

Abstract

Dioxygen reacts with the gold(I) hydride (IPr)AuH under insertion to give the hydroperoxide (IPr)AuOOH, a long-postulated reaction in gold catalysis and the first demonstration of O2 activation by Au–H in a well-defined system. Subsequent condensation gave the peroxide (IPr)Au–OO–Au(IPr) (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene). The reaction kinetics are reported, as well as the reactivity of Au(I) hydrides with radical scavengers.
Gold hydrides and gold–O2 interactions have been invoked as key species in catalytic reactions on the surface of nanoparticle gold catalysts.1 The activation of O2 on the surface of gold clusters has been extensively investigated by computational and spectroscopic methods,2 and the formation of O2 adducts, such as gold superoxide and gold hydroperoxides, has been suggested. For example, it could be shown that the preadsorption of hydrogen activates the binding of O2 by small gold clusters with the formation of hydroperoxo species, while in gold-catalyzed alcohol oxidations the presence of an alcohol or water is required to convert adsorbed O2 into surface-bound gold hydroperoxides.3,4 In homogeneously catalyzed reactions as well, O2 insertion into putative gold hydride intermediates and formation of LnAu–OOH species have been suggested.5 However, the ability of well-characterized Au–H complexes to activate and insert dioxygen has, to our knowledge, never been demonstrated.
We have recently shown that gold(III) complexes stabilized by cyclometalated C∧N∧C pincer ligands allow the isolation of Au(III) peroxides and hydroperoxides and, furthermore, that these complexes undergo successive oxygen abstraction reactions to give the isolable Au(III) hydride (C∧N∧C)AuH (Scheme 1) (C∧N∧C = 2,6-(C6H3But)2pyridine).6 However, the reverse reaction, the insertion of O2 into the AuIII–H bond to give a AuIII–OOH product, was not observed. This lack of reactivity is most probably linked to the rigidity of the pincer ligand and the reluctance of Au(III) to form five-coordinate intermediates.
Scheme 1.

Two-coordinate Au(I) complexes, on the other hand, could be expected to be kinetically more accessible, even though Au(I) as a softer metal center is less oxophilic than Au(III). We have now found that this is indeed the case. We describe here the first example of an O2 insertion into the Au–H bond of a well-defined Au(I) hydride complex, (IPr)AuH, as well as the homolytic Au–H bond cleavage of (IPr)AuH by stable radicals (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene).
Exposure of a C6D6 solution of Sadighi’s gold(I) hydride7 (IPr)AuH (1) to an atmosphere of dioxygen (1–9 bar) for 4–48 h leads to the disappearance of the resonance associated with the Au–H fragment (δH 5.11) and to the formation of a new product, the gold(I) hydroperoxide (IPr)AuOOH (2) (Figure 1 and Scheme 2). The conversion of 1 into 2 can also be observed by monitoring the proton resonances of the imidazol-2-ylidene ring.
Figure 1.

Stacked plot of 1H NMR spectra (300 MHz, C6D6) showing the reaction of (IPr)AuH (1) with O2 (9 bar, 52 °C).
Scheme 2.

After the first half-life of the reaction, a resonance at δ −0.26 is clearly noticeable, which is consistent with the formation of the gold(I) hydroxide8 (IPr)AuOH (3). The spectrum of the hydroxide 3 is very similar to that of the hydroperoxide 2 except for the notable presence of the OH resonance in 3, whereas in 2 no such OH signal could be detected.
Under 1 bar of O2 at 23 °C full conversion of 1 is observed after 48 h, giving a ratio of compounds 2:3 of about 1:3.9 Eventually, upon standing at room temperature the hydroxide 3 can be obtained quantitatively. This suggests that, while 2 is readily generated under 1 atm of oxygen, there is also slow decomposition of 2 into 3, with release of O2 (Scheme 1). This decomposition pathway is of course not unexpected for hydroperoxides and mirrors that documented, for example, for PdII– and PtIV–OOH species.10 However, in these cases the disproportionation reaction could be suppressed by increasing the dioxygen concentration, whereas in the case of gold(I) we found that the conversion of 2 to 3 takes place even under 9 bar of O2. This is likely to be the consequence of the very significant difference in Au–O bond energies between Au–OH and Au–OOH complexes (>100 kJ mol–1).6,11
In contrast to the reactivity of the gold(I) hydride (1), pressurizing an NMR tube containing (IPr)AuMe with 9 bar of O2 at 60 °C gave no traces of the methyl peroxide (IPr)AuOOMe, and the starting material could be recovered unchanged.
To gain more insight into the mechanism of O2 insertion into a Au–H bond, the kinetics of the reaction were studied by 1H NMR spectroscopy. In order to establish the reaction order in [Au], the reaction was monitored at 52 °C under 4–9 bar of O2 (see the Supporting Information). The reaction is first order in both [IPrAuH] and [O2] (eq 1).
| 1 |
An Eyring plot over the temperature interval of 36–52 °C gave the activation parameters of the reaction as ΔH⧧ = 21.1(1) kJ mol–1 and ΔS⧧ = −251.5(2) J mol–1 K–1 (see Supporting Information). The negative value of ΔS⧧ suggests that the reaction proceeds via an associative mechanism.
The reaction of the deuteride (IPr)AuD (1D) with O2 (9 bar) at 52 °C resulted in a retardation of the reaction rate (kobs(1D) = [0.82(1)] × 10–4 s–1 versus kobs(1H) = [1.94(2)] × 10–4 s–1), which corresponds to a kinetic isotope effect of kH/kD = 2.4. On the basis of the Au–H IR stretching frequency in 1H (νAuH 1976 cm–1),7 a maximum kinetic isotope effect of ca. 3.6 for the homolytic cleavage could be calculated (at T = 52 °C).12 The observed value of the kinetic isotope effect is commensurate with the breaking of the Au–H bond being involved in the rate-determining step.
Radical chain reaction mechanisms have often been invoked in the case of dioxygen insertion reactions into metal–hydride bonds.13 However, in the case of 1, conducting the reaction in the presence of TEMPO did not affect the rate (kobs(1H) = [1.94(2)] × 10–4 s–1 in the absence of TEMPO vs [2.00(2)] × 10–4 s–1 in the presence of TEMPO). The more reactive galvinoxyl radical was found to react directly with 1 via H abstraction (vide infra). These results support the view that the reaction of O2 with (IPr)AuH (1) does not proceed via a radical chain mechanism.
The formation of the Au(I) hydroperoxide 2 was further supported by its reaction with phosphines, which leads to O transfer. For Au(III) hydroperoxides this reaction had previously been shown to proceed stepwise in a well-controlled manner, to generate first the Au(III) hydroxide, followed by a second phosphine oxidation and conversion to the Au(III) hydride.6
The addition of P(p-tolyl)3 (1.2 equiv, δ31P 8.1) to (IPr)AuH (2) at room temperature followed by pressurizing with O2 (4–10 bar) led to phosphine oxidation, as revealed by 31P{1H} spectroscopy (δ31P((p-tolyl)3P=O) +30.6), alongside (IPr)AuOH.14 However, unlike the AuIII–OH precedent, the Au(I) hydroxide could not be deoxygenated further, even with a large excess of phosphine. Evidently the hydroxide to hydride transformation is thermodynamically favorable for Au(III) but not for Au(I) (at least for the ligands investigated so far) (Scheme 3).
Scheme 3.

Attempts to crystallize the hydroperoxide 2 under an atmosphere of oxygen gave single crystals of the binuclear peroxide [(IPr)Au]2(μ-κ1:κ1-O2) (4) instead. The structure of 4 was identified by single-crystal X-ray crystallography (Figure 2). Despite positional disorder of the carbene ligands and peroxo fragment, the connectivity could be assigned unambiguously and confirms the identity of 4 as peroxide bridging two gold atoms (Figure 2). The C–Au–O fragment is linear, which is a typical geometry around a gold(I) center. Unfortunately the low crystal quality precludes any detailed discussion of the bond lengths.
Figure 2.

Molecular connectivity in the peroxo complex 4·2C6H6. For disordered imidazolyl rings and peroxo groups, only one orientation is shown.
Compound 4 is most probably formed by condensation of 2 with the hydroxide 3 (Scheme 4). This behavior is reminiscent of the Au(III) hydroperoxide (C∧N∧C)AuIIIOOH, which also reacts with the corresponding hydroxide to give the bridging (μ-κ1:κ1) peroxide [(C∧N∧C)Au]2(μ-κ1:κ1-O2).6
Scheme 4.

Attempts to obtain better quality crystals of 4 by crystallizing a THF solution under an air atmosphere led to the isolation of the carbonate [{(IPr)Au}3(μ3-CO3)]OH·3THF (5·3THF), evidently the product of the reaction of in situ generated (IPr)AuOH with atmospheric CO2 (Scheme 5).
Scheme 5.
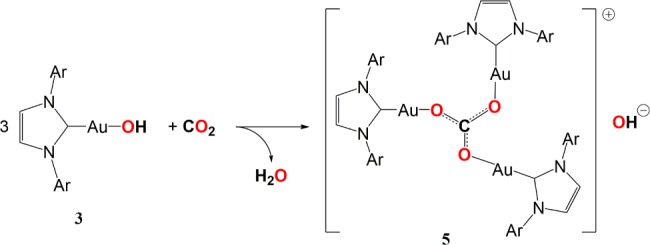
Compound 5 was identified by single-crystal X-ray diffraction. The cation consists of three (NHC)Au ions coordinated to the carbonate oxygen atoms (see the Supporting Information); the anion, most probably HO, was disordered and could not be reliably located. The cation is identical with that prepared recently by Sadighi by a different route,15 as well as by Nolan et al.16
A second reaction pathway of gold hydrides is through H radical abstraction by radical acceptors. We have previously shown that the Au(III) hydride (C∧N∧C)AuH reacts with galvinoxyl to give the Au(II) compound (C∧N∧C)Au–Au(C∧N∧C), whereas there was no reaction with TEMPO. These reactions allowed us to estimate the AuIII–H bond strength as being between 291 and 329 kJ mol–1, in good agreement with the calculated bond dissociation energy of 317 kJ mol–1.6 In the present case the intention was to explore whether H abstraction might provide a convenient and mild route to Au(0) carbene complexes.
Treating (IPr)AuIH 1 with galvinoxyl gave rise to a color change from dark blue (galvinoxyl in C6D6) to purple. This initial reaction in an NMR tube in C6D6 also revealed the rapid disappearance of the hydride resonance of 1 (δH 5.11) and the formation of a new complex. No reaction was observed with TEMPO after 48 h, which leads to an estimate of the AuI–H bond dissociation energy of between 291 and 329 kJ mol–1, similar to the case of AuIII–H. Conducting the reaction on a larger scale in CH2Cl2 and removing excess galvinoxyl and the byproduct, galvinoxylH, afforded a purple powder. The 1H NMR spectrum of this product (6) confirmed the absence of an Au–H resonance and revealed a carbene to galvinoxide ratio of 2:1.
Crystals of 6·3C6H6 were obtained by the slow evaporation of a benzene solution of 6 at room temperature. The solid-state structure of the complex was determined by X-ray crystallography (Figure 3). Despite the modest quality of the crystals, unequivocal connectivity could be established and confirms complex 6 as a linear gold(I) cation (∠C1–Au–C28 177.3(5)°) paired with a galvinoxide anion. The isopropyl substituents are arranged in such a fashion as to minimize steric repulsion between the two carbene fragments, leading to a torsion angle between the two imidazolyl planes of 46.6°. The structural parameters of the cation resemble those reported for [(IPr)2Au]BF4.17
Figure 3.

Molecular structure of [(IPr)2Au]+[galvinoxide]− (6·3C6H6). Hydrogen atoms and the molecules of benzene of crystallization are omitted. Selected bond distances (Å) and angles (deg): Au–C(1) 2.030(9), Au–C(28) 2.061(7), C(1)–Au–C(28) 177.33(45), N1–C1–C28–N3 46.6.
A plausible mechanism for the formation of 6 is given in Scheme 6. It is envisaged that abstraction of H• by galvinoxyl gives initially rise to a short-lived Au(0) species, (IPr)Au, which then undergoes ligand rearrangement to give Au(0) and Au(IPr)2. The latter is oxidized by further galvinoxyl to generate [(IPr)2Au]+[galvinoxide]− (6). The present results show that conventional NHCs are not sufficiently strongly bonded to stabilize gold(0) species.18
Scheme 6.

In summary, we have shown the first example of an insertion reaction of O2 into a gold–hydrogen bond. The reaction proceeds under mild conditions to give, initially, the unstable gold(I) hydroperoxide (IPr)Au(OOH), which readily transfers an oxygen atom to suitable reductants, such as phosphines. These key steps, which have long been postulated for catalytic cycles of homogeneous and heterogeneous gold catalysts, have therefore been demonstrated for the first time on the basis of a well-defined homogeneous system. This reactivity of gold(I) hydrides is in contrast with that of the Au(III) hydride (C∧N∧C)AuH, which does not insert O2 under comparable conditions. The reaction of gold(I) hydrides with radical abstractors allows an experimental estimate of the AuI–H bond energy.
Acknowledgments
This work was supported by the ERC (Advanced Investigator Award 338944 - GOCAT), the Leverhulme Trust, and Johnson Matthey PLC. D.-A.R. thanks the University of East Anglia for a studentship.
Supporting Information Available
Text, figures, tables, and CIF files giving experimental, spectroscopic, kinetic, and crystallographic details. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Dedication
Dedicated to the memory of Professor Michael F. Lappert, a great pioneer of organometallic chemistry and a much-missed friend.
Supplementary Material
References
- a Hashmi A. S. K.; Hutchings G. J. Angew. Chem., Int. Ed. 2006, 45, 7896. [DOI] [PubMed] [Google Scholar]; b Campbell C. T.; Sharp J. C.; Yao Y. X.; Karp E. M.; Silbaugh T. L. Faraday Discuss. 2011, 152, 227. [DOI] [PubMed] [Google Scholar]; c Barrio L.; Liu P.; Rodriguez J. A.; Campos-Martin J. M.; Fierro J. L. G. J. Phys. Chem. C 2007, 111, 19001. [Google Scholar]; d Schmidbaur H.; Raubenheimer H. G.; Dobrzanska L. Chem. Soc. Rev. 2014, 43, 345. [DOI] [PubMed] [Google Scholar]; e Li L.; Zeng X. C. J. Am. Chem. Soc. 2014, 136, 15857. [DOI] [PubMed] [Google Scholar]
- See for example:; a Sivadinarayana C.; Choudhary T. V.; Daemen L. L.; Eckert J.; Goodman D. W. J. Am. Chem. Soc. 2004, 126, 38. [DOI] [PubMed] [Google Scholar]; b Joshi A. M.; Delgass W. N.; Thomson K. T. J. Phys. Chem. B 2006, 110, 2572. [DOI] [PubMed] [Google Scholar]; c Bobuatong K.; Karanjit S.; Fukuda R.; Ehara M.; Sakurai H. Phys. Chem. Chem. Phys. 2012, 14, 3103. [DOI] [PubMed] [Google Scholar]; d Ohta N.; Nomura K.; Yagi I. J. Phys. Chem. C 2012, 116, 14390. [Google Scholar]; e Teng B.-T.; Lang J.-J.; Wen X.-D.; Zhang C.; Fan M.; Harris H. G. J. Phys. Chem. C 2013, 117, 18986. [Google Scholar]; f Woodham A. P.; Fielicke A. Angew. Chem., Int. Ed. 2014, 53, 6554.and references cited therein. [DOI] [PubMed] [Google Scholar]
- Lang S. M.; Bernhardt T. M.; Barnett R. N.; Yoon B.; Landman U. J. Am. Chem. Soc. 2009, 131, 8939. [DOI] [PubMed] [Google Scholar]
- Chang C. R.; Yang X. F.; Long B.; Li J. ACS Catal. 2013, 3, 1693. [Google Scholar]
- Xie J.; Li H.; Zhou J.; Cheng Y.; Zhu C. Angew. Chem., Int. Ed. 2012, 51, 1252. [DOI] [PubMed] [Google Scholar]
- Roşca D.–A.; Wright J. A.; Hughes D. L.; Bochmann M. Nat. Commun. 2013, 4, 2167. [DOI] [PubMed] [Google Scholar]
- Tsui E. Y.; Müller P.; Sadighi J. P. Angew. Chem., Int. Ed. 2008, 47, 8937. [DOI] [PubMed] [Google Scholar]
- a Gaillard S.; Slawin A. M. Z.; Nolan S. P. Chem. Commun. 2010, 46, 2742. [DOI] [PubMed] [Google Scholar]; b Gómez-Suárez A.; Ramón R. S.; Slawin A. M. Z.; Nolan S. P. Dalton Trans. 2012, 41, 5461. [DOI] [PubMed] [Google Scholar]
- Given that the presence of high concentrations of O2 leads to some line broadening, such that the NHC ring signals of 2 and 3 overlap, this ratio estimate is based on a comparison of the signal intensities for NHC-H3 and OH.
- a Denney M. C.; Smythe N. A.; Cetto K. L.; Kemp R. A.; Goldberg K. I. J. Am. Chem. Soc. 2006, 128, 2508. [DOI] [PubMed] [Google Scholar]; b Wick D. D.; Goldberg K. I. J. Am. Chem. Soc. 1999, 121, 11900. [Google Scholar]
- A very similar trend in bond strengths has been estimated for Au–OH and Au–OOH species on the Au(111) surface of heterogeneous catalysts.1b
- Atkins P. W.Physical Chemistry; Oxford University Press: Oxford, U.K., 1998; p 833. [Google Scholar]
- Boisvert L.; Goldberg K. I. Acc. Chem. Res. 2012, 45, 899. [DOI] [PubMed] [Google Scholar]
- A control experiment revealed that pressurizing an NMR tube containing (p-tolyl)3P in C6D6 with 4–9 bar of O2 at 52 °C does not lead to phosphine oxidation and the starting materials could be recovered unchanged.
- Robilotto T. J.; Bacsa J.; Gray T. G.; Sadighi J. P. Angew. Chem., Int. Ed. 2012, 51, 12077. [DOI] [PubMed] [Google Scholar]
- Collado A.; Gómez-Suárez A.; Webb P. B.; Kruger H.; Bühl M.; Cordes D. B.; Slawin A. M.; Nolan S. P. Chem. Commun. 2014, 50, 11321. [DOI] [PubMed] [Google Scholar]
- Gaillard S.; Nun P.; Slawin A. M. Z.; Nolan S. P. Organometallics 2010, 29, 5402. [Google Scholar]
- While this work was in progress, Bertrand showed that strongly donating cyclic (alkyl)(amino)carbenes fulfil the electronic requirements for such complexes:Weinberger D. S.; Melaimi M.; Moore C. E.; Rheingold A. L.; Frenking G.; Jerbek P.; Bertrand G. Angew. Chem., Int. Ed. 2013, 52, 8964. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.