

Abstract

Allylic alcohols can be transformed into γ,δ-unsaturated α,α-dibromo esters through a two-step process: formation of a bromal-derived mixed acetal, followed by tandem dehydrobromination/Claisen rearrangement. The scope and selectivity of both steps have been investigated. The product α,α-dibromo esters were subjected to various carbon–carbon bond-forming reactions, oxidations, and lactonizations.
α,α-Dibromo esters are a class of halogenated esters that have not been studied extensively in the field of synthetic organic chemistry. With the exception of the works of Shindo, who used α,α-dibromo esters as precursors to reactive ynolate species, few reports exist regarding their preparation or application.1 Shindo has demonstrated that they can be formed through twostep bromination processes from either a corresponding acid halide or a corresponding ester (Scheme 1). The first of these processes (method A) involves use of a reactive acid halide as the starting material, as well as the harsh conditions of the Hell–Volhard–Zelinsky reaction to install the first α-bromide.2 The second (method B) requires the repeated use of the brominating reagent 1,2-dibromotetrachloroethane. Neither method is suitable for the preparation of relatively complex α,α-dibromo esters, especially those having additional enolizable hydrogen atoms.
Scheme 1. Preparation of α,α-Dibromo Esters.

During our synthetic endeavor toward the indole alkaloid reserpine, a robust method to form a γ,δ-unsaturated α,α-dibromo ester was required.3 This compound was obtained through the reaction sequence displayed in Scheme 2. The sequence begins with the addition of an allylic alcohol into the chlorinated ether 2, facilitated by halophilic silver(I) salts. The resulting mixed acetal 3 then transforms through dehydrobromination into a dibromoketene acetal 4. This reactive intermediate readily undergoes Claisen rearrangement at −78 °C to generate a γ,δ-unsaturated α,α-dibromo ester 5. Although a few halogenated allyl vinyl ethers have been employed previously in Claisen rearrangements, to the best of our knowledge none of these transformations has used such a dibromoketene acetal intermediate.4 The halogen substituent of the vinyl group typically allows the reaction to occur at lower temperature, a rate enhancement that has been described and studied by Carpenter.5
Scheme 2. Dibromoketene Acetal Claisen Rearrangement.

While working on reserpine, we were intrigued by the high efficiency of this unprecedented dibromoketene acetal rearrangement. The reaction sequence was robust, suitable for gram-scale syntheses in a complex total synthesis setting, and required simple starting materials. There are many methods available for the preparation of allylic alcohols 1, and the chlorinated tribromoethyl ether 2 is operationally simple to synthesize on a large scale.6 Consequently, we wished to investigate the generality of this method and explore the potential synthetic applications of the α,α-dibromo esters.
First, a general method was needed to convert allylic alcohols into mixed acetals. Reaction conditions were screened for the transformation of 2-methylprop-2-enol (1a), a simple allylic alcohol, into the mixed acetal 3a (Table 1). Among tested solvents, the use of toluene at −15 °C was optimal for acetal formation (entries 1–4). After screening a number of silver(I) salts, silver(I) triflate (AgOTf) was found to facilitate the transformation in useful yields (entries 4–7). Next, the choice of base was considered. To generate the mixed acetals in a mild manner, the conditions in entries 1–7 featured 4 Å molecular sieves as the basic medium, but this approach proved problematic.7 While the mixed acetals did form, the reaction conditions were less suitable for larger-scale reactions and led to decomposition to the bromal, presumably through acid-catalyzed hydrolysis of the desired mixed acetal. To circumvent this problem, a variety of bases were screened (entries 8–10), with silver(I) carbonate (Ag2CO3) being optimal to afford the desired mixed acetal. The use of a more-dissociative counterion, namely hexafluoroantimonate (SbF6−), led to complete decomposition of the starting material (entries 11 and 12).
Table 1.
Optimization of Mixed Acetal Formation

| |||||
|---|---|---|---|---|---|
| entry | silver salta | temp (°C) | basee | solvent | yield (%) |
| 1 | AgOTf | rt | MSa | CH2Cl2 | 50 |
| 2 | AgOTf | −40 | MSa | CH2Cl2 | 67 |
| 3 | AgOTf | −78 | MSa | CH2Cl2 | NRd |
| 4 | AgOTf | −15 | MSa | toluene | 0−84 |
| 5 | Ag2O | −15 | MSa | toluene | 0 |
| 6 | AgNO3 e | −15 | MSa | toluene | 31 |
| 7 | AgCOCF3 | −15 | MSa | toluene | 12 |
| 8 | AgOTf | −15 | K2CO3 | toluene | 0 |
| 9 | AgOTf | −15 | NaHCO2 | toluene | 0 |
| 10 | AgOTf | −15 | Ag2CO3 | toluene | 71 |
| 11 | AgSbF6 | −15 | Ag2CO3 | toluene | 0 |
| 12 | AgSbF6 | −78 | Ag2CO3 | toluene | 0 |
1.3 equiv of silver salt used, unless specified otherwise.
2 equiv of base used.
4 Å molecular sieves (4 wt equiv).
Starting material recovered.
2 equiv of silver salt used.
With suitable conditions determined for the conversion of allylic alcohols 1 into mixed acetals 3, the reaction’s generality was investigated (Scheme 3). The syntheses of compounds 3b–f revealed that acyclic allylic alcohols can transform into mixed acetals in good to excellent yields. Varying the degree of olefin substitution (cf. 3b, 3c, and 3e) did not affect the yield of mixed acetal formation. Cyclic allylic alcohols 1g–p also formed their mixed acetals in good to excellent yields. Even the quite complex tetrahydropyridine derivative8 1p generated its desired acetal in high yield. The cyclohexyl and cyclohexenyl alcohols 1k, 1n, and 1o formed their desired compounds efficiently and in good yields. The reaction tolerated the presence of silyl ether (3d) and sulfonamide (3p) units, as well as a variety of nonparticipating olefinic bonds.
Scheme 3. Mixed Acetal Formation.

aReactions of secondary alcohols were run at 0 °C. bCompounds isolated as a mixture of diastereoisomers.
With a variety of mixed acetals at hand, the Claisen rearrangements were investigated (Scheme 4). The presence of potassium tert-butoxide (t-BuOK) and [18]crown-6 in THF at −78 °C was optimal for the dehydrobromination to form the ketene acetal. (2,2,6,6-Tetramethylpiperidin-1-yl)oxy radical (TEMPO) was added to this reaction mixture as a radical scavenger to avoid radical-mediated debromination of the starting material. These conditions proved effective for the desired dehydrobromination/rearrangement, allowing the formation of a wide array of γ,δ-unsaturated α,α-dibromo esters (Scheme 4).
Scheme 4. Rearrangement of Mixed Acetals.
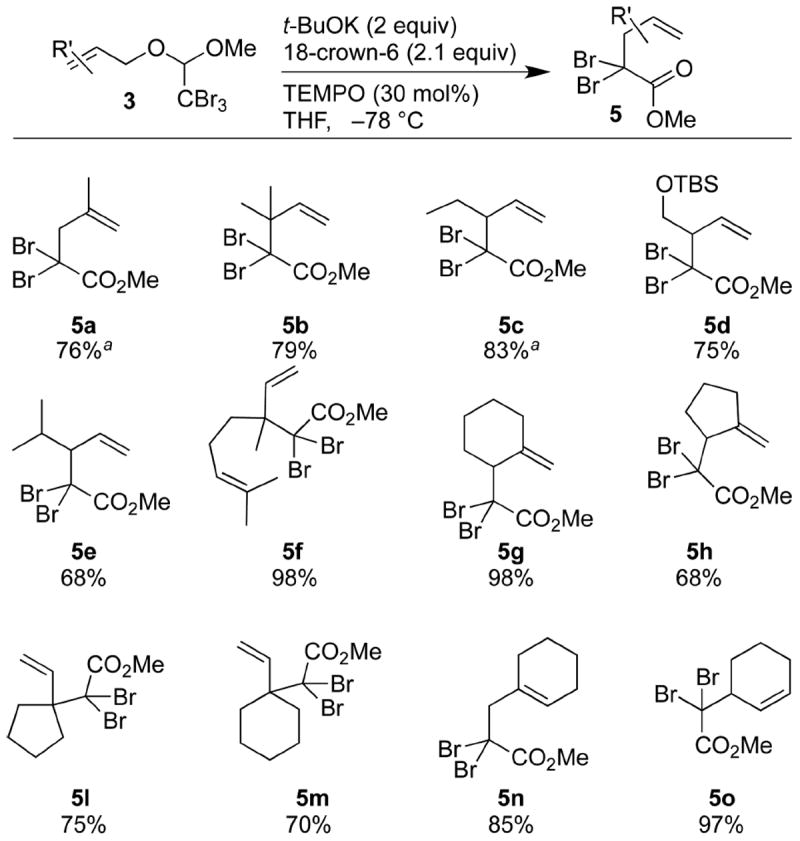
aRearrangements were performed at −90 °C.
The syntheses of compounds 5a–c and 5e illustrate the fact that increasing the steric bulk of the olefin substituent in the acetals 3a–c and 3e did not greatly impact the yield of the Claisen rearrangement. For the less-hindered acetals 3a and 3c, the rearrangements could be performed at −90 °C to increase the yields of the resulting α,α-dibromo esters 5a and 5c, respectively. While the yield of the isopropyl-substituted dibromo ester 5e was relatively moderate, the conversion was still rapid at low temperature. Hindered quaternary centers were also formed in high yields (cf. 5b, 5f, 5l, 5m); the bond-forming events occurred efficiently despite steric crowding at the dibromomethylene unit or the newly formed quaternary carbon atom. Ring-bearing acetals 3g–p rearranged to form γ,δ-unsaturated α,α-dibromo esters having various substitution patterns. Products featuring exocyclic methylene groups (5g ,5h), vinyl-substituted carbocycles (5l, 5m), and endocyclic olefinic units (5n, 5o) were formed in good to excellent yields. Mixed acetals containing silyl ether (3d) and sulfonamide (3p) moieties were transformed smoothly to their γ,δ-unsaturated α,α-dibromo esters (5d and 5p, respectively) in high yields.
The diastereoselectivity of this reaction was variable (Scheme 5). The dibromo ester 5i was not formed in a highly diastereoselective manner. When the acetal 3j was subjected to the rearrangement conditions, despite its tert-butyl group locking the cyclohexene in the half-chair conformation, the diastereoselectivity did not improve. Even decreasing the temperature to −90 °C did not greatly affect the diastereoselectivity of the rearrangement. There are two potential causes of this poor selectivity. First, there may be some steric congestion of the typically favored equatorial attack in the Claisen transition state, due to the bromo substituents on the terminal vinyl carbon.9 A similar selectivity issue was observed by Ireland in an analogous rearrangement of the alcohol 1j when a large substituent was introduced at the terminal carbon.9b Second, the high reactivity of the dibromoketene acetal would lead to poor differentiation between axial and equatorial attack.
Scheme 5. Diastereoselectivity of Rearrangement.
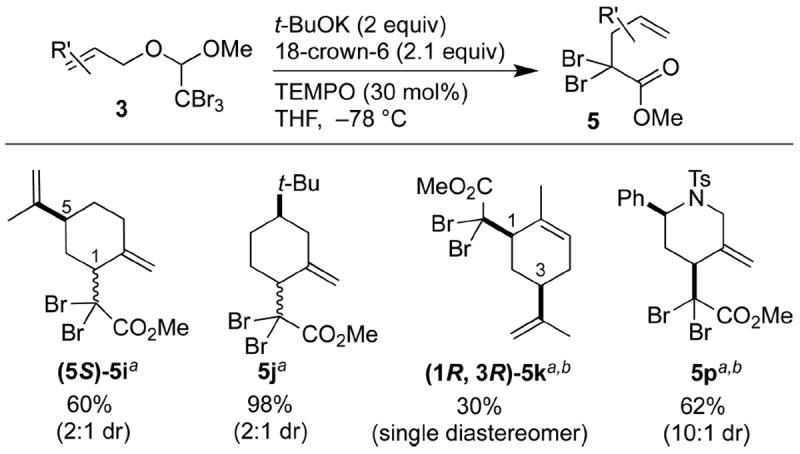
aAll diastereoisomeric ratios determined using 1H NMR spectroscopy.
bRelative stereochemistry determined through NOE experiments.
The carvone-derived α,α-dibromo ester 5k formed as a single diastereoisomer, albeit in modest yield. Another promising result was observed with the tetrahydropyridine-containing dibromo ester 5p. The rearranged product was obtained with 10:1 dr. Interestingly, these diastereoisomers formed preferably from equatorial attack, resulting in the syn product. This result can be rationalized by considering that the axial pathway of the tetrahydropyridine ring was blocked by the bulky tosyl group.
The formation of dibromo esters in this manner avoids the regioselectivity issues that would plague traditional methods based on enolate chemistry. Scheme 6 illustrates this concept for the formation of the dibromo ester 5q. The nucleophilic α-, γ-, and α′-positions of the enone 1q would have prevented the clean formation of the dibromo ester when using the conventional double-bromination sequence. In contrast, after (+)-10-hydroxycarvone 1q had been converted to the mixed acetal 3q through the chemoselective methods described herein, the α,α-dibromo ester 5q was formed cleanly upon rearrangement without complications.
Scheme 6. Synthesis of an Enone-Containing α,α-Dibromo Ester.

With a large assortment of α,α-dibromo esters available, the utility of these versatile compounds was explored (Scheme 7). As stated previously, α,α-dibromo esters have been used by Shindo in the formation of ynolates.1b This methodology is useful for the formation of vinyl ethers,10 various heterocycles,11 and tetrasubstituted olefins.12 Treating the dibromo ester 5g with 4 equiv of tert-butyllithium (t-BuLi) generated the ynolate 6. The olefination product 7 formed after the addition of acetophenone at ambient temperature. This tetrasubstituted unsaturated acid was obtained in good yield and with good E-to-Z selectivity. Notably, this transformation is the first example of ynolate formation in the presence of an alkene.
Scheme 7. Synthetic Applications of α,α-Dibromo Esters.
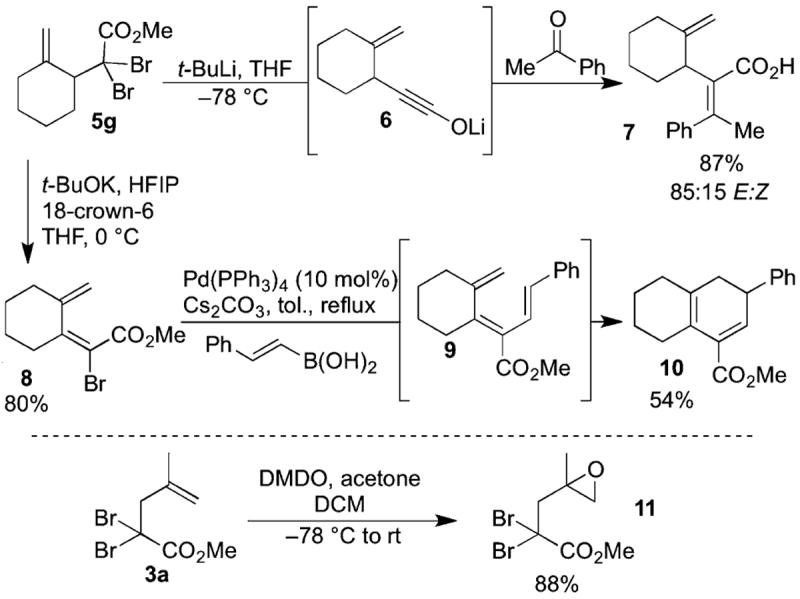
Vinyl bromides, which have many applications in crosscoupling chemistry, are also readily accessed from dibromo esters. Treatment of 5g with potassium hexafluoroisopropoxide, generated from t-BuOK and hexafluoroisopropyl alcohol (HFIP) in the presence of [18]crown-6, smoothly produced the vinyl bromide 8, which was then used to form the functionalized decalin system 10 through triene 9 in a tandem Suzuki/6π-electrocyclization.13,14 This reaction sequence might potentially be a general method for the synthesis of highly functionalized cyclohexadienes.
Both functional handles of the products of the dibromoketene acetal Claisen rearrangement can used as synthetic leverage to afford unique and synthetically important building blocks. For example, treatment of the Claisen product 3a with dimethyldioxirane (DMDO) produced the corresponding epoxide 11, which could be further functionalized.
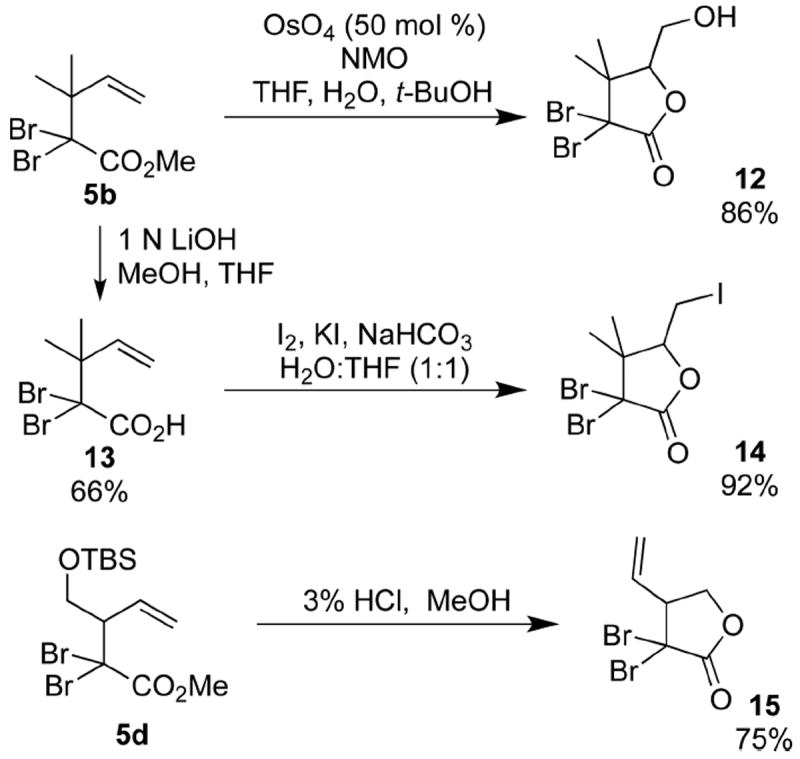
The alkene and ester units can also work in concert to form functionalized α,α-dibromobutyrolactones. Halogenated butyrolactones have gained attention for their potential as both biologically active compounds15 and as unique synthetic building blocks.16 Subjecting the α,α-dibromo ester 5b to catalytic dihydroxylation produced the dibromolactone 12 in a single step (Scheme 8).17 Compound 5b was also be converted to the carboxylic acid 13, which then underwent iodolactonization to form the functionalized lactone 14. Mild deprotection of the silyl ether unit of the α,α-dibromo ester 5d led to the vinyl-substituted lactone 15.
Scheme 8. Formation of α,α-Dibromo-γ-butyrolactones.
In summary, the Claisen rearrangements of dibromoketene acetals have been investigated. Both cyclic and acyclic allylic alcohols can be transformed into α,α-dibromo esters in a twostep process. The sequence is compatible with a variety of functionalities and can produce previously inaccessible dibromo esters. The α,α-dibromo esters produced are useful synthetic building blocks and can be used in the context of complex syntheses.
Supplementary Material
Acknowledgments
This study was funded by the NIH (R01GM071779) and the NSF Equipment Grant (CHE-1048804).
Footnotes
Supporting Information
Experimental procedures, characterization data, and 1H and 13C NMR spectra for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.(a) Shindo M, Matsumoto K, Shishindo K. Org Synth. 2007;84:11–22. [Google Scholar]; (b) Shindo M. Synthesis. 2003;15:2275–2288. [Google Scholar]; (c) Shindo M, Sato Y, Koretsune R, Yoshikawa T, Matsumoto K, Itoh K, Shishido K. Chem Pharm Bull. 2003;51:477–478. doi: 10.1248/cpb.51.477. [DOI] [PubMed] [Google Scholar]
- 2.(a) Hell C. Ber Dtsch Chem Ges. 1881;14:891–893. [Google Scholar]; (b) Volhard J. Liebigs Ann Chem. 1887;242:141–163. [Google Scholar]; (c) Zelinsky N. Ber Dtsch Chem Ges. 1887;20:2026–2026. [Google Scholar]
- 3.Barcan GA, Patel A, Houk KN, Kwon O. Org Lett. 2012;14:5388–5391. doi: 10.1021/ol302265z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Metcalf BW, Jarvi ET, Burkhart JP. Tetrahedron Lett. 1985;26:2861–2864. [Google Scholar]; (b) Welch JT, Samartino JS. J Org Chem. 1985;50:3663–3666. [Google Scholar]; (c) Yuan W, Berman RJ, Gelb MH. J Am Chem Soc. 1987;109:8071–8081. [Google Scholar]; (d) Christopher A, Brandes D, Kelly S, Minehan T. Org Lett. 2006;8:451–454. doi: 10.1021/ol052685j. [DOI] [PubMed] [Google Scholar]; (e) Teller F, Audoin M, Baudry M, Sauvêtre R. Eur J Org Chem. 2000;10:1933–1937. [Google Scholar]
- 5.Burrows CJ, Carpenter BK. J Am Chem Soc. 1981;103:6984–6986. [Google Scholar]
- 6.Neher F, Fleece CL. J Am Chem Soc. 1926;48:2416–2425. [Google Scholar]
- 7.Hithcock SA, Chu-Moyer MY, Boyer SH, Olson SH, Danishefsky SJ. J Am Chem Soc. 1995;117:5750–5756. [Google Scholar]
- 8.(a) Zhu X-F, Lan J, Kwon O. J Am Chem Soc. 2003;125:4716–4717. doi: 10.1021/ja0344009. [DOI] [PubMed] [Google Scholar]; (b) Wurz RP, Fu GC. J Am Chem Soc. 2005;127:12234–12235. doi: 10.1021/ja053277d. [DOI] [PubMed] [Google Scholar]; (c) Tran YS, Kwon O. J Am Chem Soc. 2007;129:12632–12633. doi: 10.1021/ja0752181. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kui L, Kwon O. Org Synth. 2009;86:212–225. doi: 10.1002/0471264229.os086.21. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Xiao H, Chai Z, Wang H-F, Wang X-W, Cao D-D, Liu W, Lu Y-P, Yang Y-Q, Zhao G. Chem — Eur J. 2011;17:10562–10565. doi: 10.1002/chem.201100850. [DOI] [PubMed] [Google Scholar]; (f) Tran YS, Martin TJ, Kwon O. Chem — Asian J. 2011;6:2101–2106. doi: 10.1002/asia.201100190. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zhong F, Han X, Wang Y, Lu Y. Chem Sci. 2012;3:1231–1234. [Google Scholar]; (h) Xiao H, Chai Z, Cao D, Wang H, Chen J, Zhao G. Org Biomol Chem. 2012;10:3195–3201. doi: 10.1039/c2ob25295c. [DOI] [PubMed] [Google Scholar]
- 9.(a) Ziegler FE. Acc Chem Res. 1977;10:227–232. [Google Scholar]; (b) Ireland RE, Varney MD. J Org Chem. 1983;48:1829–1833. [Google Scholar]
- 10.Shindo M, Kita T, Kumagai T, Matsumoto K, Shishido K. J Am Chem Soc. 2006;128:1062–1063. doi: 10.1021/ja0561082. [DOI] [PubMed] [Google Scholar]
- (11).Shindo M, Yoshimura Y, Hayashi M, Soejima H, Yoshikawa T, Matsumoto K, Shishido K. Org Lett. 2007;9:1963–1966. doi: 10.1021/ol0705200. [DOI] [PubMed] [Google Scholar]
- 12.Shindo M, Sato Y, Yoshikawa T, Koretsune R, Shishido K. J Org Chem. 2004;69:3912–3916. doi: 10.1021/jo0497813. [DOI] [PubMed] [Google Scholar]
- 13.Patel A, Barcan GA, Kwon O, Houk KN. J Am Chem Soc. 2013;135:4878–4883. doi: 10.1021/ja400882y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The stereochemistry of the E-vinyl bromide 8 has been established by NOESY. See ref 3 for similar isomerization during a tandem Suzuki/6π-electrocyclization.
- 15.(a) Ge P, Kirk KL. J Org Chem. 1997;62:3340–3343. doi: 10.1021/jo962394b. [DOI] [PubMed] [Google Scholar]; (b) Freer AA, Kirby WG, Rao GV, Cain RB. J Chem Soc Perkin Trans. 1996;1:2011–2116. [Google Scholar]; (c) Shah BA, Kaur R, Gupta P, Kumar A, Sethi VK, Andotra SS, Singh J, Saxena AK, Taneja SC. Bioorg Med Chem Lett. 2009;19:4394–4398. doi: 10.1016/j.bmcl.2009.05.089. [DOI] [PubMed] [Google Scholar]; (d) Guo Y, Fan L, Wang J, Yang C, Qu H, Xu X. Tetrahedron. 2013;69:774–781. [Google Scholar]
- 16.(a) Movassaghi M, Jacobsen EN. J Am Chem Soc. 2002;124:2456–2457. doi: 10.1021/ja025604c. [DOI] [PubMed] [Google Scholar]; (b) Takano S, Nishizawa S, Akiyama M, Ogasawara K. Heterocycles. 1984;22:1779–1788. [Google Scholar]; (c) Oetterli RM, Prieto L, Spingler B, Zelder F. Org Lett. 2013;15:4630–4633. doi: 10.1021/ol401572j. [DOI] [PubMed] [Google Scholar]
- 17.Larsen CH, Ridgway BH, Shaw JT, Woerpel KA. J Am Chem Soc. 1999;121:12208–12209. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.