

Abstract
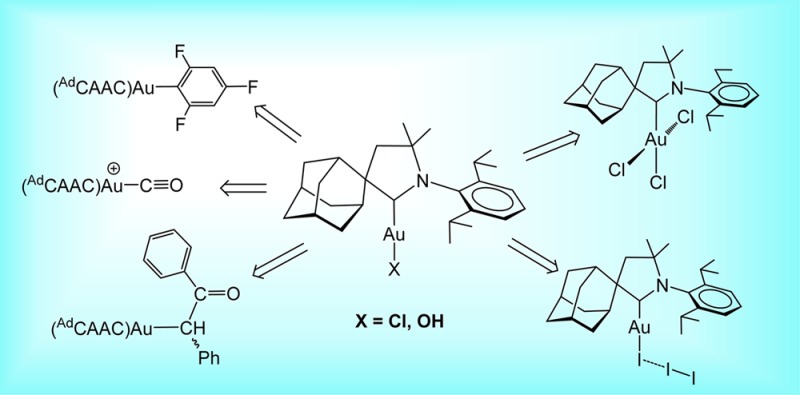
The chemistry of Au(I) complexes with two types of cyclic (alkyl)(amino)carbene (CAAC) ligands has been explored, using the sterically less demanding dimethyl derivative Me2CAAC and the 2-adamantyl ligand AdCAAC. The conversion of (AdCAAC)AuCl into (AdCAAC)AuOH by treatment with KOH is significantly accelerated by the addition of tBuOH. (AdCAAC)AuOH is a convenient starting material for the high-yield syntheses of (AdCAAC)AuX complexes by acid/base and C–H activation reactions (X = OAryl, CF3CO2, N(Tf)2, C2Ph, C6F5, C6HF4, C6H2F3, CH2C(O)C6H4OMe, CH(Ph)C(O)Ph, CH2SO2Ph), while the cationic complexes [(AdCAAC)AuL]+ (L = CO, CNtBu) and (AdCAAC)AuCN were obtained by chloride substitution from (AdCAAC)AuCl. The reactivity toward variously substituted fluoroarenes suggests that (AdCAAC)AuOH is able to react with C–H bonds with pKa values lower than about 31.5. This, together with the spectroscopic data, confirm the somewhat stronger electron-donor properties of CAAC ligands in comparison to imidazolylidene-type N-heterocyclic carbenes (NHCs). In spite of this, the oxidation of Me2CAAC and AdCAAC gold compounds is much less facile. Oxidations proceed with C–Au cleavage by halogens unless light is strictly excluded. The oxidation of (AdCAAC)AuCl with PhICl2 in the dark gives near-quantitative yields of (AdCAAC)AuCl3, while [Au(Me2CAAC)2]Cl leads to trans-[AuCl2(Me2CAAC)2]Cl. In contrast to the chemistry of imidazolylidene-type gold NHC complexes, oxidation products containing Au–Br or Au–I bonds could not be obtained; whereas the reaction with CsBr3 cleaves the Au–C bond to give mixtures of [AdCAAC-Br]+[AuBr2]− and [(AdCAAC-Br)]+ [AuBr4]−, the oxidation of (AdCAAC)AuI with I2 leads to the adduct (AdCAAC)AuI·I2. Irrespective of the steric demands of the CAAC ligands, their gold complexes proved more resistant to oxidation and more prone to halogen cleavage of the Au–C bonds than gold(I) complexes of imidazole-based NHC ligands.
Introduction
Lappert’s pioneering work in the early 1970s established N-donor-stabilized carbenes as remarkably versatile ligands across the Periodic Table, particularly for noble metals, and demonstrated the similarity of the coordination chemistry of N-heterocyclic carbenes (NHCs) and phosphines.1−3 This work also included the first examples of gold NHC complexes, the dimethylimidazolidinylidene derivatives [Au{C(NMe)2C2H4}2]X (X = Cl, BF4).4 Since then, N-heterocyclic carbenes have become one of the most successful and adaptable ligand classes in organometallic chemistry.5
A related type of saturated 5-ring carbene ligands is the family of cyclic (alkyl)(amino)carbenes (CAACs) developed by Bertrand et al.,6 which were inter alia found capable of stabilizing complexes of zerovalent gold,7 while gold(I) CAAC complexes act as catalysts for a range of interesting transformations.8 These ligands show electron affinities more negative than those of the more widely used unsaturated imidazolin-2-ylidene type carbenes and higher ligand-to-metal charge transfer ΔN values; i.e., CAAC ligands behave as stronger σ donors.9
On the other hand, it is becoming apparent that, even with NHC ligands, the π-acceptor capacity has an important influence on reactivity.10−13 As Ciancaleoni et al. showed recently,14 in contrast to the general description of NHCs as strong σ-donors, in the case of gold they donate less strongly than phosphines, and for this metal in particular there is a significant difference between NHCs with saturated and unsaturated rings; i.e. the π-acceptor capability is likely to play an important role.11−13 With this in mind, we became interested in exploring the reactivity patterns of CAAC-type carbenes, and their possible differences in comparison to more conventional types of NHCs. We report here an exploration of the reactivity of CAAC gold complexes, including oxidation reactions to Au(III) compounds. Two types of CAAC ligands were employed: the sterically less demanding dimethyl derivative Me2CAAC and the 2-adamantyl ligand AdCAAC (Chart I).6
Chart I.

Results and Discussion
Although gold(I) chloride complexes LAuCl are most commonly employed as entries into ligand exchange reactions and catalytic transformations, often in combination with silver salts, it can be synthetically advantageous to substitute the chloride ligand for a more labile oxygen-containing ligand, so that subsequent reactions benefit from the relative weakness of the Au–O bond.15 We therefore decided to prepare the corresponding CAAC gold(I) alkoxides, hydroxides, and carboxylates.
Anion Exchange Reactions
The reaction of (AdCAAC)AuCl (1) with sodium tert-butoxide in toluene generates the white alkoxide complex (AdCAAC)AuOtBu (2) in essentially quantitative yield (Scheme 1).16 The alkoxide is very sensitive to hydrolysis, and the reaction must be conducted in anhydrous solvents under inert gas. Treatment of 2 with water readily produces the air-stable hydroxide (AdCAAC)AuOH (3). Complex 3 is characterized in its 1H NMR spectrum by a broadened singlet of the OH ligand at δ −0.29 ppm (in C6D5Br).
Scheme 1.

The same product is also accessible directly from the reaction between the chloride 1 and KOH; however, this reaction proved to be very slow, requiring over 48 h to achieve a 75% conversion. On the other hand, we found that the addition of tBuOH to the mixture significantly accelerates the rate of chloride substitution and generates the hydroxide 3 cleanly within 24–36 h, evidently due to equilibrium concentrations of strongly nucleophilic tBuO–, which catalyzes chloride substitution.17 The use of CsOH, which is often found preferable in gold chloride substitution reactions, is therefore unnecessary. Both complexes 2 and 3 are soluble in polar and aromatic organic solvents (THF, toluene, 1,2-difluorobenzene) and insoluble in hexanes. Chlorinated solvents (CHCl3, CH2Cl2, and 1,2-dichloroethane) should be avoided, because their presence tends to lead to the regeneration of the gold chloride. While the hydroxide 3 can be stored at room temperature in air for months, the tert-butoxide 2 is very sensitive to hydrolysis and has to be kept under an inert atmosphere. The carbene-C resonance in the 13C NMR spectra of 2 and 3 is observed at δ 238, slightly upfield from the chloride precursor complex (AdCAAC)AuCl (δ 239.9).
During reactions of 3 with p-methoxyacetophenone (vide infra), a small crop of crystals of a condensation product of 3 was also obtained, the O-bridged cluster [{(AdCAAC)Au}3(μ3-O)]+OH–. This compound was identified crystallographically (see the Supporting Information, Figure S9). It is analogous to the well-known Nesmeyanov cation,18 and its formation indicates that, in spite of the steric bulk of AdCAAC, condensation of the hydroxide can still take place.19
Both (AdCAAC)AuOtBu (2) and (AdCAAC)AuOH (3) react cleanly with arylboronic acids in toluene under neutral conditions, i.e. without the addition of external bases, to give the corresponding gold aryls, exemplified here by the quantitative formation of (AdCAAC)Au(p-C6H4F) (4; see Scheme 1). Neutral conditions have been shown to be preferable for reactions of boronic acids with both Au(I)20 and Au(III)21 hydroxides and to lead cleanly to the corresponding gold organyl complexes in excellent yields.
The reaction of 3 with trifluoroacetic acid (tfaH) affords (AdCAAC)Au(tfa) (5). This product is also accessible directly from (AdCAAC)AuCl and Ag(tfa). Both methods give essentially quantitative yields; however, the latter approach contaminates the desired complex with traces of silver salts. The carbene carbon resonance is observed at δ(13C) 232.4. This upfield shift of the carbene 13C signal in comparison to that of the chloro complex is observed for all the complexes with Au–O bonds described here but is particularly pronounced for the trifluoroacetate. Complex 5 proved to be temperature sensitive and should be stored at −30 °C to avoid darkening of the sample.
The hydroxide 3 is a convenient starting material for the preparation of gold aryloxides and reacts with 3,5-di-tert-butylphenol to give the corresponding gold phenolate complex 6 in high yield. This synthetic method offers advantages over salt metathesis approaches, since reactions can be carried out in air and isolation of analytically pure products is straightforward. Complex 6 was isolated as a white stable solid which can be handled in air for weeks and is stable in toluene solution for months without noticeable decomposition. Like the hydroxide and alkoxide compounds, 6 is sensitive to chlorinated solvents; therefore, such solvents have to be avoided. The 13C carbene-C resonance is observed at δ 236.1 (in C6D6).
The reaction of the gold hydroxide 3 with HNTf2 in toluene is a high yield route to the Gagosz-type22 complex (AdCAAC)AuNTf2 (7), which is of interest for silver-free protocols in gold catalysis.23 Complex 7 is an air-stable white solid which is soluble in all polar organic solvents. The carbene resonance was observed at δ 233.8. The molecular structure of 7 is shown in Figure 1. The complex is linear; the Au–N and Au–C bond lengths fall in the ranges of 2.077(3)–2.094(3) and 1.969(2)–1.985(2) Å, respectively, similar to those for previously reported (NHC)AuNTf2 complexes.22
Figure 1.

Crystal structures of (left) (AdCAAC)AuNTf2 (7) and (right) (AdCAAC)AuC6HF4 (11). Ellipsoids are shown at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg): complex 7, Au–C(1) 1.977(4), Au–N(2) 2.098(3), N(2)–S(1) 1.627(4), C(1)–C(2) 1.528(5), C(1)–N(1) 1.315(5), C(1)–Au–N(2) 178.64(16), S(1)–N(2)–Au 119.48(19); complex 11, Au–C(1) 2.018(3), Au–C(28) 2.038(3), C(1)–C(2) 1.528(4), C(1)–N(1) 1.301(4), C(1)–Au–C(28) 177.54(11).
C–H Activation Reactions
The basicity of (AdCAAC)AuOH may be exploited to activate C–H bonds. Phenylacetylene and diethyl malonate give the corresponding metalation products (AdCAAC)AuC≡CPh (8) and (AdCAAC)AuCH(CO2Et)2 (9), respectively (see Scheme 2). Compounds 8 and 9 are white solids, stable in air at room temperature. Bertrand has previously reported the synthesis of complex 8 in the reaction of (AdCAAC)AuCl with the lithium salt of phenylacetylene.8a The hydroxide route allows the synthesis of 8 by a simpler procedure in air.
Scheme 2. Syntheses of Gold(I) AdCAAC Complexes.

Reaction conditions: (i) Htfa, toluene, 23 °C, 4 h; (ii) toluene, 23 °C, 18 h; (iii) toluene, 60 °C, 18 h; (iv) toluene, 80 °C, 18 h; (v) 1,4-dioxane, 75 °C, 18 h; (vi) toluene, 70 °C, 12 h; (vii) toluene, 70 °C, 18 h.
The reactivity of 3 toward a series of fluorobenzenes with decreasing degrees of F substitution enables the pKa value of the gold hydroxide to be estimated. The pKa values of a range of fluoroarenes have been calculated,24 with values of 29.0 and 23.1 for C6HF5 and 1,2,4,5-C6H2F4, respectively. As expected, 3 reacts with pentafluorobenzene at 60 °C and with 1,2,4,5-tetrafluorobenzene at 80 °C to give the corresponding aryl complexes (AdCAAC)AuC6H5–nFn (10, n = 5; 11, n = 4) in essentially quantitative yields (see Scheme 2). The structure of (AdCAAC)AuC6HF4 is shown in Figure 1. The Au–C(carbene) and Au–C(aryl) bond lengths are similar to those reported in the analogous complex (NHC)AuC6H2F3 (2.026(3) and 2.044(3) Å).25
Prolonged heating with the less C–H acidic 1,3,5-trifluorobenzene (pKa ≈ 31.5) also leads to the formation of the corresponding gold aryl complex (AdCAAC)Au(2,4,6-C6H2F3) (12); however, the reaction is slow and the product was contaminated with unreacted hydroxide 3. A higher temperature of 90 °C accelerated the gold arylation, but according to the 19F NMR spectrum this was accompanied by some decomposition. The new set of resonances for fluorine atoms in the 19F NMR spectrum was detected as multiplets centered at δ −84.60 (2F) and −116.86 (1F). To prove that these multiplets corresponded to the desired complex 12, we performed the auration of 1,3,5-trifluorobenzene with the more basic (AdCAAC)Au(OtBu), generated in situ from (AdCAAC)AuCl and NaOtBu (eq 1). These mixtures proved more reactive than pure isolated 3 and gave the desired complex 12 in 49% yield. The formation of 12 is accelerated by higher temperatures (75 °C), but since the tert-butoxide 2 is somewhat temperature sensitive, its slow decomposition may explain the reduced yield.
 |
1 |
The auration of 1,3,5-trifluorobenzene by 3 and (AdCAAC)AuCl/NaOtBu mixtures is in contrast with the lack of reactivity of (IPr)AuOH23 and is an indication for the enhanced basicity provided by the CAAC ligand. On the other hand, no reaction was observed with 1,2-difluorobenzene and with monofluorobenzene. The reactivity decreases therefore in the sequence shown in Scheme 3;24 evidently (AdCAAC)AuOH is sufficiently basic to undergo reactions with C–H bonds with pKa values of 31.5 or less. This reactivity places the (CAAC)AuOH complexes closer to that of Larossa’s systems (tBu3P)AuCl/AgSbF6 and (R3P)AuCl/NaOtBu, which also aurate 1,3,5-trifluorobenzene.25
Scheme 3.

The hydroxide 3 is a convenient starting material for the metalation of a series of functionalized C–H compounds. For example, the reaction of (AdCAAC)AuOH with p-methoxyacetophenone, deoxybenzoin, and methyl phenyl sulfone gave the corresponding gold alkyls (AdCAAC)AuR (R = CH2C(O)C6H4OMe (13), CH(Ph)C(O)Ph (14) and CH2SO2Ph (15); see Scheme 2). Related α-keto alkyls have previously been postulated as catalytic intermediates, e.g. Pd–CH(Ph)C(O)Ph species, in the α,α-diarylation of acetophenone en route to tamoxifen precursors.26 The reaction of 3 with acetophenone has a precedence in the formation of (Ph3P)Au-CH2C(O)Ph from acetophenone and Nesmeyanov’s [Au3(μ3-O)(PPh3)3]+ cation,27 while more recent alternative syntheses of gold α-keto alkyls have involved the use of silyl enolates with (Ph3P)AuCl/CsF reagents.28,29
The C–H activated products 8–15 were isolated as white air-stable solids which are soluble in all common organic solvents, with the exception of alkanes. Unlike the other compounds, the deoxybenzoin gold complex 14 possesses very low solubility in benzene and toluene. The resonances of the gold methine proton for 9 and 14 and of the gold methylene protons for 13 and 15 are shifted downfield by 1–2 ppm in the 1H NMR spectra in comparison to the signals for the free ligands. The 13C carbene-carbon resonances for 8–15 are shifted upfield relative to those for (AdCAAC)AuCl and are observed in the range δ 253.2–260.2. The Au-CHR1R2 center in the C1-symmetric complex 14 is chiral; the complex therefore shows two sets of resonances related to the AdCAAC ligand in its 1H NMR spectrum, since the CH2 and CMe2 moieties of the CAAC ligand are diastereotopic (see the Supporting Information). This is illustrated by the crystal structure of complex 14 (Figure 2), which shows that the isopropyl group C(14)–C(15)–C(16) occupies a position almost above the phenyl ring of deoxybenzoin (C36–C41), with atom C(15) oriented toward the phenyl ring plane (3.747(8) Å). This spatial orientation of C(15) explains the high-field 1H NMR chemical shift of this methyl group, at δ 0.89, due to magnetic shielding by the aryl.
Figure 2.

Crystal structure of (AdCAAC)Au(deoxybenzoinyl) (14). Ellipsoids are shown at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg): Au–C(1) 2.039(4), Au–C(28) 2.142(4), C(1)–C(2) 1.522(6), C(1)–N(1) 1.308(6), O(1)–C(29) 1.240(6), C(29)–C(28) 1.465(7), C(29)–C(30) 1.527(6), C(28)–C(36) 1.514(6), C(1)–Au–C(2) 176.89(14).
Functionalized alkyl complexes such as 13–15 should, in principle, provide access to α-keto carbenes, which have been suggested as elusive transient intermediates in a number of organic transformations.30 Preliminary tests have shown, however, that these complexes do not undergo α-hydride abstraction with standard electrophiles such as CPh3+ salts. Methods for generating functionalized gold carbene complexes are currently being investigated.
CO, CN, and Alkene Complexes
The reaction of 1 with silver salts in the presence of CO or tBuNC gives the corresponding cationic complexes [(AdCAAC)Au(L)]+ (L = CO (16); L = tBuNC (17)), which were isolated as SbF6– salts in high yields (Scheme 4). Complexes 16 and 17 are white solids, soluble in low-coordinating polar organic solvents (CH2Cl2, 1,2-difluorobenzene). Coordinating solvents such as acetone lead to immediate CO effervescence. All complexes are stable in air, but the carbonyl 16 has to be stored under a CO atmosphere.
Scheme 4.

The IR spectrum of 16 shows the CO stretching vibration at 2183 cm–1. As is characteristic for CO complexes of gold ions, the CO stretching frequency is higher than that of free CO (2143 cm–1). The CO stretch of 16 falls within the range observed for CO complexes of Au(I) with phosphine and carbene ligands;14,31 for instance, the ν(CO) value of 16 is marginally lower than those of [(Mes3P)Au(CO)][SbF6] (Mes = 2,4,6-C6H2Me3) and [(SIDipp)Au(CO)][SbF6], (by 2 and 14 cm–1, respectively).31 Similarly, the IR spectrum of the isonitrile complex 17 displays a strong vibration at 2241 cm–1 which is blue-shifted in comparison to the signal for free tert-butyl isocyanide (2135 cm–1) and almost identical with that of [(SIDipp)Au(CNtBu)][SbF6] (2244 cm–1).31
The cyano complex (AdCAAC)AuCN (18) was prepared for comparison with the CO compound, by reaction of the hydroxide (AdCAAC)AuOH with Me3SiCN or of that of (AdCAAC)AuCl and KCN. Both approaches lead to almost quantitative yields of complex 18. The complex shows a νCN frequency of 2140 cm–1. The C–N frequency of cyanide anions is relatively insensitive to the nature of the ligand in a trans position, and the value is close to that observed for a range of gold(I) CN complexes with phosphine and carbene ligands. There was no ligand rearrangement to give [Au(CN)2]− salts, as seen for [Au(PMes3)2][Au(CN)2] prepared by the (Mes3P)AuCl/KCN route.31 Complex 18 is stable in air and soluble in all polar organic solvents.
The 1H NMR spectra show that the chemical shifts for the CAAC-CH2 protons in the five-membered rings of 16–18 are about 0.3–0.8 ppm upfield of that of (AdCAAC)AuCl. The carbene-C resonances for 16–18 are observed at δ 241.1, 246.2, and 253.1, respectively. The CO 13C signal of 16 (δ 182.4) is almost identical with that of [(SIDipp)Au(CO)][SbF6] (δ 182.7). The tert-butyl isocyanide CNC(CH3)313C shifts of 17 are observed at δ 142.4 and 58.6, broadened by bonding to quadrupolar 14N. 18 shows a 13CN resonance at δ 149.5, slightly shifted upfield of that of (SIDipp)AuCN (δ 152.4).31 Overall, therefore, these data suggest that the electronic characteristics of CAAC ligands are generally comparable to those of saturated imidazolidinylidene-type NHCs.
The crystal structures of the CO, tBuCN, and CN complexes are shown in Figure 3. The carbonyl complex 16 shows the greatest deviation from linear geometry: C(1)–Au–C(28) 172.9(4)°. The Au–C(28) bond trans to the CAAC ligand elongates from 1.964(5) Å for the CO complex 16 to 2.017(5) Å for the cyanide 18, whereas the carbene–Au distances remain approximately constant throughout this series, deviating only slightly from the value of 2.031(5) Å observed for the cyanide 18. The isonitrile complex 17 crystallized with a molecule of 1,2-difluorobenzene, which exhibits a T-shaped C–F···π intermolecular interaction between carbon C(28) and one of the fluorine atoms of 1,2-difluorobenzene (C(28)···F(8) 3.090(3) Å), which falls into the range of intermolecular interactions of 2.99–3.53 Å observed for various fluoro-organic compounds.32
Figure 3.

Solid-state structures of the cations in (left to right) [(AdCAAC)Au(L)]SbF6 (L = CO (16), tBuNC (17)) and (AdCAAC)AuCN (18). Ellipsoids are shown at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg): 16, Au–C(1) 2.035(4), Au–C(28) 1.964(5), C(28)–O(1) 1.108(6), C(1)–C(2) 1.519(6), C(1)–N(1) 1.290(5), C(1)–Au–C(28) 173.97(17), Au–C(28)–O(1) 172.9(4); 17, Au–C(1) 2.022(2), Au–C(28) 1.986(3), C(28)–N(2) 1.143(3), N(2)–C(29) 1.467(3), C(1)–C(2) 1.525(3), C(1)–N(1) 1.304(3), C(1)–Au–C(28) 175.05(9), Au–C(28)–N(2) 174.2(2), C(28)–N(2)–C(29) 177.5(2); 18, Au–C(1) 2.031(5), Au–C(28) 2.017(5), C(28)–N(2) 1.127(6), C(1)–C(2) 1.516(7), C(1)–N(1)1 1.316(5), C(1)–Au–C(28) 177.33(16), Au–C(28)–N(2) 176.4(5).
In view of our earlier observation that ethylene inserts into Au(III)–trifluoroacetate bonds to give the functionalized alkyls Au–C2H4OAcF,33 (AdCAAC)AuOAcF was exposed to an atmosphere of ethylene for extended periods of time, either in CH2Cl2 with the addition of AgOAcF as catalyst or in CH2Cl2/HOAcF mixtures. However, no insertion of ethylene was observed. The intermediate in this insertion reaction is a cationic alkene complex, and such a complex is indeed easily accessible from the trifluoroacetate precursor if B(C6F5)3 is added as the anion acceptor, as exemplified by the norbornene complex 19 (Scheme 5). The compound is a white, air-stable solid which is soluble in polar organic solvents. The carbene-C signal is observed at δ 246.8.
Scheme 5.

Oxidation Reactions
Given the electron-donating nature of CAAC ligands, it might be expected that CAAC complexes should be easier to oxidize than compounds of less electron rich NHCs. It is surprising, therefore, that the oxidation chemistry of CAAC complexes does not seem to have been explored.
The oxidation of imidazolylidene-type N-heterocyclic carbene gold(I) complexes with halogens to Au(III) products is of course well precedented and proceeds smoothly in high yields with oxidants such as Br2 and PhICl2, in most cases to give products of the type (NHC)AuX3 (X = Cl, Br, I).34−41 It was therefore surprising when initial attempts at oxidizing (AdCAAC)AuX with either PhICl2 or CsBr3 in dichloromethane at room temperature proceeded with Au–C cleavage to give mixtures of products, even when the gold(I) precursor was used in excess (eq 2). The reaction of (AdCAAC)AuCl with PhICl2 in CH2Cl2 gave a yellow solution from which two types of crystals could be obtained: a small amount of colorless needles which were identified by X-ray crystallography as the dichloroaurate(I) salt [AdCAAC-Cl][AuCl2] (20a), formed by chlorination of the carbene ligand, and a larger component of yellow prisms which turned out to be the product of cocrystallization of two independent molecules of [AdCAAC-Cl][AuCl4] (20b) with one molecule of (AdCAAC)AuCl3 in the unit cell. The 1H NMR spectrum supported an approximate 2:1 ratio of these products. Lowering the temperature to −78 °C led to recovery of the starting material. The 13C NMR resonance for the iminium carbon atom C–X is shifted upfield in comparison to the signals for the starting carbene complexes and observed at δ 188.5 and 186.0 for X = Cl, Br, respectively.
 |
2 |
The mechanism of Au–C bond cleavage was not studied in detail; however, one plausible explanation may be that the primary oxidation product, (AdCAAC)AuCl3, partially undergoes photoinduced reductive elimination into Cl2 and the Au(I) complex (AdCAAC)AuCl. The eliminated chlorine could then react with either (AdCAAC)AuCl or (AdCAAC)AuCl3 to give the corresponding salts [AdCAAC-Cl][AuCl2] and [AdCAAC-Cl][AuCl4], respectively. The photochemical reductive elimination of halogens from (NHC)AuBr339a and from gold(III) phosphine complexes in the presence of olefins as halogen scavengers is known to be facile.42 In the present case the carbene C–Au bond acts as such a halogen scavenger. Similar cleavage products 21a,b are obtained using CsBr3 under ambient light conditions (eq 2). The crystal structures of the salts 20a,b are shown in Figure 4.
Figure 4.

Molecular structures of (left) [AdCAAC-Cl]+[AuCl2]− (20a) and (right) [AdCAAC-Cl]+[AuCl4]− (20b). Ellipsoids are shown at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg): 20a, Au–Cl(2) 2.2605(8), Au–Cl(3) 2.2671(8), C(1)–Cl(1) 1.696(3), C(1)–C(2) 1.506(4), C(1)–N(1) 1.288(3), Cl(2)–Au–Cl(3) 175.91(3); 20b, Au–Cl(2) 2.2828(16), Au–Cl(3) 2.2819(17), Au–Cl(4) 2.2673(13), Au–Cl(5) 2.2813(16), C(1)–Cl(1) 1.718(5), C(1)–C(2) 1.510(6), C(1)–N(1) 1.299(6), Cl(2)–Au–Cl(3) 89.84(7), Cl(2)–Au–Cl(4) 178.86(6).
However, a different course of this reaction was observed when the oxidation reactions were conducted in the absence of ambient light. This aspect was first explored using the sterically less hindered and synthetically more easily accessible Me2CAAC ligand and subsequently extended to AdCAAC gold compounds.
Stirring a mixture of [Au(Me2CAAC)2]Cl (22) and PhICl2 in dichloromethane in the dark at 0 °C to room temperature for 6 h gave a colorless complex, [AuCl2(Me2CAAC)2]Cl (23) (Scheme 5). The molecular structure was identified by X-ray diffraction (Figure 5). The gold atom occupies a special position, with the Me2CAAC and Cl ligands being related by an inversion center. The Au atom possesses the expected square-planar geometry with a trans arrangement of the ligands. The bond length Au–C(1) (2.064(2) Å) is slightly elongated in comparison to those of the analogous imidazolylidene complexes [AuCl2(NHC)2]+, while the Au–Cl(1) distance is closely similar.38b,42
Figure 5.
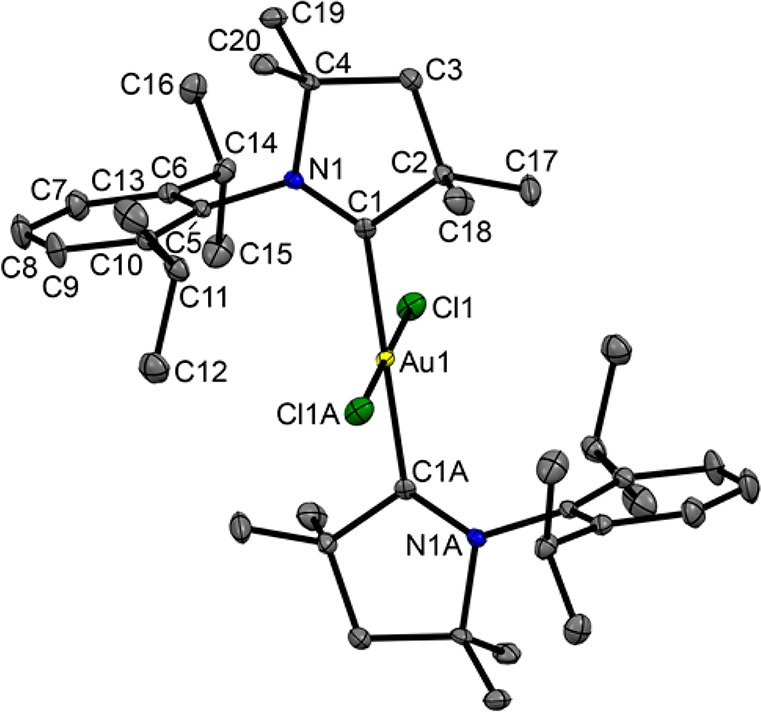
Solid-state structure of the cation in [AuCl2(Me2CAAC)2]Cl (23). Ellipsoids are shown at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg): Au–Cl(1) 2.2681(7), Au–C(1) 2.064(2), C(1)–C(2) 1.527(3), C(1)–N(1) 1.295(3), C(1)–Au–Cl(1) 87.37(7), C(1)–Au–C(1A) 92.63(7).
The crystal structure and elemental analysis show the expected composition of the desired Au(III) product, [AuCl2(Me2CAAC)2]Cl. At the same time, it is a well-known fact that the 13C NMR resonance of the carbene carbon is usually shifted upfield on oxidation of Au(I) carbene complexes to Au(III).38b However, the 1H and 13C NMR spectra in CD2Cl2 of [AuCl2(Me2CAAC)2]Cl and its precursor [Au(Me2CAAC)2]Cl are essentially identical: δ(13C) 250.6. Therefore, we cannot exclude the possibility that in solution an equilibrium exists between [AuCl2(Me2CAAC)2]Cl and its Au(I) isomer, [Au(Me2CAAC)2]Cl3, which in dichloromethane is predominantly shifted toward the Au(I) complex. It did not prove possible, however, to isolate the trichloride salt, and numerous attempts to pick out different crystals led only to unit cell measurements corresponding to the Au(III) complex [AuCl2(Me2CAAC)2]Cl.
The quality of the product strongly depends on the absence of the light during the reaction and on storage. For instance, the colorless solution of [AuCl2(Me2CAAC)2]Cl slowly turned yellow (within ca. 24 h) if left exposed to ambient light, while the 1H and 13C NMR spectra of the sample remained unchanged. The products of this reaction could not be unequivocally determined but seemed likely to contain [AuCl4]− salts.
The oxidation of 22 with CsBr3 as selective brominating agent took a somewhat different course. Under ambient conditions [Au(Me2CAAC)2]Cl reacts with CsBr3 to give a mixture of orange prisms of [Au(Me2CAAC)2]Br3 (24a) and of red crystals of [Au(Me2CAAC)2]AuBr4 (24b) (Scheme 5). In contrast, the attempted oxidation of [Au(Me2CAAC)2]Cl with iodine gave the Au(I) diiodochloride salt [Au(Me2CAAC)2]ClI2 (24c). Since 1H and 13C NMR spectra are not informative, the nature of these products was confirmed by X-ray crystallography (see the Supporting Information).
In none of these reactions did we observe the formation of gold(III) bromo or iodo complexes. The reactivity of CAAC complexes therefore differs significantly from that of unsaturated NHC complexes, where oxidation with Br2 has been shown to generate complexes of the type (NHC)AuBr3 and [AuBr2(NHC)2]+ 35a,38b,39b and where oxidation with iodine has given rise to compounds of the types (NHC)AuBrI2, [AuI2(NHC)2]+, and (NHC)AuI3.38,39a
A similar reactivity pattern was observed in the oxidation reactions of the more bulky monocarbene complexes (AdCAAC)AuX (X = Cl, Br, I). As was observed for the bis-carbene cation [Au(Me2CAAC)2]+, carbene complexes of Au(III) are only obtained if ambient light is excluded. Thus, the reaction of (AdCAAC)AuCl with PhICl2 over the temperature range from 0 °C to room temperature for 6 h in the dark led to the isolation of (AdCAAC)AuCl3 (25) as a light yellow solid in almost quantitative yield (eq 3). There was no reaction at −78 °C.
 |
3 |
The 13C NMR spectrum of 25 shows the carbene carbon signal at δ 218.8, substantially downfield of the 13C carbene signals of imidazolidine-type (NHC)AuCl3 complexes, which are typically observed in the range of δ 130–170.35−40 In comparison to Au(I) CAAC complexes, which show 13C carbene chemical shifts of ca. δ 235–240,6c the Au(III) complexes are shifted upfield by about 20 ppm. Such changes have previously been explained on the basis of increased Lewis acidity of the Au(III) center and shielding effects of the cis-halide ligands.38b,39a There was no evidence for ligand rearrangement, e.g. to [AuCl2(AdCAAC)2][AuCl4], and the solid-state structure is retained in solution. The structure of the complex is shown in Figure 6. The Au(III) atom possesses square-planar geometry. The bond lengths Au–C(1) (2.018(4) Å) and Au–Cl(2) (2.3170(13) Å in position trans to the carbene carbon) are almost identical with those observed in numerous (NHC)AuCl3 complexes.35−40
Figure 6.

Crystal structure of (AdCAAC)AuCl3 (25). Ellipsoids are shown at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg): Au–Cl(1) 2.2972(13), Au–Cl(2) 2.3170(13), Au–Cl(3) 2.2632(13), Au–C(1) 2.018(4), C(1)–C(2) 1.537(6), C(1)–N(1) 1.295(6), C(1)–Au–Cl(2) 174.25(13), C(1)–Au–Cl(1) 85.64(13), C(1)–Au–Cl(2) 95.83(13), Cl(1)–Au–Cl(2) 88.79(6).
In contrast, the reaction of (AdCAAC)AuI with iodine in dichloromethane under various reaction conditions (i.e., either protected from light or unprotected, low or ambient temperature) gave a dark red solution from which crystals of the dark red iodine adduct (AdCAAC)AuI·I2 (26) were isolated (eq 4). This is in contrast to the oxidative addition of iodine observed with other types of NHC complexes, which form gold(III) iodides.38b,45 The formation of triiodides and iodine adducts has previously been observed for phosphine and isonitrile Au(I) complexes.43,44 Indications for the redox equilibrium LAuI(I3) ⇌ LAuIII(I)3 were not detected.
 |
4 |
The structure of 26 is shown in Figure 7. According to the Cambridge Structural Database the only closely analogous compound with a triiodide moiety is [(tBuNC)2Au][AuI2]·I2, reported by Schmidbaur.43 The Au–I(1) and I(2)–I(3) bond lengths for the complex (AdCAAC)AuI·I2 (2.5684(4) and 2.7626(5) Å) are almost identical with those for [(tBuNC)2Au][AuI2]·I2 (2.553(1) and 2.738(1) Å, respectively), but the I(1)–I(2) distance is significantly shorter: 3.1655(5) vs 3.311(1) Å. At the same time the I···I distance is in accordance with typical values for polyiodide complexes.46 Analysis of intermolecular contacts shows neither aurophilic interactions (the shortest distance between gold atoms is 7.521 Å) nor polyiodide chain formation.
Figure 7.
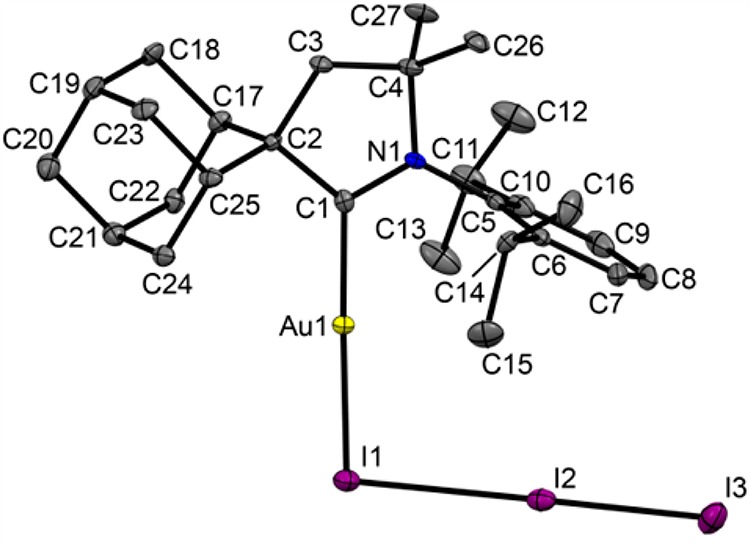
Crystal structure of (AdCAAC)AuI·I2 (26). Ellipsoids are shown at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg): Au–I(1) 2.5684(4), Au–C(1) 2.024(4), I(1)–I(2) 3.1655(5), I(2)–I(3) 2.7626(5), C(1)–C(2) 1.517(6), C(1)–N(1) 1.299(6); C(1)–Au–I(1) 177.36(12), Au–I(1)–I(2) 93.56(1), I(1)–I(2)–I(3) 177.28(2).
Conclusion
The AdCAAC ligand produces a gold(I) hydroxide with slightly increased basicity in comparison to the imidazolylidene-type complex (NHC)AuOH. It is a convenient starting material for the synthesis of a wide range of acid/base and C–H activation reactions and gives gold aryls even with 1,3,5-trifluorobenzene. Arylgold complexes of less acidic arenes are obtainable by the reactions of the corresponding arylboronic acids under neutral conditions in toluene. The oxidation reactions of CAAC-supported gold(I) complexes by halogens, on the other hand, did not conform to the expectations for electron-rich complexes, and only stronger oxidants, such as PhICl2, afforded gold(III) CAAC complexes. In bromine oxidations the CAAC ligand proved to be a halide scavenger, while iodine formed a gold(I) triiodide. The halide complexes readily decompose under the influence of light, and exclusion of light is required if cleavage of the Au–carbene bond by halogens is to be avoided. With such precautions, the first examples of gold(III) CAAC complexes could be prepared in almost quantitative yields. The reaction patterns of CAAC-type carbenes provide therefore an interesting contrast to those of more strongly π-accepting13i imidazole-based NHC carbenes.
Experimental Section
General Considerations
Unless stated otherwise, all reactions were carried out in air. Solvents were distilled and dried as required. Pentafluorobenzene, 1,2,4,5-tetrafluorobenzene, 1,3,5-trifluorobenzene, sodium tert-butoxide, diethyl malonate, trimethylsilyl cyanide, triethoxysilane, triflimide, tert-butyl alcohol, tBuNC, KCN, and norbornene were purchased from Sigma-Aldrich and used as received. (AdCAAC)AuCl6c and Me2CAAC47 were obtained according to a literature procedure. 1H, 13C{1H}, and 19F NMR spectra were recorded using a Bruker Avance DPX-300 MHz NMR spectrometer. 1H NMR spectra (300.13 MHz) and 13C{1H} (75.47 MHz) were referenced to CD2Cl2 at δ 5.32 (13C, δ 54.0), C6D6 at δ 7.16 (13C, δ 128.4), CDCl3 at δ 7.26 (δ 13C 77.2), or C6D5Br at δ 7.30 for the most downfield signal (13C, δ 122.5 for the most upfield signal) ppm. 19F NMR spectra (282.4 MHz) were referenced externally to CFCl3 and internally to C6F6 (δF −164.9). IR spectra were recorded using a PerkinElmer Spectrum One FT-IR spectrometer equipped with a diamond ATR attachment. Elemental analyses were performed by the London Metropolitan University.
Synthesis of (AdCAAC)AuOtBu (2)
An oven-dried 25 mL Schlenk flask was equipped with a stirring bar and charged with (AdCAAC)AuCl (303 mg, 0.5 mmol) and sodium tert-butoxide (48 mg, 0.5 mmol) under an argon atmosphere. Anhydrous toluene (15 mL) was added, and the resulting white suspension was stirred in the dark for 5 h and filtered through a Celite pad (2 cm), which was washed with an additional 10 mL of toluene. The volatiles were evaporated under vacuum, affording a white solid: yield 307 mg (0.475 mmol, 95.5%). 1H NMR (300 MHz, CD2Cl2): δ 7.43 (t, J = 7.6 Hz, 1H, CH-aromatic), 7.27 (d, J = 7.6 Hz, 2H, CH-aromatic), 4.12 (br d, J = 13.2 Hz, 2H, CH2), 2.77 (sept, J = 6.6 Hz, 2H, CH(CH3)2), 2.32–1.75 (m, 14H, adamantyl CH and CH2), 1.44 (d, J = 6.6 Hz, 6H, CH(CH3)2), 1.31 (s, 6H, 2CH3), 1.28 (d, J = 6.6 Hz, 6H, CH(CH3)2), 0.91 (s, 9H, OC(CH3)3) ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 238.1 (C carbene), 144.9 (o-C), 136.4 (Ar, Cipso), 129.0 (Ar, p-C), 124.8 (Ar, m-C), 75.9 (Cq), 70.2 (Cq, OC(CH)3), 63.7 (Cq), 48.7 (CH2), 39.0 (CH2), 36.8, 34.9 (OC(CH3)3), 34.8, 34.4 (CH2), 29.0 (CH), 28.9, 27.7, 27.3, 26.5, 22.9 (CH3) ppm. IR (ATR, cm–1): 2900, 2846, 1493, 1447, 1343, 1188, 958, 802, 765, 587. Anal. Calcd for C31H48AuNO (647.68): C, 57.49; H, 7.47; N, 2.16. Found: C, 57.58; H, 7.50; N, 2.18.
Synthesis of (AdCAAC)AuOH (3)
Method A
A 50 mL Schlenk flask was charged with (AdCAAC)AuCl (303 mg, 0.5 mmol), freshly ground KOH (285 mg, 5 mmol), and 10 mL of THF. To the stirred suspension was added tert-butyl alcohol (0.02 mL, 0.2 mmol), and stirring was continued for 36 h at room temperature. The dark suspension was filtered through a Celite pad (3 cm) and washed with additional THF (2 × 5 mL). Water (4 mL) was added to the THF solution, after which it was concentrated to ca. 7 mL. Water (10 mL) was added to the cloudy suspension. All volatiles were removed under vacuum (30 °C, 20 mbar). If any coloration of the solid remained, it could be redissolved in THF/H2O (4:1) and passed through Celite. The white residue was washed with hexanes (2 × 5 mL) and dried under vacuum for 1 day. Yield: 282 mg (0.48 mmol, 96%).
Method B
An excess of distilled water (5 mL) was added to the stirred solution of (AdCAAC)AuOtBu (194 mg, 0.30 mmol) in 1 mL of THF. The milky suspension was stirred for 15 min, and the volatiles were removed under vacuum. The white residue was washed with hexanes (5 mL) and dried under vacuum for 1 day. Yield: 171 mg (0.29 mmol, 97%).
1H NMR (300 MHz, C6D5Br): δ 7.21 (t, J = 7.9 Hz, 1H, CH-aromatic), 7.06 (d, J = 7.9 Hz, 2H, CH-aromatic), 4.21(br d, J = 13.2 Hz, 2H, CH2), 2.69 (sept, J = 6.6 Hz, 2H, CH(CH3)2), 1.98–1.52 (m, 14H, adamantyl CH and CH2), 1.46 (d, J = 6.6 Hz, 6H, CH(CH3)2), 1.16 (d, J = 6.6 Hz, 6H, CH(CH3)2), 0.99 (s, 6H, 2CH3), −0.29 (br s, 1H, OH) ppm. 13C{1H} NMR (75 MHz, C6D5Br): δ 238.2 (C carbene), 145.1 (o-C), 136.0 (Cipso), expected signal at ca. 129.0 (p-C) overlaps with solvent peak of C6D5Br, 125.1 (m-C), 75.8 (Cq), 63.8 (Cq), 48.6 (CH2), 39.3 (CH2), 37.1, 35.3, 34.7 (CH2), 29.2(CH), 29.0, 28.0, 27.6, 27.0, 23.3 (CH3) ppm. IR (ATR, cm–1): 3671, 3605 (br), 2960, 2900, 1500, 1448, 1358, 1098, 945, 803, 778, 540. Anal. Calcd for C27H40AuNO (591.57): C, 54.82; H, 6.81; N, 2.37. Found: C, 54.71; H, 6.84; N, 2.41.
Synthesis of (AdCAAC)Au(p-C6H4F)] (4)
Method A
Under an argon atmosphere, an oven-dried 25 mL Schlenk flask was charged with a stirring bar, (AdCAAC)AuOtBu (97 mg, 0.15 mmol), and p-fluorophenylboronic acid (22 mg, 0.15 mmol). Anhydrous toluene (5 mL) was added, and the resulting suspension was stirred overnight. The reaction mixture was filtered through a Celite pad (2 cm), which was washed with another 8 mL of toluene. The volatiles were removed under vacuum to give an off-white product, which was washed with hexanes (2 × 4 mL) and dried under vacuum. Yield: 92 mg (0.14 mmol, 92%).
Method B
A scintillation vial was charged in air with a stirring bar, (AdCAAC)AuOH (60 mg, 0.10 mmol) and p-fluorophenylboronic acid (15 mg, 0.10 mmol). Toluene (4 mL) was added and the resulting suspension was stirred overnight. The mixture was filtered through a Celite pad (2 cm) which was washed with another 8 mL of toluene. All volatiles were removed under vacuum to give an off white product which was washed with hexanes (2 × 4 mL) and dried under vacuum. Yield: 65.5 mg (0.097 mmol, 97%).
1H NMR (300 MHz, CD2Cl2): δ 7.45 (t, J = 7.6 Hz, 1H, CH-aromatic), 7.29 (d, J = 7.6 Hz, 2H, CH-aromatic), 6.99 and 6.72 (dd, 4H, JAB = ca. 9.7 Hz, 3JHa-F = ca. 7.9 Hz, 4JHb-F = ca. 5.1 Hz, p-C6H4), 4.21(br d, J = 12.6 Hz, 2H, CH2), 2.86 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.31–1.81 (m, 14H, adamantly CH and CH2), 1.45 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.35 (s, 6H, 2CH3), 1.31 (d, J = 6.7 Hz, 6H, CH(CH3)2).19F NMR (282 MHz, CD2Cl2): δ −119.9. 13C{1H} NMR (75 MHz, CD2Cl2): δ 263.9 (C carbene), 163.4 (d, 4JC–F = 3.9 Hz, Au-Cq, p-C6H4F), 160.9 (d, 1JC–F= 239 Hz, CF, p-C6H4F), 145.2 (Ar, o-C), 141.2 (d, 3JC–F= 5.5 Hz, CH, p-C6H4F), 135.6 (Cipso), 129.0 (p-C), 124.6 (m-C), 112.8 (d, 2JC–F= 17.1 Hz, CH, p-C6H4F), 77.1 (Cq), 65.2 (Cq), 48.8 (CH2), 39.0 (CH2), 37.1, 35.3, 34.4 (CH2), 29.0 (CH), 28.99 (almost overlapping with signal at 29.0), 28.2, 27.4, 26.1, 22.8 (CH3) ppm. Anal. Calcd for C28H39AuN2 (669.66): C, 59.19; H, 6.47; N, 2.09. Found: C, 59.01; H, 6.46; N, 2.13.
Synthesis of (AdCAAC)Au(OAcF)] (5)
Method A
A Schlenk flask was charged with (AdCAAC)AuOH (59 mg, 0.10 mmol) and a trifluoroacetic acid solution (9 μL, 0.11 mmol) in toluene (1 mL). The resulting solution was stirred for 4 h. All volatiles were removed under vacuum. The residue was washed with hexanes (2 × 2 mL) and dried under vacuum to give a white solid. Yield: 66.5 mg (0.096 mmol, 96%).
Method B
A Schlenk flask was charged with (AdCAAC)AuCl (59 mg, 0.1 mmol), silver trifluoroacetate (23 mg, 0.10 mmol), and CH2Cl2 (2 mL). The resulting suspension was stirred for 30 min in the dark. The mixture was filtered through a Celite pad (2 cm), which was washed with another 8 mL of CH2Cl2. All volatiles were removed under vacuum to leave an off-white product, which was dried under vacuum. Yield: 63.5 mg (0.092 mmol, 92%).
1H NMR (300 MHz, CD2Cl2): δ 7.49 (t, J = 7.7 Hz, 1H, CH-aromatic), 7.30 (d, J = 7.7 Hz, 2H, CH-aromatic), 4.00 (br d, J = 12.6 Hz, 2H, CH2), 2.74 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.39–1.81 (m, 14H, adamantyl CH and CH2), 1.38 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.35 (s, 6H, 2CH3),1.31 (d, J = 6.7 Hz, 6H, CH(CH3)2) ppm. 19F NMR (282 MHz, CD2Cl2): δ −74.5 ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 232.4 (C carbene), 160.7 (quart. 2JC–F= 37.8 Hz, CO2), 144.8 (o-C), 135.4 (Cipso), 129.8 (p-C), 125.0 (m-C), 117.7 (quart. 1JC–F= 293 Hz, CF3), 77.2 (Cq), 63.6 (Cq), 48.2 (CH2), 38.9 (CH2), 37.0, 35.1, 34.4 (CH2), 29.0 (CH), 28.9, 27.7, 27.2, 26.3, 22.6 (CH3) ppm. IR (ATR, cm–1): 2968, 2908, 1701, 1527, 1450, 1407, 1372, 1187, 1135, 1097, 841, 804, 727, 609, 520. Anal. Calcd for C29H39AuF3NO2 (687.58): C, 50.66; H, 5.72; N, 2.04. Found: C, 50.72; H, 5.80; N, 2.02.
Synthesis of (AdCAAC)Au(3,5-di-tert-butylphenolate) (6)
A scintillation vial was charged with a stirring bar, (AdCAAC)AuOH (59 mg, 0.10 mmol), and 3,5-di-tert-butylphenol (21 mg, 0.102 mmol). Toluene (3 mL) was added and the resulting yellow solution was stirred overnight. All volatiles were evaporated under vacuum, affording the product as a white solid, which was washed with hexanes (2 × 4 mL) and dried under vacuum. Yield: 75.5 mg (0.097 mmol, 97%).
1H NMR (300 MHz, C6D6): δ 7.18 (t, J = 7.7 Hz, 1H, CH-aromatic), 7.01 (d, J = 7.7 Hz, 2H, CH-aromatic), 6.98 (br d, J = 1.7 Hz, 2H, CH, phenolate), 6.93 (br t, J = 1.7 Hz, 1H, CH, phenolate), 4.40 (br d, J = 12.2 Hz, 2H, CH2), 2.67 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.15–1.60 (m, 14H, adamantyl CH and CH2), 1.51 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.40 (s, 18H, two tBu groups), 1.09 (d, J = 6.7 Hz, 6H, CH(CH3)2), 0.79 (s, 6H, 2CH3) ppm. 13C{1H} NMR (75 MHz, C6D6): δ 236.1 (C carbene), 168.6 (O-C phenolate), 150.4 (Cmeta phenolate), 144.8 (Co aromatic), 135.3 (Cipso aromatic), 129.9 (CHp aromatic), 113.9 (CHo phenolate), 108.7 (CHpara phenolate), 75.5 (Cq), 63.5 (Cq), 48.0 (CH2), 38.9 (CH2), 36.9, 35.1, 34.7 (C(CH3)3), 34.4 (CH2), 31.9 (C(CH3)3), 29.1(CH), 28.3, 27.6, 27.3, 26.7, 22.6 (CH3) ppm. IR (ATR, cm–1): 2962, 2897, 1576, 1509, 1463, 1422, 1320, 1098, 973, 804, 705, 643, 582, 474. Anal. Calcd for C41H60AuNO (779.88): C, 63.14; H, 7.75; N, 1.80. Found: C, 63.23; H, 7.81; N, 1.85.
Synthesis of (AdCAAC)Au(NTf2) (7)
A Schlenk flask was charged with a stirring bar, (AdCAAC)AuOH (59 mg, 0.10 mmol), and HNTf2 (30 mg, 0.105 mmol) under argon. Toluene (1 mL) was added, and the resulting suspension was stirred overnight. The product was precipitated with hexanes (10 mL) and dried under vacuum. The residue was washed with hexanes (2 × 2 mL) and dried under vacuum. The microcrystalline product contains half a molecule of toluene, while precipitation from dichloromethane with hexanes gives the CH2Cl2 solvate: yield 86 mg (0.091 mmol, 91%).
1H NMR (300 MHz, CD2Cl2): δ 7.45 (t, J = 7.8 Hz, 1H, CH-aromatic), 7.28 (d, J = 7.8 Hz, 2H, CH-aromatic), 3.82 (br d, J = 13.2 Hz, 2H, CH2), 2.71 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.40–1.81 (m, 14H, adamantyl CH and CH2), 1.39 (s, 6H, 2CH3),1.36 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.30 (d, J = 6.7 Hz, 6H, CH(CH3)2) ppm. 19F NMR (282 MHz, CD2Cl2): δ −75.3 ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 233.8 (C carbene), 144.5 (o-C), 135.5 (Cipso), 129.8 (p-C), 125.2 (m-C), 119.1 (quart. 1JC–F= 323.3 Hz, CF3), 77.8 (Cq), 63.8 (Cq), 48.3 (CH2), 38.8 (CH2), 37.0, 34.7, 34.4 (CH2), 29.2 (CH), 29.0, 27.5, 27.1, 26.0, 23.0 (CH3) ppm. IR (ATR, cm–1): 2907, 2852, 1532, 1450, 1397, 1375, 1192, 1131, 1053, 955, 830, 654, 607, 567, 507. Anal. Calcd for C29H39AuF6N2O4S2·CH2Cl2 (939.64): C, 38.35; H, 4.40; N, 2.98. Found: C, 38.05; H, 4.23; N, 3.18.
Synthesis of (AdCAAC)Au(CH(CO2Et)2) (8)
A scintillation vial was charged with a stirring bar, (AdCAAC)AuOH (59 mg, 0.10 mmol), and diethyl malonate (16 mg, 0.1 mmol). Toluene (2 mL) was added, and the resulting suspension was stirred overnight. All volatiles were evaporated under vacuum, affording the product as a white solid, which was washed with hexanes (2 × 2 mL) and dried under vacuum. Yield: 72 mg (0.098 mmol, 98%).
1H NMR (300 MHz, CDCl3): δ 7.38 (t, J = 7.7 Hz, 1H, CH-aromatic), 7.21 (d, J = 7.7 Hz, 2H, CH-aromatic), 3.91 (br d, J = 12.4 Hz, 2H, CH2) overlapping with 3.88 (q, J = 7.3 Hz, 2H, OCH2CH3),3.76 (q, J = 7.3 Hz, 2H, OCH2CH3), 3.41 (s, 1H, CH(CO2Et)2), 2.69 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.27–1.74 (m, 14H, adamantyl CH and CH2), 1.37 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.31 (s, 6H, 2CH3), 1.26 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.05 (t, J = 7.3 Hz, 6H, OCH2CH3) ppm. 13C{1H} NMR (75 MHz, CDCl3): δ 253.2 (C carbene), 172.7 (C=O malonate), 145.0 (o-C), 135.2 (Cipso), 129.3 (p-C), 124.7 (m-C), 77.1 (Cq, overlapping with signal from CDCl3), 64.4 (Cq), 58.4 (OCH2-malonate) 48.6 (CH2), 42.3 (CH-malonate), 39.0 (CH2), 37.0, 35.2, 34.5 (CH2), 29.2 (CH), 29.0, 27.8, 27.1, 26.6, 23.1 (CH3), 14.5 (CH3-malonate) ppm. Anal. Calcd for C34H50AuNO4 (733.73): C, 55.66; H, 6.87; N, 1.91. Found: C, 55.54; H, 6.95; N, 1.94.
Synthesis of (AdCAAC)AuC≡CPh (9)
A scintillation vial was charged with a stirring bar, (AdCAAC)AuOH (59 mg, 0.10 mmol), and phenylacetylene (17 mg, 0.166 mmol). Toluene (2 mL) was added, and the resulting suspension was stirred overnight. All volatiles were evaporated under vacuum, affording the product as a white solid, which was washed with hexanes (2 × 2 mL) and dried under vacuum. Yield: 64 mg (0.095 mmol, 95%).
1H NMR (300 MHz, CDCl3): δ 7.40 (t, J = 7.7 Hz, 1H, CH-aromatic), 7.33 (br d, J = 7.6 Hz, 2H, CH, phenylacetylide), 7.24 (d, J = 7.7 Hz, 2H, CH-aromatic), 7.12 (br t, J = 7.6 Hz, 2H, CH, phenylacetylide),7.05 (br tt, 3J = 7.6 Hz,4J = 1.4 Hz, 1H, CH, phenylacetylide), 4.02 (br d, J = 12.4 Hz, 2H, CH2), 2.77 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.28–1.78 (m, 14H, adamantyl CH and CH2), 1.47 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.31 (s, 6H, 2CH3), 1.29 (d, J = 6.7 Hz, 6H, CH(CH3)2) ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 257.5 (C carbene), 144.9 (o-C), 134.8 (Cipso aromatic), 132.3 (Ph acetylide), 129.4 (p-C), 127.5 (Ph acetylide), 127.0 (C acetylide), 126.2 (C aromatic acetylide), 125.7 (Ph acetylide), 124.8 (m-C), 106.8 (C acetylide), 77.1 (Cq), 65.2 (Cq), 48.7 (CH2), 39.0 (CH2), 37.0, 35.6, 34.5 (CH2), 29.1(CH), 29.0, 27.6, 27.2, 27.0, 23.0 (CH3) ppm. IR (ATR, cm–1): 2968, 2892, 2115 (C≡C), 1508, 1447, 1368, 1098, 910, 805, 754, 693, 527. Anal. Calcd for C35H44AuN (675.69): C, 62.21; H, 6.56; N, 2.07. Found: C, 62.13; H, 6.62; N, 2.18.
Synthesis of (AdCAAC)Au(C6F5) (10)
A Schlenk flask was charged with (AdCAAC)AuOH (118 mg, 0.2 mmol) and a pentafluorobenzene solution (42 μL, 0.4 mmol) in toluene (2 mL). The resulting mixture was heated to 60 °C for 18 h. The slightly pink solution was filtered through a Celite pad (1 cm) which was washed with an additional 6 mL of toluene. The solution was concentrated to ca. 0.3 mL under vacuum and the white residue precipitated with hexanes (10 mL). The resulting suspension was centrifuged. The solid was washed with hexanes (2 × 4 mL) and dried under vacuum to give an off-white solid. Yield: 140 mg (0.19 mmol, 95%).
1H NMR (300 MHz, CDCl3): δ 7.44 (t, J = 7.8 Hz, 1H, CH-aromatic), 7.25 (d, J = 7.8 Hz, 2H, CH-aromatic), 4.09 (br d, J = 13.2 Hz, 2H, CH2), 2.79 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.33–1.81 (m, 14H, adamantyl CH and CH2), 1.38 (s, 6H, 2CH3), 1.36 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.29 (d, J = 6.7 Hz, 6H, CH(CH3)2)ppm. 19F NMR (282 MHz, CDCl3): δ −115.3 to −115.5 (m, 2F), −160.5 (t, J = 20.7 Hz, 1F), −163.43 to −163.70 (m, 2F) ppm. 13C{1H} NMR (75 MHz, CDCl3): δ (Au–C from C6F5 was not observed) 258.5 (C carbene), 150.5–147.3 (d m,1JC–F = 226 Hz, CF), 144.9 (Co aromatic), 139.6–136.5 (d m, 1JC–F = 239 Hz, CF), 138.2–134.9 (d m, 1JC–F = 252 Hz, CF), 135.2 (Cipso), 129.4 (p-C), 124.8 (m-C), 77.2 (Cq), 64.9 (Cq), 48.9 (CH2), 39.0 (CH2), 37.2, 35.2, 34.5 (CH2), 29.3 (CH), 29.0, 27.8, 27.2, 26.3, 23.2 (CH3) ppm. Anal. Calcd for C33H39AuF5N (741.62): C, 53.44; H, 5.30; N, 1.89. Found: C, 53.59; H, 5.39; N, 1.93.
Synthesis of (AdCAAC)Au(p-C6HF4) (11)
The compound was made in a fashion similar to that for 10 from (AdCAAC)AuOH (59 mg, 0.10 mmol), and 1,2,4,5-tetrafluorobenzene (22 μL, 0.20 mmol) solution in toluene (2 mL). The resulting solution was heated to 80 °C for 18 h. The slightly pink solution was filtered through Celite pad (1 cm) which was washed with an additional with 6 mL of toluene. Concentration and precipitation with hexanes gave an off-white solid. Yield: 67 mg (0.093 mmol, 93%).
1H NMR (300 MHz, CD2Cl2): δ 7.46 (t, J = 7.6 Hz, 1H, CH-aromatic), 7.29 (d, J = 7.6 Hz, 2H, CH-aromatic), 6.55 (tt, JH–F = 9.5 and 6.9 Hz, 1H), 4.09 (br d, J = 12.9 Hz, 2H, CH2), 2.83 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.35–1.83 (m, 14H, adamantyl CH and CH2), 1.38 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.38 (s, 6H, 2CH3),1.31 (d, J = 6.7 Hz, 6H, CH(CH3)2) ppm. 19F NMR (282 MHz, CD2Cl2): δ −117.4 to −117.6 (m, 2F), −141.9 to −142.1 (m, 2F) ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 258.4 (C carbene), 151.0–146.7 (d m, 1JC–F= 228 Hz, CF), 145.1 (o-C), 147.1–143.4 (d m, 1JC–F= 250 Hz, CF), 140.4 (t m, 2JC–F= 59 Hz, C tetrafluoroaryl), 135.4 (Cipso), 129.2 (p-C), 124.7 (m-C), 102.0 (t, 2JC–F = 23.7 Hz, CH tetrafluoroaryl), 77.5 (Cq), 65.0 (Cq), 48.7 (CH2), 39.0 (CH2), 37.2, 35.2, 34.4 (CH2), 29.1 (CH), 29.0, 28.1, 27.4, 26.0, 22.9 (CH3) ppm. Anal. Calcd for C33H40AuF4N (723.63): C, 54.77; H, 5.57; N, 1.94. Found: C, 55.17; H, 5.78; N, 2.09.
Reaction of (AdCAAC)Au(OH) with 1,3,5-Trifluorobenzene
A J. Young NMR tube was loaded with (AdCAAC)AuOH (30 mg, 0.05 mmol) and 1,3,5-trifluorobenzene (15 μL, 0.152 mmol) solution in toluene-d8 (0.4 mL) and sealed. The resulting solution was heated to 90 °C for 18 h. The first signals for the product appear after 2 h of heating. The yellow solution with some black precipitate was cooled to room temperature. The major product crystallizing from the solution after 8 h was the O-bridged cluster [(AdCAAC)3Au3(μ-O)]OH (identified by X-ray crystallography as the C6H3F3 solvate) (11 mg). The yellow solution was decanted and filtered through a pipet filled with Celite (1 cm), which was washed with an additional 4 mL of toluene. Concentration and precipitation with hexanes gave a yellow solid (11 mg) as a mixture of products. Attempts to increase the reaction time to 48 h led to significant formation of decomposition products. 19F NMR (282 MHz, CD2Cl2): δ −84.60 (m, 2F), −116.81 to −116.92 (m, 1F) ppm.
Synthesis of (AdCAAC)Au(2,4,6-C6H2F3) (12)
A Schlenk flask was loaded with (AdCAAC)AuCl (60 mg, 0.1 mmol), 1,3,5-trifluorobenzene (60 μL, 0.608 mmol), NaOtBu (29 mg, 0.3 mmol), and 1,4-dioxane (0.8 mL) and sealed. The resulting suspension was heated to 75 °C for 18 h. After the suspension was cooled to room temperature, the solid was extracted with CH2Cl2 and the extract filtered through a pad of Celite (1 cm). The solution was concentrated, the product precipitated with hexanes, and the solvent decanted. All volatiles were evaporated to give an off-white solid. An analytically pure sample was obtained after flash chromatography (CH2Cl2/hexane 30/70). Evaporation of all volatiles gave a white solid: yield 34 mg (0.049 mmol, 49%).
1H NMR (300 MHz, CD2Cl2): δ 7.44 (t, J = 7.6 Hz, 1H, CH-aromatic), 7.27 (d, J = 7.6 Hz, 2H, CH-aromatic), 6.40–6.34 (m, 2H, C6H2F3), 4.13 (br d, J = 12.0 Hz, 2H, CH2), 2.82 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.33–1.81 (m, 14H, adamantyl CH and CH2), 1.37 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.35 (s, 6H, 2CH3), 1.29 (d, J = 6.7 Hz, 6H, CH(CH3)2) ppm. 19F NMR (282 MHz, CD2Cl2): δ −84.60 (m, 2F), −116.81 to −116.92 (m, 1F) ppm. 13C NMR (75 MHz, CD2Cl2): δ (C-F from C6H2F3 was not observed) 259.7 (C carbene), 170.6–169.9 (m, Au-Cq, trifluoroaryl), 145.1 (o-C), 135.4 (Cipso), 129.1 (p-C), 124.6 (m-C), 97.6 (ddd, JC–F = 36.5, 23.3, 4.8 Hz, CH trifluoroaryl), 77.3 (Cq), 65.0 (Cq), 48.7 (CH2), 39.1 (CH2), 37.2, 35.1, 34.3 (CH2), 29.05 (CH), 29.0, 28.1, 27.4, 26.0, 22.6 (CH3) ppm. Anal. Calcd for C33H41AuF3N (705.64): C, 56.17; H, 5.86; N, 1.98. Found: C, 56.39; H, 5.98; N, 1.87.
Synthesis of (AdCAAC)Au(CH2C(O)-p-methoxyphenyl) (13)
A scintillation vial was charged with (AdCAAC)AuOH (59 mg, 0.10 mmol) and p-methoxyacetophenone (20 mg, 0.13 mmol) in toluene (2 mL). The resulting mixture was heated to 70 °C for 12 h. The slightly yellow solution was filtered through a Celite pad (1 cm) which was washed with an additional 6 mL of toluene. The solution was concentrated to ca. 0.3 mL under vacuum and the white residue precipitated with hexanes (10 mL). The resulting suspension was centrifuged. The solid was washed with hexanes (2 × 4 mL) and dried under vacuum to give an off-white solid. Yield: 60 mg (0.083 mmol, 83%).
1H NMR (300 MHz, CD2Cl2): δ 8.21 (d, 2H, AA′BB′, JAB = ca. 8.2 Hz, p-C6H4), 7.13 (t, J = 7.6 Hz, 1H, CH-aromatic), 6.95 (d, J = 7.6 Hz, 2H, CH-aromatic), 6.76 (d, 2H, AA′BB′, JAB = ca. 8.2 Hz, p-C6H4), 4.17 (br d, J = 12.9 Hz, 2H, CH2), 3.31 (s, 3H, OCH3), 3.25 (s, 2H, CH2Au), 2.63 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 1.97–1.54 (m, 14H, adamantyl CH and CH2), 1.48 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.08 (d, J = 6.7 Hz, 6H, CH(CH3)2), 0.79 (s, 6H, 2CH3) ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 260.2 (C carbene), 200.9 (C=O), 161.1 (MeOCipso methoxyphenyl), 144.7 (o-C), 135.3 (Cipso), 134.1 (Cipso methoxyphenyl), 129.9 (m-C methoxyphenyl), 129.4 (p-C), 124.7 (m-C), 112.6 (o-C methoxyphenyl), 76.3 (Cq), 64.5 (Cq), 54.5 (CH3O methoxyphenyl), 48.3 (CH2), 38.9 (CH2), 37.0, 35.0, 34.3 (CH2), 33.8 (CH2Au), 29.0 (CH), 28.4, 27.8, 27.3, 26.3, 22.9 (CH3) ppm. IR (ATR, cm–1): 2968, 2904, 1625 (C=O), 1598, 1507, 1464, 1369, 1306, 1245, 1163, 1097, 1023, 840, 805, 589. Anal. Calcd for C36H49AuNO2 (724.74): C, 59.66; H, 6.81; N, 1.93. Found: C, 59.75; H, 6.92; N, 1.95.
Synthesis of (AdCAAC)Au(deoxybenzoinyl) (14)
A scintillation vial was charged with (AdCAAC)AuOH (76 mg, 0.128 mmol) and deoxybenzoin (28 mg, 0.142 mmol) in toluene (2 mL). The resulting mixture was heated to 70 °C for 12 h. All volatiles were removed from the gray suspension. The product was extracted with CH2Cl2 and passed through a Celite pad (1 cm) which was washed with an additional 6 mL of CH2Cl2. The solution was concentrated to ca. 0.3 mL under vacuum and the white residue precipitated with hexanes (10 mL). The resulting suspension was centrifuged. The residue was washed with hexanes (2 × 4 mL) and dried under vacuum to give a white solid. Yield: 87 mg (0.116 mmol, 92%).
1H NMR (300 MHz, CD2Cl2): δ 7.73–7.70 (m, 2H, C6H5), 7.41 (t, J = 7.6 Hz, 1H, CH-aromatic), 7.31 (tt, J = 7.6 and 1.8 Hz, 1H, C6H5), 7.24 (d, J = 7.6 Hz, 2H, CH-aromatic) overlapping with 7.22–7.20 (m, 1H, C6H5), 7.12–7.70 (m, 5H, C6H5), 6.84 (tt, J = 7.6 and 1.8 Hz, 1H, C6H5), 4.80 (s, 1H, Au-CH), diastereotopic signals for carbene ligand 3.75 (br d, J = 12.9 Hz, 1H, CH2), 3.43 (br d, J = 12.9 Hz, 1H, CH2), 2.64 (sept, J = 6.7 Hz, 1H, CH(CH3)2), 2.57 (sept, J = 6.7 Hz, 1H, CH(CH3)2), 2.22–1.55 (m, 14H, adamantyl CH and CH2), 1.29 (d, J = 6.7 Hz, 3H, CH(CH3)2), 1.26 (s, 3H, C-CH3), 1.25 (d, J = 6.7 Hz, 3H, CH(CH3)2) overlapping with 1.25 (s, 3H, C–CH3), 1.16 (d, J = 6.7 Hz, 3H, CH(CH3)2), 0.89 (d, J = 6.7 Hz, 3H, CH(CH3)2) ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 254.6 (C carbene), 194.9 (C=O), 144.7 (o-Co aromatic), 144.6, 144.5, 141.2, 135.6 (Cipso), 129.4 (p-C), 129.3, 128.2, 127.5, 126.9, 126.9, 124.9 (m-C),124.8, 121.2, 77.0 (Cq), 64.2 (Cq), 56.3 (CH-Au), 48.6 (CH2), 38.9 (CH2), 37.0, 36.8, 34.9, 34.6, 34.3 (CH2), 34.2 (CH2), 29.0 (CH), 28.9, 28.8, 27.7, 27.2, 26.0, 25.4, 22.9 and 22.7(CH3) ppm. IR (ATR, cm–1): 2968, 2900, 1614 (CO), 1574, 1519, 1449, 1368, 1282, 1195, 1097, 1039, 931, 847, 807, 695, 579. Anal. Calcd for C39H50AuNO (745.78): C, 62.81; H, 6.76; N, 1.88. Found: C, 62.95; H, 6.86; N, 1.93.
Synthesis of (AdCAAC)Au(CH2SO2Ph) (15)
A scintillation vial was charged with (AdCAAC)AuOH (94 mg, 0.16 mmol) and methyl phenyl sulfone (30 mg, 0.19 mmol) in toluene (2 mL). The resulting mixture was heated to 70 °C overnight. The slightly yellow solution was filtered through a Celite pad (1 cm) which was washed with an additional 6 mL of toluene. The solution was concentrated to ca. 0.3 mL under vacuum and the white residue precipitated with hexanes (10 mL). The resulting suspension was centrifuged. The solid was washed with hexanes (2 × 4 mL) and dried under vacuum to give an off-white solid. Yield: 98 mg (0.134 mmol, 85%).
1H NMR (300 MHz, CD2Cl2): δ 7.77–7.74 (m, 2H, sulfone C6H5), 7.43 (t, J = 7.8 Hz, 1H, CH-aromatic) overlapping with 7.42–7.36 (m, 3H, sulfone C6H5), 7.26 (d, J = 7.8 Hz, 2H, CH-aromatic), 4.05 (br d, J = 12.0 Hz, 2H, CH2), 2.79 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.55 (s, 2H, CH2Au), 2.34–1.80 (m, 14H, adamantyl CH and CH2), 1.37 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.32 (s, 6H, 2CH3), 1.29 (d, J = 6.7 Hz, 6H, CH(CH3)2) ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 258.2 (C carbene), 147.5 (Cipso sulfone Ph), 145.1 (o-C), 135.3 (Cipso), 130.7 (p-C sulfone), 129.2 (p-C), 128.3 (m-C sulfone), 125.9 (o-C sulfone), 124.6 (m-C), 77.3 (Cq), 65.1 (Cq), 49.7 (CH2Au, sulfone), 48.6 (CH2), 39.0 (CH2), 37.1, 35.2, 34.4 (CH2), 29.0 (CH), 28.9, 27.9, 27.4, 26.3, 22.7 (CH3) ppm. Anal. Calcd for C34H46AuNSO2 (729.76): C, 55.96; H, 6.35; N, 1.92. Found: C, 56.13; H, 6.47; N, 1.99.
Synthesis of [(AdCAAC)Au(CO)]SbF6 (16)
A Schlenk flask was charged with (AdCAAC)AuCl (60.5 mg, 0.1 mmol), AgSbF6 (35 mg, 0.1 mmol), and CH2Cl2 (2 mL). The resulting suspension was stirred for 1 h in the dark. The mixture was filtered through a Celite pad (2 cm), which was washed with another 8 mL of CH2Cl2. The colorless solution was concentrated to ca. 3 mL, cooled to −20 °C, and saturated by bubbling with CO for 1 min followed by stirring at room temperature for 2 h. Precipitating with an excess of hexanes (15 mL), decanting the solvents, and removing volatiles under vacuum for 0.5 min afforded the product as a white solid. Yield: 80.0 mg, 0.095 mmol, 95%. The compound was stored under an atmosphere of CO.
1H NMR (300 MHz, CD2Cl2): δ 7.57 (t, J = 7.7 Hz, 1H, CH-aromatic), 7.38 (d, J = 7.7 Hz, 2H, CH-aromatic), 3.23 (br d, J = 12.7 Hz, 2H, CH2), 2.70 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.47–1.85 (m, 14H, adamantyl CH and CH2), 1.44 (s, 6H, 2CH3), 1.35 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.33 (d, J = 6.7 Hz, 6H, CH(CH3)2) ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 241.1 (C carbene), 182.4 (s, CO), 144.7 (o-C), 134.2 (Cipso), 131.2 (p-C), 125.7 (m-C), 80.8 (Cq), 65.4 (Cq), 47.9 (CH2), 38.4 (CH2), 36.9, 36.8, 33.8 (CH2), 29.1 (CH), 29.0, 27.7, 27.1, 26.6, 22.7 (CH3) ppm. IR (ATR, cm–1): 2968, 2903, 2183 (C≡O), 1541, 1450, 1387, 1262, 1195, 1097, 805, 651, 610, 583. Anal. Calcd for C28H39AuF6NOSb (838.33): C, 40.12; H, 4.69; N, 1.67. Found: C, 40.01; H, 4.59; N, 1.63.
Synthesis of [(AdCAAC)Au(CNtBu)]SbF6 (17)
A Schlenk flask was charged with (AdCAAC)AuCl (60.5 mg, 0.1 mmol), AgSbF6 (35 mg, 0.10 mmol), and CH2Cl2 (2 mL). The resulting suspension was stirred for 1 h in the dark. The mixture was filtered through a Celite pad (2 cm), which was washed with another 8 mL of CH2Cl2. The colorless solution was concentrated to ca. 2 mL, and an excess of tBuNC (22 μL, 0.2 mmol) was added, followed by stirring at room temperature for 2 h. The product was precipitated with an excess of hexanes (15 mL), centrifuged, and washed with hexanes (5 mL). All volatiles were removed under vacuum to give the complex as a white solid. Yield: 83.5 mg (0.094 mmol, 94%).
1H NMR (300 MHz, CD2Cl2): δ 7.52 (t, J = 7.7 Hz, 1H, CH-aromatic), 7.33 (d, J = 7.7 Hz, 2H, CH-aromatic), 3.42 (br d, J = 12.6 Hz, 2H, CH2), 2.71 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.40–1.83 (m, 14H, adamantyl CH and CH2), 1.48 (s, 9H, C(CH3)3), 1.39 (s, 6H, 2CH3), 1.33 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.31 (d, J = 6.7 Hz, 6H, CH(CH3)2) ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 246.2 (C carbene), 144.8 (o-C), 142.4 (br s, CNtBu) 134.5 (Cipso), 130.4 (p-C), 125.3 (m-C), 79.5 (Cq), 65.1 (Cq), 58.6 (br s, CNCMe3), 48.0 (CH2), 38.5 (CH2), 36.9, 36.1, 34.0 (CH2), 29.5 (CNC(CH3)3), 29.0 (CH), 28.9, 27.8, 26.8, 26.6, 22.7 (CH3) ppm. IR (ATR, cm–1): 2973, 2899, 2241 (CNtBu), 1538, 1450, 1373, 1194, 1147, 1097, 803, 776, 654, 523. Anal. Calcd for C32H48AuF6N2Sb (893.45): C, 43.02; H, 5.41; N, 3.14. Found: C, 43.13; H, 5.49; N, 3.19.
Synthesis of (AdCAAC)AuCN (18)
Method A
Trimethylsilyl cyanide (20 μL, 0.150 mmol) was added to the solution of (AdCAAC)AuOH (59 mg, 0.1 mmol) in 2 mL of toluene. The mixture was stirred at room temperature overnight and concentrated under vacuum. The white residue was precipitated with hexanes (6 mL). The resulting suspension was centrifuged. The solid was washed with hexanes (3 × 5 mL) and dried under vacuum to give a while solid. Yield: 57.5 mg, 0.095 mmol, 95%.
Method B
(AdCAAC)AuCl (45 mg, 0.075 mmol), KCN (5 mg, 0.076 mmol) and 10 mL of ethanol were charged in a scintillation vial and stirred overnight. All volatiles were evaporated, and the white residue was extracted with CH2Cl2 (3 × 5 mL). The combined extracts were filtered through a glass frit and concentrated to ca. 0.5 mL. The product was precipitated with hexanes (10 mL) and dried under vacuum. Yield: 42 mg (0.07 mmol, 92%).
1H NMR (300 MHz, CD2Cl2): δ 7.49 (t, J = 7.8 Hz, 1H,CH-aromatic), 7.30 (d, J = 7.8 Hz, 2H, CH-aromatic), 3.71(br d, J = 13.1 Hz, 2H, CH2), 2.72 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.32–1.79 (m, 14H, adamantyl CH and CH2), 1.37 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.34 (s, 6H, 2CH3), 1.30 (d, J = 6.7 Hz, 6H, CH(CH3)2) ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 253.1 (C carbene), 149.5 (CN), 144.9 (o-C), 134.8 (Cipso), 129.8 (p-C), 124.9 (m-C), 78.0 (Cq), 64.9 (Cq), 48.3 (CH2), 38.8 (CH2), 37.0, 35.6, 34.2 (CH2), 29.0 (CH), 28.9, 27.8, 27.1, 26.4, 22.7 (CH3) ppm. IR (ATR, cm–1): 2969, 2900, 2140 (C≡N), 1530, 1447, 1370, 1097, 934, 808, 727. Anal. Calcd For C28H39AuN2 (600.58): C, 56.00; H, 6.54; N, 4.66. Found: C, 56.16; H, 6.61; N, 4.72.
Synthesis of [(AdCAAC)Au(norbornene)][tfaB(C6F5)3] (19)
A Schlenk
flask was charged with (AdCAAC)AuOAcf (60 mg,
0.088 mmol), B(C6F5)3 (90 mg, 0.176
mmol), norbornene (16.5 mg, 0.176 mmol), and dry CH2Cl2 (2 mL) under an argon atmosphere. The resulting suspension
was stirred for 1 h at −78 °C and left to warm to room
temperature while stirring overnight. The mixture was filtered through
a Celite pad (1 cm), which was washed with another 8 mL of CH2Cl2. The colorless solution was concentrated to
ca. 1 mL and the oily residue precipitated with an excess of hexanes
(15 mL). The solvents were decanted, and the residue was dissolved
in 0.5 mL of CH2Cl2 and precipitated with hexane
(15 mL). The oily colorless residue after decantation was dried under
vacuum to afford a white powder which was additionally dried under
vacuum overnight. Yield: 96 mg (0.074 mmol, 85%).
1H NMR (300 MHz, CD2Cl2): δ 7.50 (t, J = 7.8 Hz, 1H, CH-aromatic), 7.33 (d, J = 7.7 Hz, 2H, CH-aromatic), 5.83 (br s, 2H, Ha norbornene), 3.27 (br d, J = 12.2 Hz, 2H, CH2), 3.00 (br s, 2H, Hb norbornene), 2.71 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.42–1.75 (m, 18H, adamantyl CH and CH2 overlapping with 4Hd norbornene), 1.44 (s, 6H, 2CH3), 1.32 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.29 (d, J = 6.7 Hz, 6H, CH(CH3)2), 0.60 (br d, 1H, 1J = 10.2 Hz, Hc norbornene), 0.39 (br d, 1H, 1J = 10.2 Hz, Hc norbornene) ppm. 19F NMR (282 MHz, CD2Cl2): δ −76.9 (s, 3F, CF3), −135.09 (br d, 6F, JF–F = 19.5 Hz, o-C6F5), −161.7 (br t, 3F, JF–F = 19.5 Hz, p-C6F5), −166.7 (br t, 6F, JF–F = 19.5 Hz, m-C6F5) ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ (B–Cipso from C6F5 and C=O signals were not observed) 246.8 (C carbene), 149.5–146.3 (d m,1JC–F= 248 Hz, CF), 144.6 (o-C), 140.6–137.3 (d m, 1JC–F= 248 Hz, CF), 138.2–135.0 (d m, 1JC–F= 252 Hz, CF), 135.3 (Cipso), 130.7 (p-C), 125.5 (m-C), 123.7 (CHa, norbornene), 115.4 (quart. 1JC–F= 288 Hz, CF3), 79.6 (Cq), 64.5 (Cq), 48.3 (CH2), 44.6 (CHb, norbornene), 43.9 (CH2c, norbornene), 38.4 (CH2), 36.9, 35.8, 33.9 (CH2), 29.1 (CH), 29.0, 27.8, 26.7, 26.5, 23.7 (CH2d, norbornene), 22.8 (CH3) ppm. IR (ATR, cm–1): 2906, 1749 (C=O), 1643 (C=C norbornene), 1514, 1465, 1374, 1281, 1187, 1153, 1093, 977, 851, 807, 680. Anal. Calcd for C54H49AuBF18NO2 (1293.71): C, 50.13; H, 3.82; N, 1.08. Found: C, 50.32; H, 3.61; N, 1.01.
Reaction of (AdCAAC)AuCl with PhICl2 at 20 °C without Light Protection
A mixture of (AdCAAC)AuCl (61 mg, 0.10 mmol) and PhICl2 (30 mg, 0.11 mmol) in 5 mL of CH2Cl2 was stirred overnight at room temperature. The yellow solution was concentrated to ca. 0.3 mL. Adding Et2O (10 mL) gave a yellow precipitate, which was washed with Et2O (2 × 5 mL) and dried under vacuum: yield 63 mg. Crystallization by layering a CH2Cl2 solution with hexanes led to the formation of two types of crystals, the structures of which were confirmed by X-ray diffraction. A small amount of colorless needles was identified as the dichloroaurate(I) salt [(AdCAAC-Cl)][AuCl2] (20a), while the major component of yellow prisms turned out to be the cocrystallization product {2[(AdCAAC-Cl)][AuCl4]·(AdCAAC)AuCl3}. NMR spectroscopy showed two sets of ligand signals in an approximate 2:1 ratio, which were assigned on the basis of the known signals for the pure salt [(AdCAAC-Cl)]AuCl4 (20b) and the complex AdCAACAuCl3 (25). The 1H and 13C NMR spectra of the two salts [(AdCAAC-Cl)]AuCl2 and [(AdCAAC-Cl)]AuCl4 are essentially identical. Elemental analysis was not carried out due to formation of a product mixture.
Synthesis of [(AdCAAC-Cl)][AuCl4] (20b)
A solution of (AdCAAC)AuCl (61 mg, 0.1 mmol) and PhICl2 (58 mg, 0.21 mmol) in 5 mL of CH2Cl2 was stirred for 3 h without light protection. A yellow solution resulted, which was concentrated to ca. 0.3 mL. The addition of Et2O (10 mL) gave a yellow precipitate, which was washed with Et2O (2 × 5 mL) and dried under vacuum. Yield: 74 mg, 0.098 mmol, 98%.
1H NMR (300 MHz, CD2Cl2): δ 7.66 (t, J = 7.7 Hz, 1H, CH-aromatic), 7.46 (d, J = 7.7 Hz, 2H, CH-aromatic), 2.92 (s, 2H, CH2, adamantyl), 2.67 (br d, J = 13.0 Hz, 2H, CH2), 2.45 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.39–1.89 (m, 12H, adamantyl CH and CH2), 1.58 (s, 6H, 2CH3), 1.38 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.20 (d, J = 6.7 Hz, 6H, CH(CH3)2) ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 188.5 (C–Cl), 143.8 (o-C), 132.7 (p-C), 128.0 (Cipso), 126.5 (m-C), 79.2 (Cq), 60.8 (Cq), 47.8 (CH2), 38.0 (CH2), 37.1, 34.1, 32.1 (CH2), 29.9 (CH), 28.5, 26.4, 25.8, 23.1 (CH3) ppm. Anal. Calcd for C27H39AuCl5N (751.83): C, 43.13; H, 5.23; N, 1.86. Found: C, 42.96; H, 5.28; N, 2.00.
Synthesis of (AdCAAC)AuBr
A suspension of (AdCAAC)AuCl (61 mg, 0.10 mmol) and LiBr (88 mg, 1 mmol) in 10 mL of acetone was stirred for 24 h at room temperature. The solvent was removed under vacuum. The white residue was extracted with CH2Cl2 (2 × 10 mL) and filtered through Celite (1 cm). All volatiles were removed under vacuum to give an off-white solid with one solvate molecule of CH2Cl2. Yield: 72.5 mg (0.098 mmol, 98%).
1H NMR (300 MHz, CDCl3): δ 7.41 (t, J = 7.7 Hz, 1H, CH-aromatic), 7.24 (d, J = 7.7 Hz, 2H, CH-aromatic), 5.29 (s, 2H, solvent molecule CH2Cl2), 4.02 (br d, J = 12.6 Hz, 2H, CH2), 2.74 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.35–1.78 (m, 14H, adamantyl CH and CH2), 1.42 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.34 (s, 6H, 2CH3), 1.29 (d, J = 6.7 Hz, 6H, CH(CH3)2) ppm. 13C{1H} NMR (75 MHz, CDCl3): δ 242.6 (C carbene), 144.8 (o-C), 135.1 (Cipso), 129.7 (p-C), 124.9 (m-C), 76.7 (Cq), 63.7 (Cq), 53.4 (solvent molecule CH2Cl2), 48.6 (CH2), 38.9 (CH2), 37.0, 35.1, 34.5 (CH2), 29.1 (CH), 29.0, 27.5, 27.1, 26.8, 23.0 (CH3) ppm. Anal. Calcd for C27H39AuBrN·CH2Cl2 (739.40): C, 45.48; H, 5.59; N, 1.89. Found: C, 45.57; H, 5.67; N, 2.01.
Reaction of (AdCAAC)AuBr with CsBr3
A suspension of (AdCAAC)AuBr·CH2Cl2 (74 mg, 0.10 mmol) and CsBr3 (38 mg, 0.10 mmol) in 5 mL of CH2Cl2 was stirred for 20 min at −78 °C and warmed to room temperature with stirring for 1 h. The suspension was filtered through a glass frit and the filtrate concentrated to ca. 0.3 mL. An orange solid was precipitated with hexanes (10 mL) and dried under vacuum. Yield: 80 mg. Recrystallization by layering a CH2Cl2 solution with hexanes led to the formation of two types of crystals, which were identified by X-ray diffraction: a larger amount of colorless prisms of [(AdCAAC-Br)]+[AuBr2]− (21a) and a small amount of red prisms of [(AdCAAC-Br)]+[AuBr4]− (21b). Both give identical 1H and 13C NMR spectra.
1H NMR (300 MHz, CD2Cl2): δ 7.65 (t, J = 7.7 Hz, 1H, CH-aromatic), 7.46 (d, J = 7.7 Hz, 2H, CH-aromatic), 2.93 (s, 2H, CH2, adamantyl) overlapping with 2.91 (br d, J = 13.0 Hz, 2H, CH2), 2.44 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.36–1.89 (m, 12H, adamantyl CH and CH2), 1.59 (s, 6H, 2CH3), 1.38 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.26 (d, J = 6.7 Hz, 6H, CH(CH3)2) ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 186.0 (C–Br), 143.6 (o-C), 132.5 (p-C), 130.4 (Cipso), 126.7 (m-C), 80.8 (Cq), 62.7 (Cq), 48.0 (CH2), 38.3 (CH2), 37.1, 34.3, 31.9 (CH2), 29.8 (CH), 28.7, 26.5, 25.9, 23.5 (CH3) ppm. Elemental analysis was not carried out due to formation of a product mixture.
Synthesis of [(Me2CAAC)2Au]Cl (22)
A Schlenk flask was charged with Me2CAAC (0.39 g, 1.36 mmol), (Me2S)AuCl (0.195 g, 0.66 mmol), and 20 mL of THF under an argon atmosphere. The mixture was stirred at room temperature for 18 h. All volatiles were removed under vacuum and the residue was washed with hexanes (3 × 10 mL). The product was dissolved in CH2Cl2 (3 mL) and precipitated with hexanes (40 mL). All volatiles were evaporated. The residue was dried under vacuum to give a white solid. Yield: 510 mg (0.635 mmol, 96%).
1H NMR (300 MHz, CD2Cl2): δ 7.42 (t, J = 7.7 Hz, 1H, CH-aromatic), 7.23 (d, J = 7.7 Hz, 2H, CH-aromatic), 2.59 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.04 (br s, 2H, CH2), 1.31 (s, 6H, 2CH3), 1.24 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.20 (s, 6H, 2CH3), 1.01 (br d, J = 6.7 Hz, 6H, CH(CH3)2). 13C{1H} NMR (75 MHz, CD2Cl2): δ 250.6 (C carbene), 144.6 (o-C), 133.5 (Cipso), 130.1 (p-C), 125.1 (m-C), 82.3 (Cq), 54.6 (Cq), 49.3 (CH2), 28.9 (CH), 28.8, 28.4, 26.7, 22.6 (CH3). Anal. Calcd for C40H62AuClN2 (803.35): C, 59.80; H, 7.78; N, 3.49. Found: C, 59.49; H, 7.57; N, 3.31.
Synthesis of [AuCl2(Me2CAAC)2]Cl (23)
A mixture of [Au(Me2CAAC)2]Cl (80 mg, 0.10 mmol) and PhICl2 (28 mg, 0.10 mmol) in 5 mL of CH2Cl2 was stirred in the dark for 6 h at 0 °C and warmed to room temperature. The colorless solution was concentrated to ca. 0.3 mL. Addition of Et2O (10 mL) gave an off-white precipitate, which was washed with Et2O (2 × 5 mL) and dried under vacuum. Yield: 91 mg (0.94 mmol, 94%). Crystallization by layering a CH2Cl2 solution with hexanes in the dark led to the formation of large colorless prisms and negligible amounts of yellow prisms, which were identified by X-ray diffraction: the colorless prisms as [AuCl2(Me2CAAC)2]Cl·CH2Cl2 (23·CH2Cl2) and yellow prisms as [Au(Me2CAAC)2]AuCl4.
1H NMR (300 MHz, CD2Cl2): δ 7.42 (t, J = 7.7 Hz, 1H, CH-aromatic), 7.23 (d, J = 7.7 Hz, 2H, CH-aromatic), 2.58 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.04 (br s, 2H, CH2), 1.31 (s, 6H, 2CH3), 1.23 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.19 (s, 6H, 2CH3), 1.01 (br s, 6H, CH(CH3)2) ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 250.6 (C carbene), 144.6 (o-C), 133.4 (Cipso), 130.1 (p-C), 125.1 (m-C), 82.3 (Cq), 54.6 (Cq), 49.3 (CH2), 28.9 (CH), 28.8, 28.4, 26.7, 22.6 (CH3) ppm. Anal. Calcd for C40H62AuCl3N2·CH2Cl2 (959.18): C, 51.34; H, 6.73; N, 2.92. Found: C, 51.64; H, 6.96; N, 3.14.
Reaction of [Au(Me2CAAC)2]Cl with CsBr3 at −78 °C
A mixture of [Au(Me2CAAC)2]Cl (89 mg, 0.10 mmol) and CsBr3 (38 mg, 0.10 mmol) in 5 mL of CH2Cl2 was stirred for 20 min at −78 °C and warmed to room temperature with stirring for 1 h. The orange-red solution was concentrated to ca. 0.3 mL. An orange solid was precipitated with hexanes (10 mL) and dried under vacuum. Yield: 95 mg. Crystallization by layering a CH2Cl2 solution with hexanes gave two type of crystals, which were identified by X-ray diffraction as orange prisms of [Au(Me2CAAC)2]Br3 (24a) and red prisms of [Au(Me2CAAC)2]AuBr4 (24b). Both give identical 1H NMR spectra.
1H NMR (300 MHz, CD2Cl2): δ 7.43 (t, J = 7.7 Hz, 1H, CH-aromatic), 7.25 (d, J = 7.7 Hz, 2H, CH-aromatic), 2.61 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.06 (br s, 2H, CH2), 1.33 (s, 6H, 2CH3), 1.26 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.22 (s, 6H, 2CH3), 1.03 (br d, J = 6.7 Hz, 6H, CH(CH3)2) ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 250.6 (C carbene), 144.6 (o-C), 133.5 (Cipso), 130.1 (p-C), 125.1 (m-C), 82.4 (Cq), 54.6 (Cq), 49.5 (CH2), 28.9 (CH), 28.8, 28.5, 26.7, 22.6 (CH3) ppm. Elemental analysis was not carried out due to formation of a product mixture.
Synthesis of [Au(Me2CAAC)2]ClI2 (24c)
A solution of [Au(Me2CAAC)2]Cl (80 mg, 0.10 mmol) and I2 (26 mg, 0.10 mmol) in 3 mL of CH2Cl2 was stirred for 1 h at 0 °C and warmed to room temperature with stirring for 1 h. Addition of hexanes (15 mL) led to the precipitation of a brown product, which was centrifuged, washed with 5 mL of Et2O, and dried under vacuum. Yield: 102.5 mg (0.097 mmol, 97%).
1H NMR (300 MHz, CD2Cl2): δ 7.44 (t, J = 7.7 Hz, 1H, CH-aromatic), 7.24 (d, J = 7.7 Hz, 2H, CH-aromatic), 2.59 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.06 (br s, 2H, CH2), 1.32 (s, 6H, 2CH3), 1.25 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.21 (s, 6H, 2CH3), 1.02 (br d, J = 6.7 Hz, 6H, CH(CH3)2) ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 250.6 (C carbene), 144.6 (o-C), 133.5 (Cipso), 130.1 (p-C), 125.1 (m-C), 82.4 (Cq), 54.6 (Cq), 49.4 (CH2), 28.9 (CH), 28.8, 28.5, 26.7, 22.6 (CH3) ppm. Anal. Calcd for C40H62AuClI2N2 (1057.16): C, 45.45; H, 5.91; N, 2.65. Found: C, 45.37; H, 5.83; N, 2.59.
Synthesis of (AdCAAC)AuCl3 (25)
All operations have to be performed with minimum exposure to light. A scintillation vial was charged with (AdCAAC)AuCl (61 mg, 0.10 mmol) and PhICl2 (29 mg, 0.105 mmol) and wrapped in aluminum foil. Chilled CH2Cl2 (5 mL) was added and resulting solution stirred for 6 h at 0 °C in the dark. A slightly yellow solution resulted, which was concentrated to ca. 0.3 mL. The addition of Et2O (15 mL) gave a pale yellow precipitate which was washed with Et2O (2 × 5 mL) and dried under vacuum. Yield: 64 mg (0.095 mmol, 95%). 1H NMR (300 MHz, CD2Cl2): δ 7.52 (t, J = 7.7 Hz, 1H, CH-aromatic), 7.38 (d, J = 7.7 Hz, 2H, CH-aromatic), 3.33 (br d, J = 13.2 Hz, 2H, CH2), 3.04 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.54–1.82 (m, 14H, adamantyl CH and CH2), 1.55 (s, 6H, 2CH3), 1.48 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.27 (d, J = 6.7 Hz, 6H, CH(CH3)2) ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 218.8 (C carbene), 146.2 (o-C), 133.5 (p-C), 130.9 (Cipso), 126.7 (m-C), 82.1 (Cq), 69.1 (Cq), 47.6 (CH2), 38.0 (CH2), 37.7, 35.3, 34.5 (CH2), 30.1 (CH), 29.0, 27.1, 26.6, 26.1 (CH3). Anal. Calcd for C27H39AuCl3N (680.92): C, 47.62; H, 5.77; N, 2.06. Found: C, 47.69; H, 5.72; N, 2.12.
Synthesis of (AdCAAC)AuI
A mixture of [Au(AdCAAC)(Cl)] (61 mg, 0.10 mmol) and NaI (150 mg, 1 mmol) in 10 mL of acetone was stirred for 24 h at room temperature. The solvent was removed under vacuum. The white residue was extracted with CH2Cl2 (2 × 10 mL) and the solution filtered through Celite (1 cm). All volatiles were removed under vacuum to give an off-white solid with 0.5 CH2Cl2 as a solvate molecule. Yield: 73.5 mg (0.098 mmol, 98%).
1H NMR (300 MHz, CD2Cl2): δ 7.48 (t, J = 7.7 Hz, 1H, CH-aromatic), 7.29 (d, J = 7.7 Hz, 2H, CH-aromatic), 5.33 (s, 1H, solvent molecule 0.5CH2Cl2) 3.98 (br d, J = 12.6 Hz, 2H, CH2), 2.78 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.38–1.81 (m, 14H, adamantyl CH and CH2), 1.41 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.36 (s, 6H, 2CH3), 1.30 (d, J = 6.7 Hz, 6H, CH(CH3)2) ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 247.5 (C carbene), 144.9 (o-C), 135.1 (Cipso), 129.6 (p-C), 124.8 (m-C), 77.1 (Cq), 63.9 (Cq),, 48.4 (CH2), 38.8 (CH2), 37.0, 34.9, 34.3 (CH2), 29.0 (CH), 28.9, 27.8, 27.2, 26.4, 22.8 (CH3) ppm. Anal. Calcd for C27H39AuIN·0.5CH2Cl2 (743.94): C, 44.40; H, 5.42; N, 1.88. Found: C, 44.33; H, 5.48; N, 1.93.
Synthesis of (AdCAAC)AuI·I2 (26)
A mixture of (AdCAAC)AuI·0.5CH2Cl2 (74 mg, 0.10 mmol) and I2 (26 mg, 0.10 mmol) in 5 mL of CH2Cl2 was stirred for 20 min at −78 °C and warmed to room temperature with stirring for 2 h to give a dark red solution. The solvent was removed under vacuum and the residue washed with hexanes (3 × 6 mL) and dried under vacuum to afford a dark red solid. Yield: 92 mg (0.097 mmol, 97%).
1H NMR (300 MHz, CD2Cl2): δ 7.50 (t, J = 7.7 Hz, 1H, CH-aromatic), 7.29 (d, J = 7.7 Hz, 2H, CH-aromatic), 3.96 (br d, J = 12.6 Hz, 2H, CH2), 2.77 (sept, J = 6.7 Hz, 2H, CH(CH3)2), 2.38–1.82 (m, 14H, adamantyl CH and CH2), 1.41 (d, J = 6.7 Hz, 6H, CH(CH3)2), 1.36 (s, 6H, 2CH3), 1.30 (d, J = 6.7 Hz, 6H, CH(CH3)2) ppm. 13C{1H} NMR (75 MHz, CD2Cl2): δ 246.6 (C carbene), 144.8 (o-C), 135.1 (Cipso), 129.8 (p-C), 125.0 (m-C), 77.2 (Cq), 63.9 (Cq), 48.4 (CH2), 38.8 (CH2), 37.0, 35.1, 34.3 (CH2), 29.0 (CH), 28.9, 27.8, 27.2, 26.5, 22.8 (CH3) ppm. Anal. Calcd for C27H39AuI3N (955.28): C, 33.95; H, 4.11; N, 1.47. Found: C, 33.81; H, 4.02; N, 1.39.
X-ray Crystallography
Crystals suitable for X-ray study were obtained by layering of a CH2Cl2 solution with hexanes, with the exception of complex 17 (1,2-difluorobenzene/hexanes). Crystals were mounted in oil on glass fibers and fixed in the cold nitrogen stream on a diffractometer. X-ray diffraction experiments were carried out with an Oxford Diffraction Xcalibur-3/Sapphire3-CCD diffractometer, using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at 140 K. Data were processed using the CrystAlisPro-CCD and -RED software.48 The principal crystallographic data and refinement parameters are given in Table S1 (Supporting Information). The CH2 (C3 atom) and methyl groups (C17 and C18 atoms) were disordered over two positions with occupancies of 0.65/0.35 and 0.62/0.38, respectively. The Cl1 and I2 atoms for complex 24c were disordered into two positions linked by a center of inversion with equal occupancies. The complex [{(AdCAAC)Au}3(μ3-O)]+OH– crystallizes with two independent molecules and half of a benzene molecule. The oxygen atoms of the hydroxide counteranion for [{(AdCAAC)Au}3(μ3-O)]+OH– were not refined anisotropically, due to disorder problems masked by the presence of the disordered solvent molecules. For the final refinement, the contribution of severely disordered CH2Cl2 molecules in crystals of 7, 14, and [{(AdCAAC)Au}3(μ3-O)]+OH– were removed from the diffraction data with PLATON/SQUEEZE.49,50 The structures were solved by direct methods and refined by the full-matrix least squares against F2 in an anisotropic (for non-hydrogen atoms) approximation. All hydrogen atom positions were refined in an isotropic approximation in the “riding” model with the Uiso(H) parameters equal to 1.2[Ueq(Ci)] and for methyl groups equal to 1.5[Ueq(Cii)], where U(Ci) and U(Cii) are respectively the equivalent thermal parameters of the carbon atoms to which the corresponding H atoms are bonded. The hydrogen atom of the hydroxide counteranion in [{(AdCAAC)Au}3(μ3-O)]+OH– was not located. All calculations were performed using the SHELXTL software.51 Intensities for complex 14 and [{(AdCAAC)Au}3(μ3-O)]+OH– were collected at 100(2) K on a Bruker-Nonius Roper CCD diffractometer, equipped with Mo Kα radiation and a graphite monochromator at the EPSRC National Crystallography service, Southampton, U.K.52 Data were processed using CrystalClear-SM Expert 3.1 b21 (Rigaku, 2012) programs.
Acknowledgments
This work was supported by the European Research Council (Framework 7 ERC Advanced Investigator Award 338944-GOCAT).
Supporting Information Available
Tables, figures, and CIF files giving crystallographic data for compounds 7, 11, 14, 16–18, 20a/24, 20b, 21a,b, 23, 24a–c, 26, (AdCAAC)AuX (X = Br, I), and [{(AdCAAC)Au}3(μ3-O)]+OH– and 1H and 13C{1H} NMR spectra for complex 14. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Dedication
Dedicated to the memory of Professor Michael F. Lappert, a superbly inventive pioneer of organometallic chemistry and a much-missed friend.
Supplementary Material
References
- Cardin D. J.; Cetinkaya B.; Lappert M. F.; Manojlović-Muir L.; Muir K. W. An Electron-rich Olefin as a Source of Co-ordinated Carbene; Synthesis of trans-PtCl2[C(NPhCH2)2]PEt3. J. Chem. Soc., Chem. Commun. 1971, 400. [Google Scholar]
- Cardin D. J.; Cetinkaya B.; Lappert M. F. Transition Metal-Carbene Complexes. Chem. Rev. 1972, 72, 545. [Google Scholar]
- Lappert M. F. Contributions to the chemistry of carbenemetal chemistry. J. Organomet. Chem. 2005, 690, 5467. [Google Scholar]
- Cetinkaya B.; Dixneuf P.; Lappert M. F. J. Chem. Soc., Dalton Trans. 1974, 1827. [Google Scholar]
- See for example:; a Herrmann W. A.; Köcher C. Angew. Chem., Int. Ed. Engl. 1997, 36, 2162. [Google Scholar]; b Bourissou D.; Guerret O.; Gabbaï F. P.; Bertrand G. Chem. Rev. 2000, 100, 39. [DOI] [PubMed] [Google Scholar]; c Hahn F. E.; Jahnke M. C. Angew. Chem., Int. Ed. 2008, 47, 3122. [DOI] [PubMed] [Google Scholar]; d Chem. Rev. 2009, 109, 3209–3884 (carbene themed issue).19642638 [Google Scholar]; e Nelson D. J.; Nolan S. P. Chem. Soc. Rev. 2013, 42, 6723. [DOI] [PubMed] [Google Scholar]; f N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis; Cazin C. S. J., Ed.; Springer: Heidelberg, Germany, 2011; Catalysis by Metal Complexes Vol. 32. [Google Scholar]
- a Lavallo V.; Canac Y.; Prasang C.; Donnadieu B.; Bertrand G. Angew. Chem., Int. Ed. 2005, 44, 5705. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lavallo V.; Canac Y.; DeHope A.; Donnadieu B.; Bertrand G. Angew. Chem., Int. Ed. 2005, 44, 7236. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Frey G. D.; Dewhurst R. D.; Kousar S.; Donnadieu B.; Bertrand G. J. Organomet. Chem. 2008, 693, 1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger D. S.; Melaimi M.; Moore C. E.; Rheingold A. L.; Frenking G.; Jerabek P.; Bertrand G. Angew. Chem., Int. Ed. 2013, 52, 8964. [DOI] [PubMed] [Google Scholar]
- a Lavallo V.; Frey G. D.; Kousar S.; Donnadieu B.; Bertrand G. Proc. Nat. Acad. Sci. 2007, 104, 13569. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lavallo V.; Frey G. D.; Donnadieu B.; Soleilhavoup M.; Bertrand G. Angew. Chem., Int. Ed. 2008, 47, 5224. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zeng X.; Frey G. D.; Kousar S.; Bertrand G. Chem. Eur. J. 2009, 15, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Zeng X.; Frey G. D.; Kinjo R.; Donnadieu B.; Bertrand G. J. Am. Chem. Soc. 2009, 131, 8690. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Zeng X.; Soleilhavoup M.; Bertrand G. Org. Lett. 2009, 11, 3166. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Zeng X.; Kinjo R.; Donnadieu B.; Bertrand G. Angew. Chem., Int. Ed. 2010, 49, 942. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Kinjo R.; Donnadieu B.; Bertrand G. Angew. Chem., Int. Ed. 2011, 50, 5560. [DOI] [PubMed] [Google Scholar]; h Ung G.; Bertrand G. Angew. Chem., Int. Ed. 2013, 52, 11388. [DOI] [PubMed] [Google Scholar]; i Hirner J. J.; Roth K. E.; Shi Y.; Blum S. A. Organometallics 2012, 31, 6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukov A. A.; Normand A. T.; Nechaev M. S. Dalton Trans. 2009, 7015. [DOI] [PubMed] [Google Scholar]
- Alcarazo M.; Stork T.; Anoop A.; Thiel W.; Fürstner A. Angew. Chem., Int. Ed. 2010, 49, 2542. [DOI] [PubMed] [Google Scholar]
- Hartwig J.Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Sausalito, CA, 2010; p 41 ff. [Google Scholar]
- Bochmann M.Organometallics and Catalysis-An Introduction; Oxford University Press: Oxford, U.K., 2014; pp 102–107. [Google Scholar]
- For investigations of the electronic characteristics of carbene ligands see for example:; a Hu X.; Castro-Rodriguez I.; Olsen K.; Meyer K. Organometallics 2004, 23, 755.(DFT). [Google Scholar]; b Khramov D. M.; Lynch V. M.; Bielawski C. W. Organometallics 2007, 26, 6042.(IR, 1H NMR). [Google Scholar]; c Jacobsena H.; Correa A.; Poater A.; Costabile C.; Cavallo L. Coord. Chem. Rev. 2009, 253, 687.(DFT). [Google Scholar]; d Gusev D. G. Organometallics 2009, 28, 6458.(IR). [Google Scholar]; e Tonner R.; Frenking G. Organometallics 2009, 28, 3901.(DFT). [Google Scholar]; f Liske A.; Verlinden K.; Buhl H.; Schaper K.; Ganter C. Organometallics 2013, 32, 5269.(77Se NMR). [Google Scholar]; g Back O.; Henry-Ellinger M.; Martin C. D.; Martin D.; Bertrand G. Angew. Chem., Int. Ed. 2013, 52, 2939.(31P NMR). [DOI] [PubMed] [Google Scholar]; h Nelson D. J.; Collado A.; Manzini S.; Meiries S.; Slawin A. M. Z.; Cordes D. B.; Nolan S. P. Organometallics 2014, 33, 2048.(IR, NMR). [Google Scholar]; i Marchione D.; Belpassi L.; Bistoni G.; Macchioni A.; Tarantelli F.; Zuccaccia D. Organometallics 2014, 33, 4200. [Google Scholar]
- Ciancaleoni G.; Scafuri N.; Bistoni G.; Macchioni A.; Tarantelli F.; Zuccaccia D.; Belpassi L. Inorg. Chem. 2014, 53, 9907.and references cited therein. [DOI] [PubMed] [Google Scholar]
- A related approach has used C-bonded acac complexes for transauration reactions; see for example:; a Vicente J.; Chicote M. T.; Guerrero R.; Jones P. G. J. Am. Chem. Soc. 1996, 118, 699. [Google Scholar]; b Vicente J.; Chicote M. T.; Saura-Llamas I.; Lagunas M. C. J. Chem. Soc., Chem. Commun. 1992, 915. [Google Scholar]; c Vicente J.; Chicote M. T.; Abrisqueta M. D.; Ramirez de Arellano M. C.; Jones P. G.; Humphrey M. G.; Cifuentes M. P.; Samoc M.; Luther-Davies B. Organometallics 2000, 19, 2968. [Google Scholar]; d Vicente J.; Chicote M. T.; Gonzalez-Herrero P.; Jones P. G. J. Chem. Soc., Dalton Trans. 1994, 3183. [Google Scholar]; e Vicente J.; Chicote M. T.; Abrisqueta M. D. J. Chem. Soc., Dalton Trans. 1995, 497. [Google Scholar]; f Vicente J.; Chicote M. T.; Guerrero R.; Saura-Llamas I. M.; Jones P. G.; Ramirez de Arellano M. C. Chem. Eur. J. 2001, 7, 638. [DOI] [PubMed] [Google Scholar]
- For other examples of gold(I) alkoxides see:; a Sutherland B. R.; Folting K.; Streib W. E.; Ho D. M.; Huffman J. C.; Caulton K. G. J. Am. Chem. Soc. 1987, 109, 3489. [Google Scholar]; b Komiya S.; Iwata M.; Sone T.; Fukuoka A. J. Chem. Soc., Chem. Commun. 1992, 1109. [Google Scholar]; c Usui Y.; Noma J.; Hirano M.; Komiya S. Inorg. Chim. Acta 2000, 309, 151. [Google Scholar]; d Laitar D. S.; Müller P.; Gray T. G.; Sadighi J. P. Organometallics 2005, 24, 4503. [Google Scholar]
- While this work was in progress, a similar effect was reported for imidazole-type NHC complexes:Nahra F.; Patrick S. R.; Collado A.; Nolan S. P. Polyhedron 2014, 84, 59. [Google Scholar]
- Nesmeyanov A. N.; Perevalova E. G.; Struchkov Y. T.; Antipin M. Y.; Grandberg K. I.; Dyadhenko V. P. J. Organomet. Chem. 1980, 201, 343. [Google Scholar]
- Related O-bridged clusters with sterically less demanding CAAC ligands have recently been made by Ag2O reactions or PPh3 displacement:Jin L.; Weinberger D. S.; Melaimi M.; Moore C. E.; Rheingold A. L.; Bertrand G. Angew. Chem., Int. Ed. 2014, 53, 9059. [DOI] [PubMed] [Google Scholar]
- a Dupuy S.; Crawford L.; Bühl M.; Slawin A. M. Z.; Nolan S. P. Adv. Synth. Catal. 2012, 354, 2380. [Google Scholar]
- a Roşca D.-A.; Smith D. A.; Bochmann M. Chem. Commun. 2012, 48, 7247. [DOI] [PubMed] [Google Scholar]; b Smith D. A.; Roşca D.-A.; Bochmann M. Organometallics 2012, 31, 5998. [Google Scholar]; c Hofer M.; Gomez-Bengoa E.; Nevado C. Organometallics 2014, 33, 1328. [Google Scholar]
- a Mezailles N.; Ricard L.; Gagosz F. Org. Lett. 2005, 7, 4133. [DOI] [PubMed] [Google Scholar]; b Ricard L.; Gagosz F. Organometallics 2007, 26, 4704. [Google Scholar]
- a Gaillard S.; Slawin A. M. Z.; Nolan S. P. Chem. Commun. 2010, 46, 2742. [DOI] [PubMed] [Google Scholar]; b Patrick S. R.; Gomez-Suarez A.; Slawin A. M. Z.; Nolan S. P. Organometallics 2014, 33, 421. [Google Scholar]
- Shen K.; Fu Y.; Li J.-N.; Liu L.; Guo Q.-X. Tetrahedron 2007, 63, 1568. [Google Scholar]
- Lu P.; Boorman T. C.; Slawin A. M. Z.; Larossa I. J. Am. Chem. Soc. 2010, 132, 5580. [DOI] [PubMed] [Google Scholar]
- Churruca F.; SanMartin R.; Carril M.; Tellitu I.; Domínguez E. Tetrahedron 2004, 60, 2393. [Google Scholar]
- Nesmeyanov A. N.; Perevalova E. G.; Lemenovskii D. A.; Dyadchenko V. P.; Grandberg K. I. Izv. Akad. Nauk SSSR, Ser. Khim. 1974, 1661. [Google Scholar]
- Ito Y.; Inouye M.; Suginome M.; Murakami M. J. Organomet. Chem. 1988, 342, C41. [Google Scholar]
- The mono- and diauration of sulfones by lithiation followed by treatment with LAuCl has been reported:; a Kneuper H.-J.; Harms K.; Boche G. J. Organomet. Chem. 1989, 364, 275. [Google Scholar]; b Djordjevic B.; Porter K. A.; Nogai S.; Schier A.; Schmidbaur H. Organometallics 2003, 22, 5336. [Google Scholar]
- a Li L.; Shu C.; Zhou B.; Yu Y.-F.; Xiao X.-Y.; Ye L.-W. Chem. Sci. 2014, 5, 4057. [Google Scholar]; b Schulz J.; Jašíková L.; škríba A.; Roithová J. J. Am. Chem. Soc. 2014, 136, 11513.and references cited therein. [DOI] [PubMed] [Google Scholar]
- Celik M. A.; Dash C.; Adiraju V. A. K.; Das A.; Yousufuddin M.; Frenking G.; Dias H. V. R. Inorg. Chem. 2013, 52, 729.and references cited therein. [DOI] [PubMed] [Google Scholar]
- Bagryanskaya I. Y.; Gatilov Y. V.; Maksimov A. M.; Platonov V. E.; Zibarev A. V. J. Fluor. Chem. 2005, 126, 1281. [Google Scholar]
- Savjani N.; Roşca D.-A.; Schormann M.; Bochmann M. Angew. Chem., Int. Ed. 2013, 52, 874. [DOI] [PubMed] [Google Scholar]; See also:; Langseth E.; Nova A.; Tråseth E. A.; Rise F.; Øien S.; Heyn R. H.; Tilset M. J. Am. Chem. Soc. 2014, 136, 10104. [DOI] [PubMed] [Google Scholar]
- Raubenheimer H. G.; Olivier P. J.; Lindeque L.; Desmet M.; Hrušak J.; Kruger G. J. J. Organomet. Chem. 1997, 544, 91. [Google Scholar]
- a De Frémont P.; Singh R.; Stevens E. D.; Petersen J. L.; Nolan S. P. Organometallics 2007, 26, 1376. [Google Scholar]; b Gaillard S.; Slawin A. M. Z.; Bonura A. T.; Stevens E. D.; Nolan S. P. Organometallics 2010, 29, 394. [Google Scholar]
- Dit Dominique F. J. B.; Gormitzka H.; Sournia-Saquet A.; Hemmert C. Dalton Trans. 2009, 340. [DOI] [PubMed] [Google Scholar]
- Pažický M.; Loos A.; Ferreira M. J.; Serra D.; Vinokurov N.; Rominger F.; Jäkel C.; Hashmi A. S. K.; Limbach M. Organometallics 2010, 29, 4448. [Google Scholar]
- a Jothibasu R.; Huynh H. V.; Koh L. L. J. Organomet. Chem. 2008, 693, 374. [Google Scholar]; b Huynh H. V.; Guo S.; Wu W. Organometallics 2013, 32, 4591. [Google Scholar]
- a Hirtenlehner C.; Krims C.; Hölbling J.; List M.; Zabel M.; Fleck M.; Berger R. J. F.; Schoefberger W.; Monkowius U. Dalton Trans. 2011, 40, 9899. [DOI] [PubMed] [Google Scholar]; b Kriechbaum M.; List M.; Berger R. J. F.; Platzschke M.; Monkowius U. Chem. Eur. J. 2012, 18, 5506. [DOI] [PubMed] [Google Scholar]
- Orbisaglia S.; Jaques B.; Braunstein P.; Hueber D.; Pale P.; Blanc A.; de Frémont P. Organometallics 2013, 32, 4153. [Google Scholar]
- Canovese L.; Visentin F.; Levi C.; Santo C. Inorg. Chim. Acta 2012, 391, 141. [Google Scholar]
- Teets T. S.; Nocera D. G. J. Am. Chem. Soc. 2009, 131, 7411. [DOI] [PubMed] [Google Scholar]
- Schneider D.; Schuster O.; Schmidbaur H. Organometallics 2005, 24, 3547. [Google Scholar]
- Schneider D.; Schier A.; Schmidbaur H. Dalton Trans. 2004, 1995. [DOI] [PubMed] [Google Scholar]
- Baron M.; Tubaro C.; Basato M.; Natile M. M.; Graiff C. J. Organomet. Chem. 2013, 723, 108. [Google Scholar]
- a Castro-Castro L. M.; Guloy A. M. Inorg. Chem. 2004, 43, 4537. [DOI] [PubMed] [Google Scholar]; b Svensson P. H.; Rosdahl J.; Kloo L. Chem. Eur. J. 1999, 5, 305. [Google Scholar]
- Jazzar R.; Dewhurst R. D.; Bourg J.-B.; Donnadieu B.; Canac Y.; Bertrand G. Angew. Chem., Int. Ed. 2007, 46, 2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Programs CrysAlisPro; Oxford Diffraction Ltd., Abingdon, U.K., 2010.
- Spek A. L. Acta Crystallogr., Sect. D 2009, D65, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Sluis P.; Spek A. L. Acta Crystallogr., Sect. A 1990, A46, 194. [Google Scholar]
- Sheldrick G. M. SHELX-97 and SHELX-2014-Programs for crystal structure determination (SHELXS) and refinement (SHELXL). Acta Crystallogr., Sect. A 2008, A64, 112.18156677 [Google Scholar]
- Coles S. J.; Gale P. Chem. Sci. 2012, 3, 683. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.