differing from ravidomycin mainly because of their neutral deoxysugar residues. Although the biological activities of these compounds have never been investigated in direct comparison, their biological activity data revealed that amino sugar containing gilvocarcin analogues show increased antibiotic/anticancer potency when compared to compounds with neutral or branched neutral sugar moities.28 Thus, investigation of the deoxysugar biosynthetic enzymes responsible for the production of d-ravidosamine, and characterization of the glycosyltransferase that attaches the sugar to the polyaromatic backbone are particularly important. An in-depth characterization of these enzymes may provide an opportunity to generate new ravidomycin analogues with altered aminosugar residues for structure–activity relationship (SAR) studies.
Fig. 1.

Structures of natural products comprising ravidosamine sugar moiety.
To understand the biosynthesis of ravidomycin V, we recently cloned and sequenced its biosynthetic gene cluster from Strepto-myces ravidus.29 The deoxysugar biosynthetic genes and associated glycosyltransferase were found in the cluster spanning a 6 kb region (Fig. 2). Bioinformatics analysis combined with heterologous expression experiments suggested that five genes, ravD, ravE, ravIM, ravAMT and ravNMT, encode enzymes involved in the conversion of D-glucose-1 -phosphate (8) to NDP-d-ravidosamine, the proposed sugar donor substrate for the glycosylation reaction. Similarly, the product of ravGT has been proposed to be the C-glycosyltransferase that links d-ravidosamine to the polyketide-derived backbone defuco-gilvocarcin V.
Fig. 2.
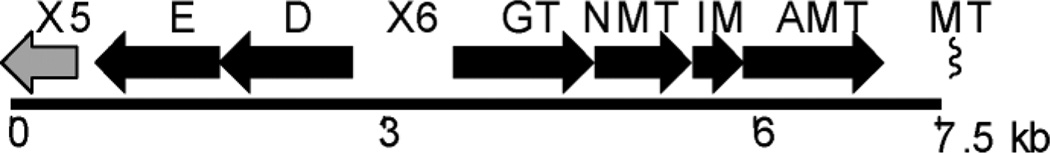
A section of the ravidomycin gene cluster spanning the putative TDP-d-ravidosamine (17) biosynthetic genes (ravD, E, IM, AMT and NMT).
Suzuki and co-workers have developed a synthetic route for the production of d-ravidosamine30 Multiple synthetic steps involved in this protocol and a low overall yield are major drawbacks of this approach. Activation of the sugar with a suitable nucleosyl-diphosphate (NDP) group poses another challenge towards the synthetic preparation of the donor substrate for RavGT. Recent development in understanding of deoxysugar biosynthetic pathways has made it possible to synthesize a target NDP-activated sugar in vitro through the activities of suitable enzymes. This approach is superior compared to the chemical total synthesis as it is concerted, less time-consuming, stereo- and regioselective, and offers high yields of product in an environmentally friendly procedure. We herein, report the preparation of thymidine diphosphate-activated ravidosamine (TDP-d-ravidosamine, 17) from glucose-1-phosphate 8 utilizing enzymes of the ravidosamine biosynthetic pathway. We demonstrate through in vitro assays and NMR analyses that ravD, ravE, ravIM and ravAMT encode TDP-d-glucose synthase, TDP-d-glucose-4,6-dehydratase, TDP-4-keto-6-deoxy-d-glucose-3,4-ketoisomerase and TDP-3-keto-6-deoxy-d-galactose-3-aminotransferase, respectively. Similarly, the product of ravNMT was characterized as TDP-3-amino-3,6-dideoxy-d-galactose- N, N-dimethyltransferase. Characterization of these five deoxysugar biosynthetic enzymes unambiguously established the biosynthetic pathway of 17. In addition, we report a one-pot enzymatic synthetic protocol for the production of 17. The results presented in this study lay the foundation for the in vitro characterization of the glycosyltransferase RavGT involved in ravidomycin biosynthesis.
Results and discussion
Expression and characterization of RavD and RavE
Basic local sequence alignment search tool (BLAST) analysis of RavD revealed high sequence similarity to several NDP-d-glucose synthases from deoxysugar biosynthetic pathways. RavD has the highest sequence homology to the proposed NDP-glucose synthase ChryD (83% identity/90% similarity) from the chrysomycin biosynthetic pathway29 The other proteins that displayed high amino acid homology with RavD were NDP-d-glucose synthases GilD (75%/86%),31 MtmD (76%/86%),32 SsfS1 (74%/85%)33 and StrD (71%/83%)34 from the gilvocarcin, mithramycin, SF2575 and streptomycin biosynthetic pathways, respectively. These proteins are proposed/proven to catalyze the attachment of TDP at C-1 of glucose utilizing 8 and TTP (25). These similarity results indicated that RavD catalyzes the conversion of 8→9. To validate this proposed function, the enzyme was expressed in E. coli. SDS-PAGE analysis revealed a dominant band for His6-RavD (Fig. S1†). Incubation of the purified enzyme with 8 and 25 resulted in the production 9 (calculated 98% product yield). The conversion was confirmed through HPLC analysis of the products with the addition of commercially available 9 and also through comparison of the 1H NMR data of the product with data reported in the literature (Fig. S6, S11 †).35 Compared to TTP, there was less than 5% turnover observed when using adenosine-5′-triphosphate (ATP), uracil-5′-triphosphate (UTP), guanosine-5′-triphosphate (GTP) or cytidine-5′-triphosphate (CTP) as substrates.
The amino acid sequence of the product of ravE displayed strong similarities (70%–75% amino acid identities) to NDP-d-glucose-4,6-dehydratases involved in deoxysugar biosynthetic pathways from various secondary metabolites such as RmbB (accession no ACR46364) from the doxorubicin, ChryE from the chrysomycin, GilE from the gilvocarcin, MtmE from the mithramycin, StrE from the streptomycin, and SsfS2 from the SF2575 biosynthetic pathway. These similarity results indicated that RavE could serve as a TDP-d-glucose-4,6-dehydratase, which catalyzes the deoxygenation and oxidation at C-6 and C-4 positions of 9, respectively. To test this assumption, RavE was heterologously produced in E. coli and purified. The size of the protein band observed on SDS-PAGE was in agreement with the calculated value for His6-RavE. Incubation of 9 in the presence of NAD+ did not reveal any noticeable change in the HPLC profile when compared to the control without enzyme except for the broadening of the TDP-d-glucose peak (Fig. 3, trace b). The preparative scale reaction was carried out by incubating 8, 25 and NAD+ with RavD and RavE. Separation of the crude products through gel filtration led to the isolation of 10 with 90% product yield. The structure of 10 was confirmed through comparison of its 1H NMR data with those reported in the literature (see ESI for the 1H NMR spectrum†). These results demonstrated that RavD and RavE catalyze the conversion of 8→9 and 9→10, respectively, during TDP-d-ravidosamine (17) biosynthesis.
Fig. 3.

HPLC traces demonstrating the conversion of TDP-d-glucose (9) to TDP-3-amino-3,6-dideoxy-d-galactose (15). Trace a: 9 generated from TTP (25) and glucose-1-phosphate (8) using RavD; trace b: reaction mixture following the incubation of 9 with RavE; trace c: reaction mixture including 10, PLP, l-glutamate, RavIM and RavAMT; trace d: TDP-3-amino-3,6-dideoxy-d-galactose (15) purified through gel filtration. The compounds for 14, 23 and 24 represents yet uncharacterized compound (preferably (2R,3S)-2-methyl-3,5-dihy-droxy-4-keto-2,3-dihydropyran), TMP and TDP, respectively.
Functional assignment of RavIM and RavAMT as isomerase and aminotransferase
BLAST analysis of the amino acid sequence of RavIM revealed a modest sequence similarity (40%–50% amino acid identities) with putative enzymes belonging to cupin2-superfamily. These include hypothetical proteins from Beggiatoa sp. PS (accession ZP_01998770), Limnobacter sp. MED105 (ZP_01913949) and C-terminal domains of WxcM domain-containing proteins from various organisms. WxcM from Xanthomonas compestris was proposed to catalyze two steps, i) isomerization of 10→12 by its C-terminal region and ii) N-acetylation of 15 by the N-terminal region, during the biosynthesis of the TDP-N-acetyl-3-amino-3,6-dideoxy-d-galactose (18) subunit of surface lipopolysaccharide (LPS)36 RavIM also showed considerable amino acid sequence identities (42% identity/62% similarity) to the recently characterized 3,4-ketoisomerase FdtA from Aneurinibacillus ther-moaerophilus.37 FdtA catalyzes the conversion of 10→12 during the biosynthesis of the S-layer glycan chain. Closer analysis of the RavIM amino acid sequence revealed the presence of the characteristic signature [RGXHAH(K/R)X(L/I)XQXGS] identified in FdtA (Fig. S3†). Analyses of active-site residues in the FdtA crystal structure and subsequent site-directed mutagenesis studies identified several active-site residues in this signature region.38 These results support the aforementioned FdtA-like role of RavIM in the biosynthesis of 17.
To assign the exact functional role of RavIM, the enzyme was heterologously produced in E. coli, and the protein was purified using immobilized metal affinity chromatography (IMAC) (Fig. S1†). Size exclusion chromatography analyses of RavIM with known proteins suggested that the enzyme exists in dimeric form. The enzyme was then subjected to a series of enzyme assays. No significant change was observed in the HPLC chromatogram of the assay mixture, when RavIM was incubated with 10 for 30 min. However, the incubation of 10 with RavIM for 24 h resulted in the consumption of 10 and the production of a new unknown product observable in the HPLC at 2.9 min (presumably (2R, 3S)-2-methyl-3,5-dihydroxy-4-keto-2,3-dihydropyran 14), TDP (at 51 min) and a minor product (at 43 min). The UV spectrum of the minor compound observed at 43 min indicated a thymine chromophore. Thus, the unknown compound peak at 43 min could be the product (12) of RavIM. Despite several attempts we were unable to isolate and characterize 12 due to its poor yield (< 5%) and instability. However, these results indicate RavIM might be catalyzing the isomerization reaction (10→12), and that product 12 might be spontaneously decomposing to TDP (24) and dihydromaltol derivative 14 in the absence of the follow-up enzyme, aminotransferase RavAMT.
BLAST analyses revealed that the product of ravAMT shows strong sequence similarity to aminotransferases involved in the biosynthesis of D-desosamine such as EryC139 (60%/75%), OleN2 40 (61%/73%) and DesV 41 (59%/71%) from the erythromycin A, oleandomycin and pikromycin bio synthetic pathways, respectively. All of these homologous enzymes were pyridoxal-5-phosphate (PLP)-dependent and were proposed/characterized to catalyze amino transfer from an amino acid, preferably l-glutamate, to the carbonyl carbon of a deoxysugar intermediate.41,42 Therefore, we assumed that RavAMT might serve as an aminotransferase which converts the product of RavIM (12) to TDP-3-amino-3,6-dideoxy-d-galactose (15).
To validate the function of RavAMT, the enzyme was over-expressed in E. coli with a His6-tag and was purified using IMAC. Analysis of the structure by size exclusion chromatography indicated that the enzyme exists as a dimer. Incubation of 10 with RavIM and RavAMT in the presence of sodium glutamate and pyridoxal-5-phosphate (PLP) resulted in the formation of a new compound showing a peak (calculated 20% yield) at 12.8 min in the HPLC analysis. Production of TDP (24) in minor amounts was also noticed (Fig. 3, trace c and d). The new peak was collected from the HPLC and desalted through gel filtration. Low resolution ESI mass analyses revealed a molecular ion peak at 546 Daltons, which matched with the molecular weight of 15 (molecular formula C16H27N3O14P2, calcd. [M - H]− = 546.0968, Fig. S12†).
The negative-mode high-resolution ESI-MS revealed a molecular ion peak at 546.0922 which further confirmed the calculated molecular formula of 15. The 1H NMR of this compound revealed C-5 methyl protons as a doublet at δ 1.16. The proton at C-5 position appeared as a quartet at δ 4.25. The small coupling constant (J = 3.0 Hz) observed for the C-4 proton (δ 3.93) indicated its equatorial orientation. The proton at C-3 position appeared as a double doublet at δ 3.63 with the coupling constants J = 11.0 Hz and 3.0 Hz. This further confirmed the presence of an equatorial proton at C-4 and of an axial hydrogen atom at C-2. As observed for 10, the proton at C-2 and C-1 of 15 appeared as a double triplet at δ 3.88 and a double doublet at δ 5.54, respectively. These assignments were made by analyzing the coupling constants of the 1H NMR in addition to 1H-1H COSY correlations (Table 1). As a positive control, FdtA and FdtB were also expressed in E. coli and the purified enzymes were used to replace RavIM and RavAMT, respectively. Incubation of 10 with FdtA resulted in the gradual consumption of substrate with the concomitant production of a new compound, likely 12 (eluted at 43 min) (see Figs. S2 and S4†). This peak continuously grew for 60 min and then remained unchanged from 60 through 120 min. However, a continuous production of TDP (24) and a new peak (1.8 min) was observed throughout the assay period indicating the decomposition of 12 (Fig. S5†). The specific activity of FdtA (201 µmol mg−1 min−1) was found to be ~6-fold higher than that of RavIM (31.3 µmol mg−1 min−1). Co-incubation of FdtA and FdtB with TDP-4-keto-6-deoxy-d-glucose, pyridoxal-5-phosphate and l-glutamate resulted in the production of 15 as observed earlier through the co-assay of RavIM and RavAMT. These results also clearly established that RavIM and RavAMT act as 3,4-keto-isomerase and aminotransferase, respectively, which catalyze the conversion of 10→12 and 12→15, respectively.
Table 1.
1H NMR (500 MHz) data of TDP-3-amino-3,6-dideoxy-d-galactose (15) and TDP-d-ravidosamine (17) in D2O
| 15 |
17 |
|||
|---|---|---|---|---|
| Position | δH [ppm] | Multiplicity (J/Hz) |
δH [ppm] | Multiplicity (J/Hz) |
| 1 | 5.54 | dd (6.9, 3.5) | 5.64 | dd (6.9, 3.6) |
| 2 | 3.88 | td (11.1, 3.5) | 4.21 | m |
| 3 | 3.63 | dd (11.1, 3.1) | 3.60 | dd (11.4, 2.7) |
| 4 | 3.93 | br, d(3.1) | 4.21 | m |
| 5 | 4.25 | q(6.4) | 4.25 | q(6.6) |
| 5-CH3 | 1.16 | d (6.4) | 1.19 | d (6.6) |
| 1′ | 6.28 | t(6.8) | 6.33 | t(6.8) |
| 2′ | 2.36–2.28 | m | 2.29–2.38 | m |
| 3′ | 4.55 | br, m | 4.60 | br, m |
| 4′ | 4.10–4.16 | m | 4.10–4.16 | m |
| 5′ | 4.10–4.16 | m | 4.10–4.16 | m |
| 5″-CH3 | 1.87 | s | 1.91 | d(1.1) |
| 6″ | 7.66 | br, s | 7.70 | d (1.1) |
| 3-N(CH3)2 | — | — | 2.94 | s |
Heterologous production and characterization of RavNMT
The encoded amino acid sequence of ravNMT showed strong similarity to various S-adenosylmethionine (SAM)-dependent methyltransferases (AdoMet-MTase). These homologous enzymes include N-methyltransferases such as SnogX (56%/71%) and SnogA (57%/70%) from the TDP-l-nogalamine biosynthetic pathway.43 Similarly, N,N-dimethytransferases such as DesVI (55%/71%) and OleM1 (53%/68%) from the TDP-d-desosamine pathway 42,44 represent the other homologous enzymes in the database. Both enzymes are involved in the biosynthesis of TDP-d-desosamine, and transfer two methyl groups in an iterative fashion from SAM to the amino nitrogen. Since the nitrogen at C-3′ position of ravidosamine contains two methyl groups, and there was no other deoxysugar-N-methyltransferase encoding candidate gene identified in the ravidomycin V gene cluster, we reasoned that RavNMT serves as a dimethyltransferase catalyzing the conversion of 15→17.
To validate the proposed function of RavNMT, the enzyme was heterologously produced in E. coli, and purified through IMAC (Fig. S1†). Size exclusion chromatography analysis of RavNMT along with standard proteins suggested that RavNMT exists as a dimer. Incubation of 15 with RavNMT in the presence of SAM resulted in the complete consumption of 15 with concomitant production of a new peak (calculated ~ 90% product yield) at 4.8 min in the HPLC (Fig. 4). The compound pertaining to the new peak was isolated and purified through HPLC and gelfiltration. High resolution ESI mass of the isolated compound revealed a quasi molecular ion peak of 574.1227 Dalton, which is in agreement with the molecular formula C18H31N3O14P2 of the proposed product 17 (calcd. MW, [M - H]− = 574.1281). Moreover, the 1H NMR analysis of the compound revealed a new singlet peak at 8 2.94 indicating the presence of two N-methyl groups. To prove that RavNMT catalyzes the transfer of two methyl groups sequentially, the reaction was stopped after 10 min and the products subjected to MS analyses. The negative-mode high-resolution ESI mass spectra of the reaction mixture revealed molecular ion peaks of 574.1227, 560.1073 and 546.0922 Daltons, which corresponded to the molecular ions of 17, 16 and 15, respectively. These results showed that RavNMT catalyzes the transfer of methyl groups from SAM to the amino nitrogen of 15 in a sequential manner.
Fig. 4.

HPLC traces demonstrating the conversion of TDP-3-amino-3,6-dideoxy-d-galactose (15) to TDP-d-ravidosamine (17). Trace a: control assay mixture including 15 and S-adenosylmethionine (SAM); trace b: products formed after addition of RavNMT with the mixture shown in trace a; trace c: TDP-d-ravidosamine (17) purified through gel filtration. Peaks 23 and 24 represent TMP and TDP produced through the spontaneous decomposition of 15, respectively. The peak marked with an asterisk was not characterized, as its UV spectrum did not indicate a thymidine chromophore.
One-pot enzymatic synthesis of TDP-d-ravidosamine
One of the major goals of this study was to generate the anticipated sugar donor substrate 17 of RavGT, necessary for future in vitro glycosylation assays. Such assays require an easy, reproducible and an efficient way to generate 17. Having all of the biosynthetic enzymes in hand, we attempted to set up a protocol to generate 17 in a large-scale one-pot setup. However, the fairly expensive starting material TTP (25) would limit a larger-scale production of 17. Therefore, we cloned the enzymes necessary for the biosynthesis of TTP from E. coli following a reported protocol.45 Thymidine monophosphate kinase (TMK) generates 24 from 23, and acetate kinase (ACK) further phosphorylates 24 to TTP (25). ATP serves as a phosphate donor thereby converting into ADP, acetate kinase also regenerates ATP by transferring the phosphate group of acetyl phosphate to ADP (Scheme 2).
Scheme 2.

Enzymatic synthesis of thymidine-triphosphate (TTP).
Both proteins were overexpressed in E. coli and purified through IMAC (Fig. S1†). Incubation of 23 with acetyl phosphate and catalytic amount of ATP resulted in the production of 24 and 25 in quantitative yields. Since these reactions were reversible, the reaction was driven forward by adding TDP-d-glucose synthase and 8 to the assay mixture (Fig. 5). TDP-d-glucose synthase from Salmonella typhimurium (RfbA) was heterologously produced in E. coli and was used in place of relatively unstable RavD. Considering the fact that 24 can serve as an inhibitor for the downstream pathway enzymes, overall TDP-ravidosamine (17) biosynthesis was divided into two stages in the same pot. In the first stage, 23, 8 and ATP were incubated with ACK, TMK, RfbA and RavE/RfbB/RmlB that resulted in the production of 10 efficiently (~95% yield) in 5 min (for details, see Experimental section). Because of its instability RavE was substituted by the TDP-d-glucose-4,6-dehydratase (RfbB) from Salmonella typhimurium. RfbB has been routinely used for an efficient conversion of 9→10.8,35,46 However, following several batches of protein production, the recombinant RfbB was expressed mostly as insoluble protein. Therefore, an additional TDP-d-glucose-4,6-dehydratase (encoded by the gene rmlB) was cloned from E. coli, and the protein was expressed and purified. MS analysis of the products revealed that addition of RmlB in the assay mixture resulted in the complete conversion of 9→10 within five minutes. Following the complete conversion of 8→10, the enzymes were removed from the assay mixture and the second stage enzymes (RavIM, RavAMT, and RavNMT) along with their co-factors (PLP, l-glutamate, and S-adenosyl-methionine (SAM)) were then added to the assay mixture to complete the synthesis of 17. More than 95% of 10 was consumed in 65 min (Fig. 6) with the concomitant productions of 15 and a mixture of 16 and 17. The presence of 16 and 17 in the assay mixture was confirmed through HR-MS analysis. However, we were unable to determine their exact quantities, as both compounds were eluted at identical retention times in the HPLC. The production of these compounds remained unchanged from 60 to 180 min. This was a clear indication of the lack of SAM in the assay mixture. Addition of excess SAM resulted in a dramatic increase in production of 17 with the consumption of 15 as well as of 16 in additional 60 min, as evident from 1H NMR analyses of the products. The overall yield of 17 generated was estimated to be 85%.
Fig. 5.

Enzymatic total synthesis of TDP-d-ravidosamine. Trace a: Thymidine monophosphate (TMP, 23) incubated with acetate kinase (ACK), thymidine monophosphate kinase (TMK) and adenosine triphosphate (ATP), 24 = TDP, 25 = TTP; trace b: assay mixture of trace a incubated with glucose-1-phosphate (G-1-P) and RavD, 9 = TDP-d-glucose; trace c: assay mixture of trace b supplemented with RavE, 10 = TDP-4-keto-6-deoxy-d-glucose; trace d: assay mixture of trace c supplemented with RavIM, RavAMT, l-glutamate and pyridoxal-phosphate (PLP), 15 = TDP-3-amino-3,6-dideoxy-d-galactose; trace e: assay mixture of trace d supplemented with S-adenosyl methionine (SAM) and RavNMT, 17 = TDP-d-ravidosamine. Reactions were carried out for 1 h prior to the HPLC analyses. The compounds on the HPLC traces for a, b, c and d remained unchanged when the commercial TTP was used in place of the TTP generated in situ by ACK, TMK, acetyl phosphate, TMP (23) and ATP. The peaks with numbers 9, 10, 15, 17, 23, 24 and 25 represent TDP-d-glucose, TDP-4-keto-6-deoxy-d-glucose, TDP-3-amino-3,6-dideoxy-d-galactose, TDP-d-ravidosamine; TMP, TDP and TTP, respectively. The peak marked with an asterisk was not characterized, as its UV spectrum did not indicate a thymidine chromophore. SAM represents S-adenosyl methionine.
Fig. 6.

Conversion profile of TDP-4-keto-6-deoxy-d-glucose (10) to TDP-d-ravidosamine (17) at the second stage. Lines with empty circles, solid triangles and empty squares represent the concentration profile of TDP-4-keto-6-deoxy-d-glucose (10), TDP-3-keto-6-deoxy-d-galactose (12), and TDP-3-amino-3,6-dideoxy-d-galactose (15), respectively. The line with filled squares from 0 through 180 min represents concentrations of the mixture of TDP-d-ravidosamine (17) and TDP-3-N-methyl-3-amino-3,6-dideoxy-d-galactose (16). Further addition of SAM at 180 min (arrow) resulted in the complete conversion of 15 and 16→17 in 240 min.
Discussion
Bioactive glycosylated natural products containing amino sugars have the advantage that they can be solubilized through formulation as salts, or packaged into nano-liposome delivery vehicles.47–49 Salt forms of such products often exhibit better bioavailability in addition to their increased hydrophilicity. Therefore, the study of dedicated glycosyltransferases which utilize amino sugars is promising for the generation of new pharmaceutically useful natural products through glycodiversification. C-GTs which establish stable C–C bonds between sugar and aglycone are considered special, since C-glycosylated compounds cannot be destroyed by hydrolysis through glycosidases. Although dozens of C-GTs have been identified from secondary metabolite biosynthetic pathways, none of those that utilize deoxyaminosugars has been characterized in vitro. Deacetylravidomycin V (2) is of particular interest as it contains a C-glycosidically linked aminosugar and is one of the most potent drugs among the gilvocarcin family of anticancer compounds. It was our hope that by producing the anticipated sugar donor substrate for RavGT, presumably TDP-d-ravidosamine (17), we would be able to further investigate the glycosylation activity of RavGT in vitro.
We had previously demonstrated that expression of the cosmid cosRav32 in S. lividans TK64 accumulates 2 and deacetylravidomycin M (4). This indicated that the hypothesized enzymes RavD, RavE, RavIM, RavAMT and RavNMT are sufficient to biosynthesize 17. Biosynthesis of all deoxysugars begins with the activation of hexose-1-phosphate utilizing a particular NTP. Although TTP is most commonly used by NDP-hexose synthases, there are also examples where GTP or CTP are utilized for activation.50–53 Therefore, various NTPs were tested for the activation of 8. In vitro assays clearly demonstrated that RavD preferentially utilizes TTP (25) to generate 9 which converts into 10 through the activity of RavE. These two steps are common to a variety of deoxysugar biosynthetic pathways, such as TDP-d-olivose,54 TDP-l-olivose,55 TDP-d-mycarose,14 TDP-l-mycarose,46 TDP-d-fucose 56 and TDP-l-rhamnose.57 Isomerization of 10→12, catalyzed by RavIM, was the unique step of the TDP-ravidosamine (17) biosynthetic pathway. Such a conversion was further confirmed by the formation of an identical product in a parallel assay involving RavIM’s homologue FdtA. To the best of our knowledge, no such isomerase has been previously reported from secondary metabolite biosynthetic pathways. FdtA from Aneurinibacillus thermoaerophillus L420-91 was the only enzyme characterized to catalyze such a transformation during the biosynthesis of 3-acetamido-3,6-dideoxy-d-galactose (18) during cell wall biosynthesis.37,38 The TDP-d-mycaminose (22) biosynthetic pathway is identical to the ravidosamine (17) pathway except for the isomerization step where Tyl1a converts 10→19 with an equatorial hydroxyl group at the C-4 position (Scheme 1).35 Incorporation of ravidosamine in place of the mycaminose moiety of a pikromycin derivative through the expression of FdtA in the Tyl la-deletion mutant has demonstrated a close resemblance of these pathways.58 Comparative studies of RavIM and FdtA revealed that the former enzyme is less active compared to the latter (specific activity of RavIM appeared to be ~6.4-fold less than that of FdtA). Such a small turnover of the recombinant RavIM hindered us from studying the isomerization step in detail. Therefore, FdtA was used to monitor the 3,4-ketoisomerization reaction.
Scheme 1.

Biosynthetic pathways to TDP-d-ravidosamine (17), TDP-d-mycaminose (22), and TDP-3-N-acetamido-3,6-dideoxy-d-galactose (18).
Isolation and characterization of 12 would provide direct evidence for the activity of FdtA and RavIM. However, the newly formed compound was spontaneously decomposed to TDP (24) and presumably (2R, 3S)-2-methyl-3,5-dihydroxy-4-keto-2,3-dihydropyran (14); however, the structure of the latter is yet to be confirmed. In a time course study, a continuous production of 24 with the consumption of 10 was observed, but the concentration of a new peak pertaining to 12 remained unchanged after 55 min. This suggested an existence of two parallel reactions: (i) the enzyme-catalyzed isomerization 10→12 and (ii) the spontaneous decomposition of 12→24 and 14. The product 12 could easily undergo keto–enol tautomerism leading to the spontaneous decomposition as shown in Scheme 1. There is precedence for such a transformation, observed in the D-mycaminose pathway where the Tyl1a product 19 spontaneously degraded to TDP and (2R, 3R)-2-methyl-3,5-dihydroxy-4-keto-2,3-dihydropyran (20).35
Comparable conversions of 10→15 using RavIM-RavAMT or FdtA-FdtB were in agreement with previous reports using Tyl1a-TylB, where the isomerization reaction is reversible and transamination drives the equilibrium forward.35 The conversion of 15 to 17 through the activity of RavNMT unambiguously proved the enzyme to be an N, N-dimethyltransferase. Identification of both 17 and 16 in the assay mixture suggested a sequential transfer of methyl groups from SAM to the amino nitrogen. DesVI and TylM1 from desosamine and mycaminose biosynthetic pathways, respectively, are other examples of such N,N-dimethyltransferases which catalyze dimethylation of a sugar amino group in a sequential manner.44,59
Along with a high demand of NDP-activated sugars for in vitro glycosylation studies, reports on one-pot enzymatic syntheses of NDP-sugars are rapidly growing.46,57,60 With all of the functional biosynthetic enzymes in hand, we also devised a one-pot enzymatic synthesis protocol for the quick and efficient production of TDP-d-ravidosamine (17) in a cost-efficient manner. Instead of using expensive commercial TTP (25), the compound was generated in situ from TMP and acetyl phosphate using acetate kinase (ACK) and thymidine monophosphate kinase (TMK) from E. coli. An identical strategy was applied earlier for the enzymatic synthesis of TDP-l-rhamnose,57 TDP-4-keto-6-deoxy-d-glucose (10),45 and TDP-4-amino-4,6-dideoxy-d-glucose.60 In the first stage, intermediate 10 was produced in satisfactory yields (~95%). Further incubation of the first-stage product with the remaining biosynthetic enzymes resulted in the efficient production of TDP-d-ravidosamine (17), with ~85% yield for the second-stage process. Suzuki et al. had developed an elegant chemical synthesis of ravidosamine and some of its derivatives.30 However, like the syntheses of other deoxysugars, particularly when TDP-activated, the reported procedure involved lengthy synthetic steps with overall poor yields. In addition, the published ravidosamine synthesis lacks the TDP-activation, and is therefore unsuited to provide 17 for glycosyltransferase studies. The enzymatic total synthesis described here is also likely the better method to mass-produce ravidosamine derivatives in general.
In summary, all of the proposed ravidosamine biosynthetic enzymes were characterized in vitro. The results unambiguously established a pathway for the biosynthesis of the sugar moiety of deacetylravidomycin V (2) and deacetylravidomycin M (4). The one-pot enzymatic synthesis protocol developed in this study is a valuable tool for producing TDP-d-ravidosamine (17) in large scale for in vitro glycosylation studies involving RavGT as well as other suitable GTs.
Experimental
Cloning and preparation of expression constructs
Oligonucleotide primers used in this study are summarized in Table S1†. Strain E. coli XL1 Blue MRF was used for the routine amplification of plasmid DNA. Genomic DNA of Salmonella enterica serovar. typhimurium LT2, E. coli XL1 Blue and Aneurini-bacillus thermoaerophillus L420-91 were prepared following a standard protocol.61 Nucleotide sequences for the genes encoding TDP-d-glucose-4,6-dehydratase (rmlB), acetate kinase (ack) and thymidine monophosphate kinase (tmK) were amplified from the genomic DNA of E. coli. Similarly, the genes encoding TDP-d-glucose thymidyltransferase (rfbA), TDP-d-glucose-4,6-dehydratase (rfbB), and TDP-4-keto-6-deoxy-d-glucose-3,4-keto-isomerase (fdtA) and aminotransferase fdtB were amplified from the genomic DNA of Salmonella typhimurium LT2 and Aneurini-bacillus thermoaerophillus L420-91, respectively. All of the TDP-d-ravidosamine biosynthetic genes were amplified from cosmid cosRav3229
Expression and purification of proteins
Pfu polymerase (Stratagene) was used for PCR amplifications, and the obtained products were cloned into the ZeroBlunt-TOPO vector (Invitrogen). Sequencing of the TOPO clones were carried out to ascertain the fidelity of the PCR amplifications. Nucleotide fragments covering ack, ravD, ravE, ravIM, ravAMT, ravNMT and rfbA genes were isolated from their respective TOPO-clones using NdeI and EcoRI restriction enzymes and subsequently ligated to identical sites of the pET28a(+) expression vector (Novagen). Similarly, gene sequences for tmk, rfbB and rmlB were ligated at EcoRI/HindIII, BamHI/HindIII and BamHI/XhoI sites of pET28a(+), respectively. The generated constructs were then transformed into E. coli BL21(DE3). A single colony was inoculated into 20 mL of LB liquid supplemented with kanamycin sulfate and was grown for 4 h to prepare a seed culture. One liter of LB (100 mL × 10 flasks) was inoculated with 10 mL of the seed culture and grown at 37 °C until OD600 reached ~0.5. The culture was then grown at 18 °C for 12 h following addition of IPTG (0.5 mmol final concentration). The cell pellets were collected by centrifugation (4000 × g, 15 min) and were washed twice with 20 mL of lysis buffer (50 mmol KH2PO4, 300 mmol NaCl, 10 mmol imidazole, pH 7.6). Disruption of the pellets was carried out using a French Press (Thermo electron corporation), and the crude soluble enzyme fraction was collected through centrifugation (17 000 × g). N-terminal His-tagged enzymes were purified through immobilized metal affinity chromatography (IMAC). The purified proteins were further desalted using a Profinia protein purification system (Bio-Rad). The Bradford protein assay method was used to determine the concentration of the enzymes.62 The purities of the enzymes were evaluated using dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The yields of purified proteins (mg per L culture) which slightly varied from batch to batch are as follows: ACK, 8–10; TMK, 18–20; RfbA, 18–20; Rm1B, 21–24; FdtA, 6–8; FdtB, 10–13; RavD, 18–20; RavE, 8–10; RavNMT, 12–15; RavAMT, 12–15 and RavIM, 4–6.
Substrates and co-factors
Thymidine-5′-phosphate (TMP, 23), thymidine-5′-diphosphate (TDP, 24), thymidine-5′-triphosphate, (TTP, 25), uridine-5′-triphosphate (UTP), guanosine-5′-triphosphate (GTP), cytidine-5′-triphosphate(CTP), adenosine-5′-triphosphate (ATP), glucose-1-phosphate (8), TDP-d-glucose (9), acetyl phosphate, pyridoxal-5′-phosphate (PLP), l-glutamic acid and S-adenosyl methionine (SAM) were purchased from Sigma-Aldrich.
Enzyme assay of RavD and preparation of TDP-d-glucose
Enzyme assays for TDP-d-glucose synthases (RavD or RfbA) were carried out in 200 µL mixture composed of 50 mmol phosphate buffer (pH 7.5), 20 mmol MgCl2, 5 mmol glucose-1-P (8), 5 mmol of each NTP (ATP, UTP, TTP, GTP and CTP, respectively) and 3 µmol enzyme. The mixture was incubated for 30 min in a 37 °C water bath. The reaction was stopped by heating the mixture at 90 °C for 3 min. The precipitated enzymes were removed by centrifugation and the clear supernatant fractions were passed through an amicon filter (MW cut-off 3000 Da). HPLC analyses of the products were carried out at 25 °C using Waters HPLC system equipped with photodiode array detector. Compounds were monitored at 267 nm wavelength. The filtrate (50 µL) was injected in the HPLC. CarboPack™ PA1 (4 × 250 mm) column (Dionex) was used to separate compounds following the previously reported procedure.35 A gradient of ammonium acetate (500 mmol, solvent B) and water (solvent A) was used to separate the assay products: 5–20% solvent B from 0–15 min, 20–60% solvent B from 15–35 min, 60–100% solvent B from 35–37 min, 100% solvent B from 37–40 min, 100–5% solvent B from 40–45 min and 5% solvent B from 45–60 min. A control sample without enzyme and standard TDP-d-glucose (9) was also injected for the identification of the product peak. For large-scale production of 9, TMP (5 mmol), 8 (5.1 mmol), acetyl phosphate (20 mmol) and ATP (5 µmol) were mixed in a 20 mL solution composed of 50 mmol phosphate buffer (pH 7.5), 20 mmol MgCl2, 4 µmol ACK, 6 µmol TMK and 5 µmol RavD/RfbA. The mixture was incubated for 1 h, and the reaction was quenched by heating the mixture at 90 °C for 5 min. The mixture was centrifuged at 10 000 × g for 10 min and the supernatant was lyophilized. The dry powder was dissolved in 5 mL of water and the solution was loaded into the pre-equilibrated Bio-Gel P2 gel filtration column 100 cm × 25 mm. Separation was carried out at 4 °C with water at flow rate of 0.2 mL min−1. Aliquots of 4 mL were collected and the purity of compounds was monitored by HPLC at 267 nm. Pure fractions were combined, lyophilized and analyzed by NMR. About 80 mg of pure 9 was obtained along with an additional 125 mg of 9 with minor TMP (23) and TDP (24) impurities. The purity 9 was assayed through HPLC and 1H NMR (Fig. S6†). 1H NMR of 9(500 MHz, D2O): δ 1.90 (3H, s, 5″-Me), 2.31–2.38 (2H, m, 2′-H), 3.43 (1H, t, J 9.8 Hz, 3-H), 3.51 (1H, td, J 9.8, 3.5 Hz, 2-H), 3.72–3.78 (2H, m, 4-H, 5-H), 3.81–3.90 (2H, m, 6-H2), 4.12–4.16 (1H, m, 4′-H), 4.12–4.16 (2H, m, 5′-H), 4.62 (1H, m, 3′-H), 5.58 (1H, dd, J 7.4, 3.5 Hz, 1-H), 6.33 (1H, t, J 6.98 Hz, 1′-H), 7.72 (1H, s, br, 6″-H).
Enzyme assay of RavE and preparation of TDP-4-keto-6-deoxy-d-glucose
To carry out TDP-d-glucose-4,6-dehydratase assays, the enzymes (4 µmol RavE or 3 µmol RfbB or 3 µmol RmlB) were added to 200 µL solution composed of phosphate buffer (50 mmol, pH. 7.5), MgCl2 (20 mmol), NAD+ (2 µmol) and 9 (5 mmol). The mixture was incubated for 30 min and the reaction was stopped. An enzyme-free solution was analyzed by HPLC. For large-scale production, a one-pot enzymatic synthesis was carried out in 50 mL volume composed of phosphate buffer (50 mmol, pH 7.5), MgCl2 (20 mmol), 8 (150 mmol), TMP (23, 98.3 mmol), acetyl phosphate (400 mmol), ATP (1 mmol), ACK (0.8 µmol), TMK (2.1 µmol) and RfbA (2.0 µmol). The mixture was incubated for 1 h at 37 °C, and then RavE (final concentration of 2.73 µmol) or RfbB (2.3 µmol) and NAD+ (2 µmol) were added and incubation was continued for an additional 2 h. Surprisingly, when RavE and RfbB was expressed for additional reactions they appeared exclusively in the form of inclusion bodies. Therefore, these enzymes were replaced by RmlB (6 µmol) in the following batches. The reaction mixture was heated to denature the enzymes. The precipitated fractions were removed by centrifugation. The crude product was lyophilized to powder and dissolved in 10 mL water. Separation of the products was carried out in Bio-gel P2 following the protocol described above. The purity of each fraction was analyzed by HPLC. Pure fractions were combined and lyophilized to powder. This afforded 700 mg of TDP-4-keto-6-deoxy-d-glucose (10) with ~95% purity and an additional 630 mg of 10 with >70% purity. The purity and identity of the compound was further confirmed through the comparison of 1H NMR data reported in the literature (Fig. S7†). 1H NMR of 10 (500 MHz, D2O): δ 1.06 (3H, d, J 6.50 Hz, 5-Me), 1.77 (3H, s, J 0.97 Hz, 5″-Me), 2.23 (2H, dd, J 6.90, 4.85 Hz, 2′-H2), 3.51 (1H, td, J 10.07, 3.3 Hz, 2-H), 3.63 (1H, d, J 10.07 Hz, 3-H), 3.93 (1H, q, J 6.5, 5-H), 3.99–4.06 (3H, m, 4′-H, 5′-H), 4.43–1.47 (1H, m, 3′-H), 5.40 (1H, dd, J 6.8, 3.3 Hz, 1-H), 6.19 (1H, t, J 6.90 Hz, 1′-H), 7.57 (1H, d, 6″-H).
Assay of RavIM alone or coupled with RavAMT
RavIM (20 µmol final concentration) was incubated at 37 °C with 10 (1 mmol) in a 500 µL solution composed of phosphate buffer (50 mmol, pH 7.5) and MgCl2 (20 mmol) for 60 min. An aliquot (60 µL) was drawn from the incubation mixture and flash-frozen in dry ice–acetone. The enzyme was removed by further heating the mixture at 90 °C for 3 min. The solution was filtered through a centricon (MW cut-off 3000 Da) and the filtrate (50 µL) was analyzed by HPLC. In the coupled enzyme assays, RavAMT (30 µmol), l-glutamate (10 mmol) and PLP (60 µmol) were added in the RavIM assay mixture and progress of the reaction was monitored until 60 min. A new peak appeared in the coupled assays at 12.8 min which was collected and analyzed by high-resolution ESI-MS. Similarly, FdtA and FdtA-FdtB coupled assays were also carried out in parallel as positive controls. For the preparative scale production of TDP-3-amino-3,6-dideoxy-d-galactose (15), the substrate and enzyme concentrations were doubled and the assay volume was increased to 20 mL. After 6 h of incubation, enzymes were removed and the volume was reduced to 1 mL. Separation of the compounds was carried out in analytical-scale HPLC. The compound eluted at 12.8 min as before and was collected and separated from the ammonium acetate buffer using a Bio-Gel P2 column. The pure fractions were combined and lyophilized. Overall, 12 mg powder of 15 was purified. The structure of 15 was confirmed through MS and NMR data analyses.
HPLC analyses of FdtA-catalysed isomerization reaction
TDP-4-keto-6-deoxy-d-glucose (10, 1 mmol) was incubated at 37 °C with 24.9 µmol enzyme in 400 µL solution containing 50 mmol phosphate buffer and 20 mmol magnesium chloride. Aliquots (60 µL) were drawn from the assay mixture at various time points and quenched in a dry ice–acetone bath. The samples were boiled for 4 min and centrifuged to remove the precipitated proteins. The clear supernatant (50 µL) was injected into the HPLC and the peak areas under the curve were normalized (Fig. S4 and S5†).
In vitro assay of RavNMT
For an analytical scale assay, a 200 µL assay mixture containing 50 mmol phosphate buffer, 20 mmol MgCl2, 1 mmol of 15, 4.0 mmol SAM and 10 µmol RavNMT were incubated for two hours. An enzyme-free solution was prepared as discussed earlier and injected in an HPLC. A new peak appeared at 4.8 min and was collected and analyzed by HR-ESI mass spectrometry. For large-scale production, a coupled assay was carried out in 20 mL using 10 (2 mmol), l-glutamate (14 mmol), PLP (80 µmol), SAM (4.0 mmol) RavIM (4.8 µmol), RavAMT (6.5 µmol) and RavNMT (8.0 µmol). After two hours of incubation, the enzymes were removed and the solution was lyophilized. The dried powder was dissolved in 1 mL water and separated by HPLC. The compound eluted at 4.8 min and was collected and desalted using a Biogel -P2 column. Lyophilization of the pure fractions afforded 5 mg of TDP-d-ravidosamine (17) and 8 mg of a 1:1 mixture of TDP-3-N-methyl-3-amino-3,6-dideoxy-d-galactose(16) and 17, respectively. The structure of 17 was determined through NMR and HR-MS analyses.
Two-stage one-pot enzymatic synthesis of TDP-d-ravidosamine
Overall enzymatic synthesis of 17 was divided into two stages. For the first stage, a 20 mL solution containing 8 (0.5 mmol), 23 (0.48 mmol), ATP (45 µmol), acetyl phosphate (2 mmol), ACK (5 µmol), TMK (10 µmol), RfbA (8 µmol), RmlB (5.8 µmol), phosphate buffer (50 mmol, pH 7.5) and MgCl2 (20 mmol) was incubated at 37 °C for 30 min. HPLC analyses of the assay mixture revealed the production of 10 (0.47 mmol) in the assay mixture. Enzymes were precipitated by heating the solution to 95 °C and removed through centrifugation. In the second stage, RavIM (2.3 µmol), AMT (3.9 µmol), RavNMT (4.5 µmol), l-glutamate (7 mmol), PLP (42 µmol) and SAM (0.7 mmol) were then added to the assay mixture and incubation was continued for 5 h. Considering the fact that SAM degrades in water, an additional 0.7 mmol (final concentration) of SAM was supplied after 3 h. The reaction was monitored using HPLC as discussed above. A calibration curve was generated varying the concentration of 23 from 62 nM to 15 mmol. The concentrations of TDP-d-hexoses were determined using this calibration curve. The reaction was stopped by heating at 90 °C for 4 min. The denatured protein was removed through centrifugation and the liquid fraction was lyophilized. The peak corresponding to 17 was separated through gel filtration. The fractions including 17 were lyophilized and then analyzed by NMR spectroscopy.
Supplementary Material
Acknowledgements
This work was supported by the US National Institutes of Health (grant CA 102102 to JR). The NMR and mass spectrometry core facilities of the University of Kentucky are acknowledged for the access to their instruments. Dr Guy Carter, Wyeth, is acknowledged for providing the ravidomycin producer Streptomyces ravidus.
Footnotes
Electronic supplementary information (ESI) available: Full details of in vitro experimentation and NMR data.
References
- 1.Johnson DA, Liu H-W. In: Occurrence, genetics, and mechanisms of biosynthesis Comprehensive Natural Products Chemistry. Barton DHR, Nakanishi K, editors. Vol. 3. Amsterdam: Elsevier; 1999. pp. 311–365. [Google Scholar]
- 2.Hermann T. Curr. Opin. Struct. Biol. 2005;15:355–366. doi: 10.1016/j.sbi.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Kren V, Martinkova L. Curr. Med. Chem. 2001;8:1303–1328. doi: 10.2174/0929867013372193. [DOI] [PubMed] [Google Scholar]
- 4.Weymouth-Wilson AC. Nat. Prod. Rep. 1997;14:99–110. doi: 10.1039/np9971400099. [DOI] [PubMed] [Google Scholar]
- 5.Blanco G, Patallo EP, Brana AF, Trefzer A, Bechthold A, Rohr J, Mendez C, Salas JA. Chem. Biol. 2001;8:253–263. doi: 10.1016/s1074-5521(01)00010-2. [DOI] [PubMed] [Google Scholar]
- 6.He XM, Liu HW. Curr. Opin. Chem. Biol. 2002;6:590–597. doi: 10.1016/s1367-5931(02)00367-8. [DOI] [PubMed] [Google Scholar]
- 7.He XMM, Liu HW. Annu. Rev. Biochem. 2002;71:701–754. doi: 10.1146/annurev.biochem.71.110601.135339. [DOI] [PubMed] [Google Scholar]
- 8.Hong L, Zhao Z, Melancon CE, 3rd, Zhang H, Liu HW. J. Am. Chem. Soc. 2008;130:4954–4967. doi: 10.1021/ja0771383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lhibodeaux CJ, Melancon CE, Liu HW. Angew. Chem., Int. Ed. 2008;47:9814–9859. doi: 10.1002/anie.200801204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lrefzer A, Salas JA, Bechthold A. Nat. Prod. Rep. 1999;16:283–299. doi: 10.1039/a804431g. [DOI] [PubMed] [Google Scholar]
- 11.Lrefzer A, Blanco G, Remsing L, Kunzel E, Rix U, Lipata F, Brana AF, Mendez C, Rohr J, Bechthold A, Salas JA. J. Am. Chem. Soc. 2002;124:6056–6062. doi: 10.1021/ja017385l. [DOI] [PubMed] [Google Scholar]
- 12.Lrefzer A, Pelzer S, Schimana J, Stockert S, Bihlmaier C, Fiedler HP, Welzel K, Vente A, Bechthold A. Antimicrob. Agents Chemother. 2002;46:1174–1182. doi: 10.1128/AAC.46.5.1174-1182.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melancon CE, 3rd, Liu HW. J. Am. Chem. Soc. 2001;129:4896–4897. doi: 10.1021/ja068254t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perez M, Baig I, Brana AF, Salas JA, Rohr J, Mendez C. ChemBioChem. 2008;9:2295–2304. doi: 10.1002/cbic.200800299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blanchard S, Lhorson JS. Curr. Opin. Chem. Biol. 2006;10:263–271. doi: 10.1016/j.cbpa.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Liu T, Kharel MK, Zhu L, Bright S, Rohr J. ChemBioChem. 2009;10:278–286. doi: 10.1002/cbic.200800348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang CS, Fu Q, Albermann C, Li LJ, Lhorson JS. ChemBioChem. 2001;8:385–390. doi: 10.1002/cbic.200600509. [DOI] [PubMed] [Google Scholar]
- 18.Findlay JA, Liu JS, Radics L. Can. J. Chem. 1983;61:323–327. [Google Scholar]
- 19.Findlay JA, Liu JS, Radics L, Rakhit S. Can. J. Chem. 1981;59:3018–3020. [Google Scholar]
- 20.Itoh T, Kinoshita M, Aoki S, Kobayashi M. J. Nat. Prod. 2003;66:1373–1377. doi: 10.1021/np030212k. [DOI] [PubMed] [Google Scholar]
- 21.Itoh L, Kinoshita M, Wei H, Kobayashi M. Chem. Pharm. Bull. 2003;51:1402–1404. doi: 10.1248/cpb.51.1402. [DOI] [PubMed] [Google Scholar]
- 22.Lakahashi KY, Lomita M, Shirahata F. J. Antibiot. 1981;43:271–275. doi: 10.7164/antibiotics.34.271. [DOI] [PubMed] [Google Scholar]
- 23.Weiss U, Yoshihira K, Highet RJ, White RJ, Wei TT. J. Antibiot. 1982;35:1194–1201. doi: 10.7164/antibiotics.35.1194. [DOI] [PubMed] [Google Scholar]
- 24.Nakashima T, Ladashi F, Kazuya S, Lomohiro S, Hiroyuki K, Lakeo Y. US Pat. 1999 09/308710. [Google Scholar]
- 25.Nakajima S, Kojiri K, Suda H, Okanishi M. J. Antibiot. 1991;44:1061–1064. doi: 10.7164/antibiotics.44.1061. [DOI] [PubMed] [Google Scholar]
- 26.Kojiri K, Arakawa H, Satoh F, Kawamura K, Okura A, Suda H, Okanishi M. J Antibiot. 1991;44:1054–1060. doi: 10.7164/antibiotics.44.1054. [DOI] [PubMed] [Google Scholar]
- 27.Li YQ, Huang XS, Ishida K, Maier A, Kelter G, Jiang Y, Peschel G, Menzel KD, Li MG, Wen ML, Xu LH, Grabley S, Fiebig HH, Jiang CL, Hertweck C, Sattler I. Org. Biomol. Chem. 2008;6:3601–3605. doi: 10.1039/b808633h. [DOI] [PubMed] [Google Scholar]
- 28.Rakhit S, Eng C, Baker H, Singh K. J. Antibiot. 1983;36:1490–1494. doi: 10.7164/antibiotics.36.1490. [DOI] [PubMed] [Google Scholar]
- 29.Kharel MK, Nybo SE, Shepherd MD, Rohr J. ChemBioChem. 2010;11:523–532. doi: 10.1002/cbic.200900673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu DS, Matsumoto T, Suzuki K. Synlett. 2005:801–804. [Google Scholar]
- 31.Fischer C, Lipata F, Rohr J. J. Am. Chem. Soc. 2003;125:7818–7819. doi: 10.1021/ja034781q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Remsing LL, Garcia-Bernardo J, Gonzalez A, Kunzel E, Rix U, Brana AF, Bearden DW, Mendez C, Salas JA, Rohr J. J. Am. Chem. Soc. 2002;124:1606–1614. doi: 10.1021/ja0105156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pickens LB, Kim W, Wang P, Zhou H, Watanabe K, Gomi S, Lang Y. J Am. Chem. Soc. 2009;131:17677–17689. doi: 10.1021/ja907852c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stockmann M, Piepersberg W. FEMS Microbiol. Lett. 1992;90:185–189. doi: 10.1016/0378-1097(92)90626-y. [DOI] [PubMed] [Google Scholar]
- 35.Melancon CE, Hong L, White JA, Liu YN, Liu HW. Biochemistry. 2007;46:577–590. doi: 10.1021/bi061907y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vorholter FJ, Niehaus K, Puhler A. Mol. Genet. Genomics. 2001;266:79–95. doi: 10.1007/s004380100521. [DOI] [PubMed] [Google Scholar]
- 37.Pfoestl A, Hofinger A, Kosma P, Messner P. J. Biol. Chem. 2003;278:26410–26417. doi: 10.1074/jbc.M300858200. [DOI] [PubMed] [Google Scholar]
- 38.Davis ML, Lhoden JB, Holden HM. J Biol. Chem. 2007;282:19227–19236. doi: 10.1074/jbc.M702529200. [DOI] [PubMed] [Google Scholar]
- 39.Dhillon N, Hale RS, Cortes J, Leadlay PF. Mol. Microbiol. 1989;3:1405–1414. doi: 10.1111/j.1365-2958.1989.tb00123.x. [DOI] [PubMed] [Google Scholar]
- 40.Quiros LM, Aguirrezabalaga I, Olano C, Mendez C, Salas JA. Mol. Microbiol. 1998;28:1177–1185. doi: 10.1046/j.1365-2958.1998.00880.x. [DOI] [PubMed] [Google Scholar]
- 41.Zhao LS, Que NLS, Xue YQ, Sherman DH, Liu HW. J. Am. Chem. Soc. 1998;120:12159–12160. [Google Scholar]
- 42.Olano C, Rodriguez AM, Michel JM, Mendez C, Raynal MC, Salas JA. Mol. Gen. Genet. 1998;259:299–308. doi: 10.1007/s004380050816. [DOI] [PubMed] [Google Scholar]
- 43.Lorkkell S, Kunnari T, Palmu K, Mantsala P, Hakala J, Ylihonko K. Mol. Genet. Genomics. 2001;266:276–288. doi: 10.1007/s004380100554. [DOI] [PubMed] [Google Scholar]
- 44.Chang CW, Zhao LH, Yamase H, Liu HW. Angew. Chem., Int. Ed. 2000;39:2160–2163. doi: 10.1002/1521-3773(20000616)39:12<2160::aid-anie2160>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 45.Oh J, Lee SQ, Kim BG, Sohng JK, Liou K, Lee HC. Biotechnol. Bioeng. 2003;84:452–458. doi: 10.1002/bit.10789. [DOI] [PubMed] [Google Scholar]
- 46.Lakahashi H, Liu YN, Liu HW. J. Am. Chem. Soc. 2006;128:1432–1433. doi: 10.1021/ja0562144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang J, Li S, Li X. Recent Pat. Nanotechnol. 2009;3:225–231. doi: 10.2174/187221009789177803. [DOI] [PubMed] [Google Scholar]
- 48.Fang JY. Chang Gung Med. J. 2006;29:358–362. [PubMed] [Google Scholar]
- 49.Lassalle V, Ferreira ML. Macromol. Biosci. 2007;7:767–783. doi: 10.1002/mabi.200700022. [DOI] [PubMed] [Google Scholar]
- 50.Weitnauer G, Muhlenweg A, Lrefzer A, Hoffmeister D, Sussmuth RD, Jung G, Welzel K, Vente A, Girreser U, Bechthold A. Chem. Biol. 2001;8:569–581. doi: 10.1016/s1074-5521(01)00040-0. [DOI] [PubMed] [Google Scholar]
- 51.Palaniappan N, Ayers S, Gupta S, Habibel S, Reynolds KA. Chem. Biol. 2006;13:753–764. doi: 10.1016/j.chembiol.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 52.Aparicio JF, Caffrey P, Gil JA, Zotchev SB. Appl. Microbiol. Biotechnol. 2003;61:179–188. doi: 10.1007/s00253-002-1183-5. [DOI] [PubMed] [Google Scholar]
- 53.Lhorson JS, Lo SF, Liu HW. J Am. Chem. Soc. 1993;115:5827–5828. [Google Scholar]
- 54.Perez M, Lombo F, Baig I, Brana AF, Rohr J, Salas JA, Mendez C. Appl. Environ. Microbiol. 2006;72:6644–6652. doi: 10.1128/AEM.01266-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodriguez L, Aguirrezabalaga I, Allende N, Brana AF, Mendez C, Salas JA. Chem. Biol. 2002;9:721–729. doi: 10.1016/s1074-5521(02)00154-0. [DOI] [PubMed] [Google Scholar]
- 56.Yoshida Y, Nakano Y, Nezu L, Yamashita Y, Koga L. J. Biol. Chem. 1999;274:16933–16939. doi: 10.1074/jbc.274.24.16933. [DOI] [PubMed] [Google Scholar]
- 57.Kang YB, Yang YH, Lee KW, Lee SG, Sohng JK, Lee HC, Liou K, Kim BG. Biotechnol. Bioeng. 2006;93:21–27. doi: 10.1002/bit.20648. [DOI] [PubMed] [Google Scholar]
- 58.Melancon CE, Liu HW. J Am. Chem. Soc. 2007;129:4896. doi: 10.1021/ja068254t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen HW, Yamase H, Murakami K, Chang CW, Zhao LS, Zhao ZB, Liu HW. Biochemistry. 2002;41:9165–9183. doi: 10.1021/bi020245j. [DOI] [PubMed] [Google Scholar]
- 60.Chung YS, Kim DH, Seo WM, Lee HC, Liou K, Oh TJ, Sohng JK. Carbohydr. Res. 2007;342:1412–1418. doi: 10.1016/j.carres.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 61.Sambrook J, Russell DW. Molecular Cloning: a laboratory manual. 3rd edn. New York: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 62.Bradford MM. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.