

Abstract
At least 170 million people are chronically infected with hepatitis C virus (HCV). Due to the narrow host range of HCV and restricted use of chimpanzees, there is currently no suitable animal model for HCV pathogenesis studies or the development of a HCV vaccine. To identify cellular determinants of interspecies transmission and establish a novel immunocompetent model system, we examined the ability of HCV to infect hepatocytes from a small non-human primate, the rhesus macaque (Macaca mulatta). We show that the rhesus orthologs of critical HCV entry factors support viral glycoprotein-dependent virion uptake. Primary hepatocytes from rhesus macaques are also permissive for HCV RNA replication and particle production, which is enhanced when antiviral signaling is suppressed. We demonstrate that this may be due to the diminished capacity of HCV to antagonize MAVS-dependent innate cellular defenses. To test the ability of HCV to establish persistent replication in vivo, we engrafted primary rhesus macaque hepatocytes into immunocompromised xenorecipients. Inoculation of resulting simian liver chimeric mice with either HCV genotype 1a or 2a resulted in HCV serum viremia for up to 10 weeks. Conclusion: Together, these data indicate that rhesus macaques may be a viable model for HCV and implicate host immunity as a potential species-specific barrier to HCV infection. We conclude that suppression of host immunity or further viral adaptation may allow robust HCV infection in rhesus macaques and creation of a new animal model for studies of HCV pathogenesis, lentivirus coinfection and vaccine development.
Keywords: Hepatitis C, hepatitis virus, animal model, host tropism
Introduction
Chronic hepatitis C virus (HCV) infection frequently causes liver disease including fibrosis, cirrhosis and hepatocellular carcinoma. In most patients, liver disease progresses slowly over several decades but co-morbidities, including obesity, alcohol consumption and co-infection with HIV frequently accelerate progression and exacerbate disease. The underlying mechanisms that lead to chronic HCV infection and ultimately end-stage liver disease requiring liver transplantation are incompletely understood. This is due, in part, to the lack of a suitable animal model for pathogenesis studies and the development of a HCV vaccine.
HCV has a narrow host range, infecting only humans and chimpanzees. The determinants dictating this restriction are poorly defined (1). Some species express dominant restriction factors that limit pathogen replication. As an example, HIV-1 infects humans and chimpanzees but not old world monkeys such as rhesus or cynomolgus monkeys (2). HIV-1 can enter cells of the old world monkeys but encounters a block in replication before reverse transcription (3). It has been shown that this barrier to replication is due in part to sequence variation in TRIM5 and TRIM5 genotyping can be used to predict levels of simian immunodeficiency viral replication (4, 5). Variations in the type or intensity of the antiviral response between hosts are also known to restrict the tropism of certain viruses, such as myxoma virus, which is only permissive in human and murine cells that have impaired interferon (IFN) responses (reviewed in (6)). Dominant, cell intrinsic restriction factors have not been described for limiting HCV infection in non-permissive species. However, nonhomologous dependency factors and/or differences in the magnitude and kinetics of antiviral innate responses between species may certainly affect HCV’s ability to establish productive infection. Indeed, blunting antiviral immunity via genetic disruption of IRF1, IRF7, IFN-αβR or STAT1 in transgenic mice expressing human CD81 and occludin (OCLN; required to render murine cells permissive for HCV entry) resulted in persistent HCV replication over several weeks (7). Viral adaptation has also been utilized to enhance infection of murine cells (8) and induced pluripotent stem cell (iPSC)-derived hepatic cells from pig-tailed macaques (9) suggesting that both host cell manipulation and selection of viral variants are viable strategies to overcome species barriers.
Elucidation of the barriers for the viral life-cycle in non-permissive primate species will help delineate the determinants and key pathways responsible for viral permissiveness. Towards an improved understanding of the cellular determinants of interspecies transmission and the establishment of a novel immunocompetent model system, we sought to adapt HCV to infect small, non-human primates, specifically rhesus macaques (Macaca mulatta). Rhesus macaques are more closely related to humans than rodents, can be bred in captivity, have sufficient research reagents, can be used for terminal experiments and are excellent animal models for other human viral diseases. In the present study, we performed a systematic evaluation of the HCV life-cycle in rhesus macaque cells to identify potential species-specific incompatibilities that would preclude HCV infection. We demonstrate that rhesus macaque hepatocytes are permissive for the entire HCV life-cycle, but that virus production is enhanced with pharmacological-mediated suppression of Janus kinase (Jak) signaling. These data correlate with our finding that the HCV NS3-4A protease is unable to antagonize endogenous MAVS in rhesus macaque cells – a well-documented mechanism of viral interferon antagonism in human hepatocytes infected with HCV. We extend these studies to immunocompromised mice engrafted with rhesus macaque hepatocytes and demonstrate that persistent infection is also achieved in vivo in the context of immunodeficiency.
Experimental procedures (see also supporting information)
Human Subjects and Animal Usage
All protocols involving human tissue were reviewed and exempted by the Rockefeller University Institutional Review Board. All procedures involving mice were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Rockefeller University and Princeton University Institutional Animal Care and Use Committees (protocols 12536 and 1930, respectively).
HCV generation
Construction of J6/JFH1 (10), Jc1 (11) with an intragenotypic break point at the C3 position, Jc1[p7nsGluc2a] (12) and J6/JFH1-clone2 (13) was described elsewhere. H77-JFH1 harboring cell culture adaptive mutations was provided by Apath, LLC. HCVcc stocks were generated in Huh-7.5.1 (14) or Huh-7.5 (15) and quality controlled as described previously (10). Serum containing the H77 genotype 1a isolate was obtained from infected chimpanzees (16).
Quantification of HCV RNA
Total RNA was isolated from mouse serum using the QIAamp Viral RNA kit (Qiagen) and the HCV genome copy number was quantified by one-step rtPCR using a Multicode-RTx HCV RNA kit (Luminex Corp.) and a Roche LightCycler 480, according to manufacturer’s instructions.
Cloning of rhesus macaque CD81, SCARB1, CLDN1 and OCLN
Total RNA from Macaca mulatta liver was reverse transcribed with 50ng random hexamer primers per 5μg RNA and the Superscript III enzyme (Invitrogen) according to the manufacturer’s instructions. MmCD81, MmSCARB1, MmCLDN1 and MmOCLN were amplified from the resulting cDNA with gene specific 5′- and 3′-oligonucleotides and TOPO cloned into pCR2.1 (Invitrogen). To obtain the complete 5′ sequence 5′ RACE was performed using Clontech Marathon kit.
Pseudoparticles
All pseudoparticles were generated as described previously (17). For construct design details please see the supporting information.
Antibodies and Inhibitors
The human anti-HCV E2 antibody (AR4A) and anti-HIV (b6) (18) were kindly provided by Mansun Law (The Scripps Research Institute). Mouse anti-human CD81 (clone JS-81) and mouse IgG1 isotype control antibodies were obtained from BD Pharmingen. The mouse anti-HAV antibody (clone K2-4F2) used for flow cytometry was kindly provided by Susan Emerson (NIH).
2’C methyl adenosine (2’CMA) was the gift of D. Olsen and S. Carroll (Merck Research Laboratories, West Point, PA) and was also obtained from Carbosynth Limited. Ruxolitinib, a pan-Janus kinase (JAK) inhibitor (19) was obtained from ChemieTek.
RT-PCR quantification of HCV entry factors
To quantify expression of human and rhesus macaque entry factors, total liver RNA was isolated from human adult or fetal hepatocytes or rhesus macaque adult hepatocytes using a RNeasy isolation kit (Qiagen, Valencia, CA). cDNA was synthesized from 0.5μg RNA using a SuperScript® VILO™ cDNA Synthesis Kit (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions using gene specific primers. Quantitative PCR was performed with a Roche LightCycler 480 using an Applied Biosystems SYBR Green PCR Master Mix (Warrington, UK) and the following primer pairs:
| Human Gene | Forward Primer | Reverse Primer |
| CD81 | TGTTCTTGAGCACTGAGGTGGTC | TGGTGGATGATGACGCCAAC |
| SCARB1 | CGGATTTGGCAGATGACAGG | GGGGGAGACTCTTCACACATTCTAC |
| CLDN1 | CACCTCATCGTCTTCCAAGCAC | TCCTGGGAGTGATAGCAATCTTTG |
| OCLN | CGGCAATGAAACAAAAGGCAG | GGCTATGGTTATGGCTATGGCTAC |
| Rhesus Gene | Forward Primer | Reverse Primer |
| CD81 | GCCAAGGATGTGAAGCAGTT | CCTTCTTGAGGAGGTTGCTG |
| SCARB1 | CGGATTTGGCAGATGACAGG | GGGGGAGACTCTTCACACATTCTAC |
| CLDN1 | CCGTTGGCATGAAGTGTATG | CCAAATTCGTACCTGGCATT |
| OCLN | AAGTGGTTCAGGAGCTTCCA | AGTCCTCCTCCAGCTCATCA |
Western blotting
Cells were lysed at the indicated times using modified RIPA buffer containing 50mM Tris-HCl pH 8.0, 1% v/v NP-40, 0.5% v/w Na-deoxycholate, 150mM NaCl, 0.1% SDS. Protein lysates (5 or 15μg) were separated on 4–12% Bis/Tris NuPage polyacrylamide gels (Invitrogen). Proteins were transferred to nitrocellulose membranes and entry factors were detected using mouse anti-human CD81 (clone JS-81; BD Pharmingen; 1:200), rabbit anti-human SCARB1 (clone EP1556Y; Abcam; 1:2000), mouse anti-human OCLN (clone OC-3F10; Invitrogen; 1:500) and mouse anti-CLDN1 (clone 2H10D10; Invitrogen; 1:400). For further western blot analyses, rabbit sera against human MAVS was kindly provided by Zhijian Chen (UT-Southwestern Medical Center; 1:500), mouse anti-HCV NS3 was obtained from Virostat (1:100), mouse anti-HAV VP1 from LifeSpan Biosciences, Inc. (1:500), and HRP-conjugated mouse anti-human β-actin from Sigma-Aldrich (1:15000). Following secondary antibody staining with Peroxidase-AffiniPure Donkey Anti-Mouse IgG (H+L) (Jackson Immuno Research; 1:10000) or goat anti-rabbit IgG (H+L), HRP conjugate (Pierce; 1:10000), western blots were visualized using SuperSignal West Dura, Pico or Femto (Thermo Scientific).
Generation of human and simian liver chimeric mice
FAH−/− NOD Rag1−/− IL2RNULL mice were generated and transplanted as previously described (20) Female FNRG mice greater than 6 weeks of age were transplanted with ca. 1 × 106 cryopreserved adult rhesus macaque or human hepatocytes.
Enzyme-linked immunosorbent assays (ELISAs)
Human hepatocyte engraftment was monitored by serial human albumin determination. Serum was obtained through tail vein bleeding and diluted for measurement by home made ELISAs using goat polyclonal capture and horseradish peroxidase (HRP)-conjugated goat anti-human albumin detection antibodies (Bethyl, Montgomery, TX).
Immunohistological analysis
Immunohistochemistry was performed on formalin-fixed, paraffin-embedded human and simian chimeric murine liver tissues using mouse anti-human (clone 2) fumarylacetoacetate hydrolase (FAH) (Abcam, Cambridge, MA, USA) as described previously (21).
Graphing and statistical analysis
Statistical analysis of virological data was performed with GraphPad Prism 5. Specific tests are noted in figure legends.
Results
Rhesus macaque entry factor orthologs support HCV uptake
The cellular tropism of a given virus is determined by virus interaction with cell type specific host factors and by the virus’ ability to avoid and/or antagonize cellular antiviral pathways. However, orthologs of essential host factors for pathogen propagation may be incompatible or entirely absent. To identify potential blocks against HCV infection in rhesus macaque cells, we expressed the minimal required set of HCV entry factors (hCD81, hOCLN, hSCARB1 and hCLDN1) in LLC-MK2 or FRhk4 rhesus macaque cell lines and determined that these cells were competent for HCVpp entry, indicating that dominant negative or otherwise inhibitory factors are not present that would preclude HCV particle uptake (Fig. 1a, b). We have previously shown that species-specific differences in the entry factors CD81 and OCLN can limit infection (17). We have validated our initial in vitro observations and demonstrated that expression of human CD81 and OCLN can allow for viral uptake in mice (22). OCLN, SCARB1, CD81 and CLDN1 are expressed in rhesus macaque liver tissue (Fig. S1) and all elements of CLDN1 known to be critical for HCV uptake, in particular residues I32 and E48 within the first extracellular loop of CLDN1 (23) are conserved between species (Fig. S2). Consequently, rhesus CLDN1 can facilitate HCV entry as efficiently as human CLDN1 into 293T cells, a cell line lacking endogenous CLDN1 expression (Fig. 1c). While differences exist in amino acid sequence between human and rhesus OCLN, SCARB1 and CD81, rhesus macaque entry factor orthologs were able to rescue HCV uptake in human cells lacking endogenous expression of OCLN, SCARB1 or CD81 demonstrating that they are functionally competent for HCV entry (Fig. 1d–f, Fig. S3). To assess entry directly, we generated cultures of primary rhesus macaque hepatocytes (PRMH; Fig. 2c) and observed efficient HCV infection that was viral glycoprotein-dependent and required CD81. Pre-incubation with anti-E2 or anti-CD81 antibody resulted in up to 80% or 55% loss of infectivity, respectively (Fig. 1g, h).
Figure 1. Rhesus macaque hepatocytes support HCV uptake.
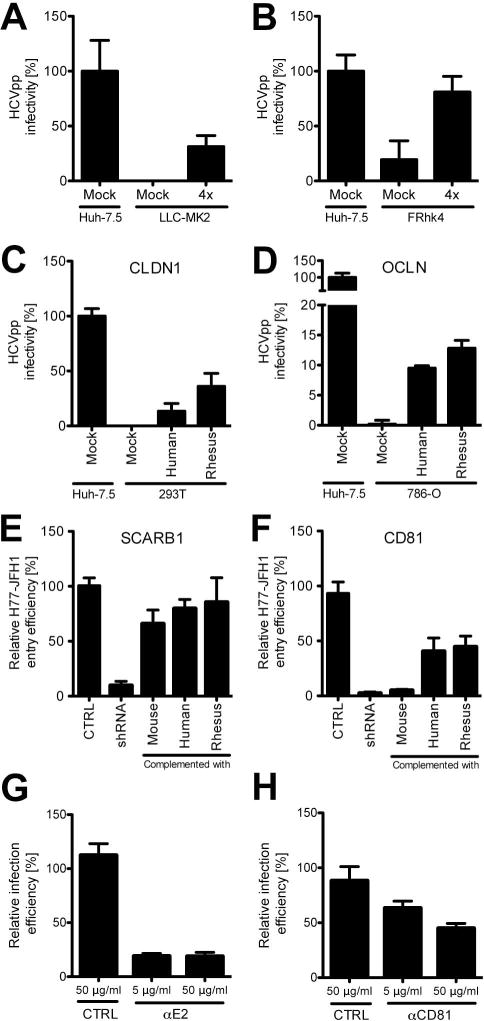
(A) Entry efficiency of HCVpp–H77 (gt1a) in LLC-MK2 or (B) FRhk4 rhesus macaque cells expressing human entry factors (hSRB1, hCD81, hCLDN and hOCLN; denoted as “4x” in the figure). HCVpp-mediated GFP expression was measured 72 hours post-transduction. The relative HCVpp infectivity after Env- subtraction and VSV-G normalization is shown. Huh-7.5 entry was set to 100%. (C) Entry efficiency of HCVpp-H77 into human cell lines expressing three human entry factors plus human or rhesus macaque CLDN1 or (D) OCLN. (E) H77-JFH1 infection of Huh-7.5 cells knocked down for endogenous SCARB1 or (F) CD81 and transduced with mouse, human or rhesus macaque SCARB1 or CD81, respectively. Entry efficiency is defined by the percentage of NS5A antigen positive cells in total live cells, normalized to cells containing control shRNA. (G) HCV (Jc1[p7nsGluc2A]) infection of primary rhesus macaque hepatocytes following pre-incubation of HCV with anti-E2 (AR4A; or IgG control (b6 anti-HIV)) or (H) cells with anti-CD81 (JS81) or IgG control. Viral infection was measured D2pi by luciferase quantification in the culture medium. Infection efficiency is defined as luciferase units normalized to IgG controls. Data shown in all panels is the mean and standard deviation (SD) of biological triplicates from a single experiment. Similar data were obtained in replicate experiments.
Figure 2. HCV replication in primary rhesus macaque hepatocytes is enhanced by suppression of the innate immune response.
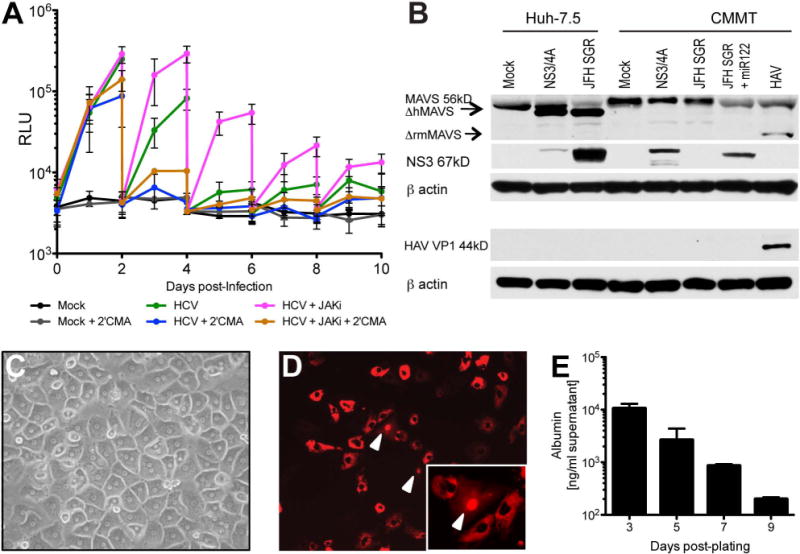
(A) Luciferase secreted into the culture medium as a measure of HCV replication in primary adult rhesus macaque hepatocytes. PRMH were infected with Jc1[p7nsGluc2a] 3 days post-plating and medium (+/− 2μM Jak inhibitor (INCB018424) and/or 5μM 2’CMA) was changed every other day. (B) Western blot analysis of naïve, NS3-4A-expressing, JFH1 subgenomic replicon-expressing or hepatitis A virus-infected Huh-7.5 or CMMT cell lysates detecting MAVS, HCV NS3, HAV VP1 and beta actin. (C) Photomicrograph of PRMH cultures 1 day post-plating. (D) Visualization of HCV infection in PRMH. PRMH were transduced with lentivirus expressing RFP-NLS-IPS1 and infected with HCVcc (J6/JFH1-clone 2) two days later. Cultures were imaged 3 days post-HCVcc infection by fluorescence microscopy. Representative pseudocolored florescence image is shown. White arrows indicate nuclear RFP, indicative of HCV replication. (E) Albumin production in PRMH quantified by ELISA. Data shown in all panels is the mean and standard deviation (SD) of biological triplicates from a single experiment. Similar data were obtained in replicate experiments.
Primary rhesus macaque hepatocytes are permissive for HCV replication
Given productive entry, we next tested PRMH for their ability to support HCV RNA replication. Previous reports suggested that several other non-human primate species, including cynomolgus, rhesus, Japanese and African green monkeys, as well as Chacma and doguera baboons, were resistant to HCV infection (24, 25). In contrast, more recent work suggests that hepatic cells derived from induced pluripotent stem cells (iPSCs) of pig-tailed macaques can support the entire HCV life-cycle (9). To assay HCV replication in PRMH, we utilized a highly sensitive HCVcc reporter virus expressing secreted Gaussia luciferase (Gluc)(Jc1[p7nsGluc2a]). Accumulation of luciferase in the medium was observed in HCV-infected cultures, but not mock-infected cultures or HCV-infected cultures treated with the NS5B viral polymerase inhibitor 2’CMA demonstrating that PRMH can support HCV RNA replication (Fig. 2a). Replication was transient, however, likely due in part to poor maintenance of hepatocyte phenotype/function as indicated by a loss of albumin production (Fig. 2e). To further characterize HCV infection in PRMH, we took advantage of an engineered fluorescence-based live cell reporter in which HCV serine protease NS3-4A-mediated cleavage of the C-terminal mitochondrial targeting domain of MAVS results in translocation of a red fluorescent protein (RFP) to the nucleus (13). Nuclear RFP signal was detected at low frequency following infection with a cell culture adapted variant of J6/JFH1 termed Clone 2 (13), confirming that HCV can infect and replicate in PRMH, but indicating HCV infection is inefficient in these cells (Fig. 2d).
HCV does not efficiently antagonize the innate immune response in rhesus macaque cells
In previous work, blunting antiviral innate defenses in mice expressing human CD81 and OCLN allowed completion of the entire HCV life-cycle, exemplifying the considerable impact of innate immune control on viral infection (7). Similar to this finding, pharmacological-mediated suppression of antiviral signaling pathways via Janus kinase (Jak) inhibition in PRMH enhanced viral replication up to 8.8 fold and promoted persistent infection, suggesting HCV may be restricted by the host response to infection in rhesus macaque hepatocytes in vitro (Fig. 2a). However, use of this inhibitor has also been shown to boost replication in primary human fetal liver cultures, thus, it is unclear if this pathway constitutes a species-specific restriction. In human hepatocytes, HCV dampens the innate immune response by cleaving mitochondrial antiviral signaling protein (MAVS) from the outer mitochondrial membrane thereby preventing downstream signaling and activation of an antiviral program triggered by PAMP activation of RLRs (26). It was recently proposed that rhesus MAVS was resistant to HCV antagonism due to sequence variation near (aa506) the virus-targeted cleavage site (aa508) (27). Thus, we aimed to determine whether NS3-4A expression in rhesus cells would result in cleavage of endogenous monkey MAVS. Consistent with this previous report (27), we observed cleavage of MAVS in human hepatoma cells (Huh-7.5), but not in CMMT cells expressing NS3-4A (Fig. 2b). By comparison, hepatitis A virus (HAV) infection of CMMT cells (Fig. S4) yielded a MAVS cleavage product of lower molecular weight (aprox. 50kDa vs. 52kDa for HCV-mediated cleavage), demonstrating that virus mediated cleavage of MAVS was possible in these cells and corroborating data indicating different cleavage sites within MAVS for these two viruses (28). Together, our data suggest that HCV is unable to tightly control the innate immune response in rhesus cells likely due, in part, to the virus’ inability to efficiently inactivate MAVS.
Primary rhesus macaque hepatocytes produce infectious HCV particles
Completion of the viral life-cycle in PRMH was demonstrated by quantification of infectious particles released into the medium by limiting dilution assay on naïve Huh-7.5 cells, a human hepatoma cell line that is highly permissive to HCV infection, using supernatants collected on day 2 post-infection. As predicted by our luciferase data, titers, which achieved a mean 284 TCID50/ml in untreated cultures, were enhanced 3.6 fold in the presence of Jak inhibitor (Fig. 3). Even in the presence of Jak inhibitor, however, infectious virus titers produced by PRMH cultures were 13 to 147-fold less that those obtained in Huh-7.5 cells on day 2 and day 4 post-inoculation, respectively. These data were consistent with lower levels of replication in PRMH as compared to human hepatoma cells and were expected given the low viral yields described for other primary hepatocyte culture systems (Fig. S5)(29, 30).
Figure 3. The HCV lifecycle is complete in primary adult rhesus macaque hepatocyte cultures.
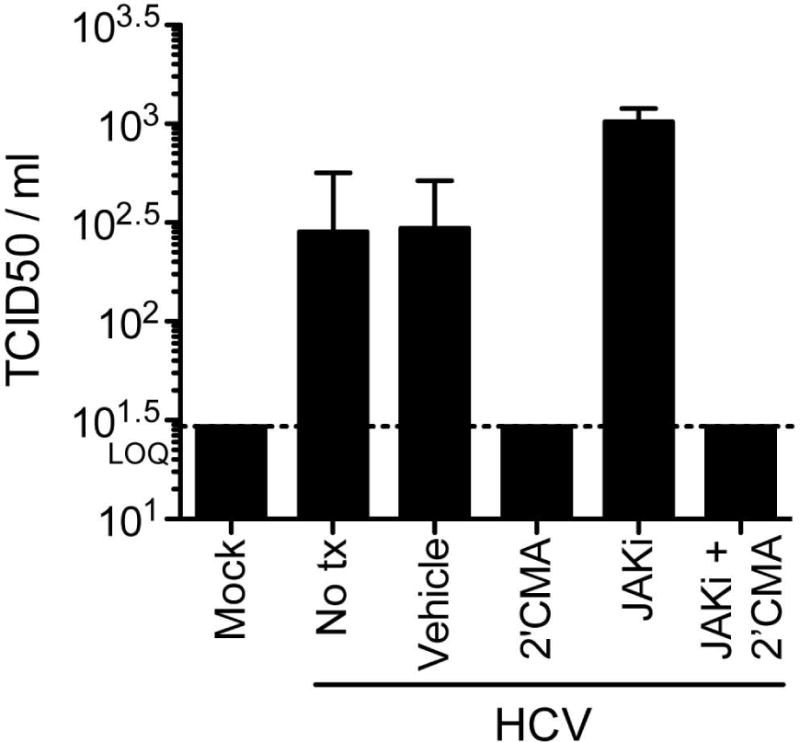
Infectious virus in supernatants from PRMH cultures two days post-infection with Jc1[p7nsGluc2a] quantified by limiting dilution assay on naïve Huh-7.5 cells. Data shown in all panels is the mean and standard deviation (SD) of biological triplicates from a single experiment. Similar data were obtained in replicate experiments. Lower limit of quantification is denoted as LOQ.
Persistent HCV replication in immunodeficient simian liver chimeric mice
To test whether HCV can establish persistent replication in vivo, we transplanted primary rhesus macaque hepatocytes into immunocompromised xenorecipients (FNRG) lacking fumaryl acetoacetate hydrolase (Fah)−/−. Engraftment and expansion of rhesus hepatocytes in these animals is mediated by withdrawal of 2-(2-nitro-4-trifluoro-methylbenzoyl)-1,3-cyclohexanedione (NTBC) that induces genetically determined toxicity in murine hepatocytes, allowing expansion of transplanted cells (13) (Fig. 4a). We monitored engraftment over time using an ELISA specific for human and rhesus macaque albumin (Fig. 4b). Successful integration of rhesus macaque hepatoyctes into the murine liver was also evident by immunohistological identification of FAH antigen positive cells in characteristic islands as described for mice repopulated with primary adult human hepatocytes (Fig. 4c) (21). Resulting simian liver chimeric mice (and human liver chimeric mice generated in parallel as controls) that achieved albumin levels of >1mg/ml were inoculated with serum derived from HCV genotype 1a (H77) or 2a (J6/JFH1) viremic human liver chimeric mice. Following viral challenge, HCV RNA was detected in the serum of all H77-challenged (n=13) and J6/JFH1-challenged (n=8) simianized mice. Infection of mouse cohort 1 was inefficient, achieving robust viremia only after reinoculation with HCV on day 54, and reaching maximal titers of 2.4×105 and 2×105 HCV RNA copies per ml for H77 and J6/JFH1, respectively. However, viremia was detected in a second cohort of simianized mice by 4 weeks post-inoculation. While viral load in this group fluctuated over the 102 day-long time course, H77 titers averaged 1.1×106 HCV RNA copies per ml after week 4 and reached a mean peak titer of 3.7×106 HCV RNA copies per ml sixty-six days after viral challenge. Similarly, J6/JFH1 infected simianized mice exhibited peak viremia at day 70 post-inoculation with 9.5×106 mean HCV RNA copies per ml although the virological set-point for this genome appeared to be closer to 1.2×105 HCV RNA copies per ml. HCV RNA was detected in the serum of these mice up to 10 weeks post-inoculation indicating that simianized mice are permissive for HCV infection and support persistent viral replication (Fig 4d, e).
Figure 4. HCV persistently replicates in simian liver chimeric mice.
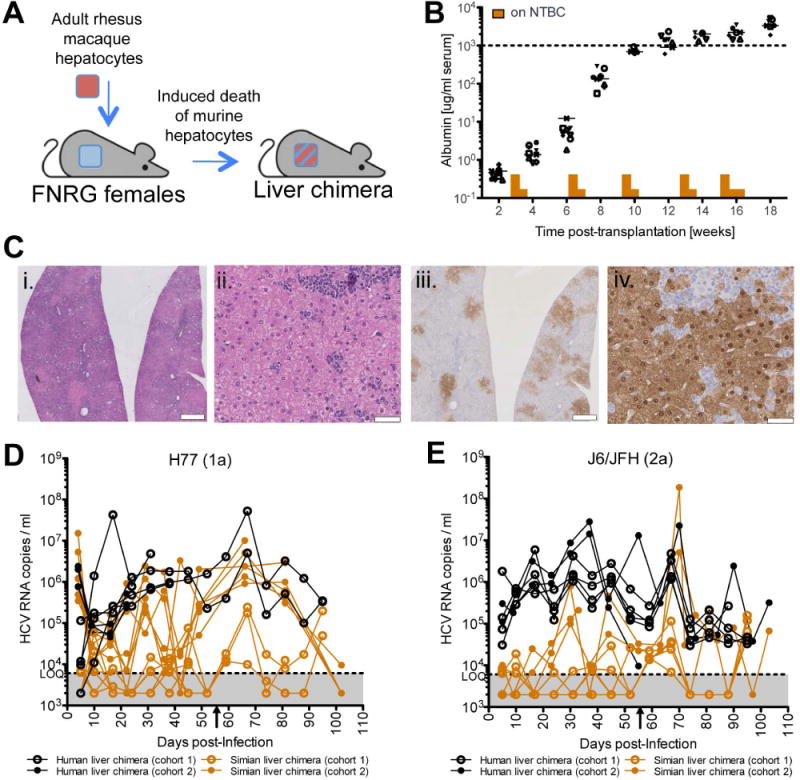
(A) Construction of simian liver chimeric mice. FNRG female mice were transplanted with 1×106 rhesus macaque adult hepatocytes and cycled on NTBC and water to promote engraftment. (B) Rhesus macaque serum albumin quantified by ELISA as a marker of hepatocyte engraftment. (C) Histological sections stained with hematoxylin and eosin (H&E; panels i and ii) or anti-FAH antibody (iii and iv) identify islands of rhesus macaque hepatocytes in the murine liver. Scale bars: 1mm (i. and iii.) and 50μm (ii. and iv.). (D) Hepatitis C viremia in simianized and humanized mice inoculated with H77 (gt1a) or (E) J6/JFH1 (gt2a) determined by qRT-PCR. Arrowhead on x-axis indicates the time point at which the simianized mice in cohort 1 were reinoculated with HCV.
Discussion
Our results demonstrate that the entire HCV life cycle can be recapitulated in PRMH and simian liver chimeric mice. This creates a strong rationale for future studies aiming at virological and immunological characterization of HCV infection in rhesus macaques. HCV infection in rhesus monkeys is an attractive model to study host response and pathogenesis because they are genetically more closely related to humans than rodents. In contrast to chimpanzees, they are more readily accessible and can be subjected to terminal experimentation. Given the susceptibility of rhesus macaques to SIV (31, 32) and simian-adapted HIV (33) mechanisms of HIV-exacerbated viral hepatitis could be modeled in co-infected animals. About one quarter of HIV-infected persons in the United States are also infected with HCV making it one of the most important co-morbidities. Co-infection with HIV frequently results in more exacerbated liver disease and more accelerated disease progression, a process that remains poorly understood. Based on these in vitro and in vivo data and prior failed attempts to detect infection in this species (24, 25), we hypothesize that suppression of both innate and adaptive immune responses in rhesus macaques may be necessary to allow efficient HCV infection, replication, and spread enabling the virus to further adapt to this non-human primate species.
MAVS (34), also known as IPS-1 (35), Cardif (26) or VISA (36), is an adaptor protein that relays signals from RIG-I-like helicases, which are cytoplasmic sensors of RNA virus infection that ultimately activate type I interferon and downstream signaling cascades. Cleavage of MAVS mediated by the HCV NS3-4A protease is thought to be an important mechanism by which HCV blunts innate immune activation in human hepatocytes (26). It was previously demonstrated that MAVS cleavage in the liver of patients with chronic hepatitis C correlates with reduced activation of the endogenous interferon system (37). Consistent with a previous report using artificial MAVS substrates (27) we demonstrate that the HCV NS3-4A protease, expressed ectopically in the context of the viral polyprotein, cannot cleave endogenous rhesus MAVS. HCV’s inability to effectively evade innate antiviral defenses may explain in part why suppression of type I interferon signaling increases permissiveness of rhesus macaque hepatocytes in vitro. Suppression of the innate immune response in human fetal liver cultures via addition of a pan-Jak inhibitor (38) or expression of paramyxovirus V proteins (30) also results in an enhancement of HCV replication in cells where HCV can efficiently cleave MAVS. This suggests that both primary human and rhesus hepatocytes cultured in vitro may exhibit an elevated baseline immune activation state that limits HCV replication. It should be noted that the NS3-4A protease has several other cellular targets (reviewed in (39)) including Toll-IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF) whose cleavage was also shown to impair cellular innate defenses (40). Furthermore, it has been shown that the viral protease NS3-4A efficiently blocks activation of IRF3, a transcription factor that is essential for IFN induction and direct activation of a subset of antiviral genes independent of IFN (41). However, it is unclear whether HCV is able to antagonize these and other – putatively unknown – antiviral mechanisms in rhesus macaque cells.
Interestingly, when engrafted into the parenchyma of liver-injury xenorecipients, rhesus macaque hepatocytes are susceptible to HCV infection and pharmacological suppression of innate responses was not necessary to establish chronicity. Homo- and heterotypic cellular interactions as well as soluble factors within the 3D microenvironment in vivo likely impact rhesus macaque hepatocyte physiology and improved maintenance of hepatocyte morphology and differentiation status is known to contribute to HCV persistence (29) while the absence of an adaptive immune response likely allows the virus to avoid clearance. Still, slower viral kinetics and lower HCV RNA copies per ml were observed in simianized as compared to humanized mice inoculated in parallel suggesting that virus replication is less efficient in rhesus hepatocytes. Thus, the failure to cleave MAVS and control early innate immune responses could still be limiting viral replication in vivo, although this was not directly assessed in this study. Future studies are aimed at identifying and characterizing putative adaptive mutations that may allow HCV to replicate more efficiently in rhesus macaque hepatocytes.
Recently, induced pluripotent stem cell-derived hepatic like cells from pig-tailed macaques (Macaca nemestrina) were shown to be permissive for HCV infection although viral entry was limited by low levels of OCLN and subpar activity of CD81. In our study, rhesus macaque entry factor orthologs were comparable to human entry factors in allowing HCV uptake suggesting that this step of the viral life-cycle would not be limiting in a rhesus macaque model. Immune modulation was not attempted in pig-tailed macaque cells and the sequence of Macaca nemestrina MAVS protein is not yet available, thus it is difficult to speculate whether HCV replication could benefit from immune suppression in this related species. Notably, the fusion of TRIM5 to cyclophilin A in pig-tailed macaques renders this monkey more susceptible to HIV-1 infection (42); however, this may conversely negatively impact HCV as CypA is an essential host factor for HCV replication.
The value of a small, non-human primate model for the study of HCV has prompted others to challenge rhesus macaques in the past, albeit unsuccessfully (24, 25). While the potential reasons for these failures are many, it is possible that a more robust HCV genome (or one that has been adapted to rhesus macaques) as compared to less well defined inocula, i.e. patient samples, and more sensitive detection methods available today will yield more favorable results. In sum, our data provide a proof-of-concept that rhesus macaque hepatocytes can support the entire HCV life-cycle and indicate that suppression of host immunity will be both necessary and sufficient to launch HCV replication in rhesus monkeys in vivo.
Supplementary Material
Acknowledgments
We thank Timothy Sheahan for generating the HAV stock, Sally Marik and Charles Gilbert (The Rockefeller University) for providing rhesus macaque liver tissue, Mansun Law (The Scripps Research Institute) for providing the AR4A and b6 antibodies, Sue Emerson (NIH) for providing the HAV encoding plasmid and HAV antibody as well as William Schneider for generating the Jc1[p7nsGluc2a] stock and the blasticidin-resistant miR122-expressing pTRIP vector. We also thank Mohsan Saeed for the HIV-1 gag-pol/delta neomycin pseudoparticle packaging plasmid. Lastly, we wish to acknowledge William Schneider and Ursula Andreo for helpful discussion as well as Santa Maria Pecoraro Di Vittorio, Joseph Palarca, Julia Sable, Mary Ellen Castillo, and Sonia Shirley for outstanding administrative and/or technical support.
Financial Support
This work was supported in part by National Institute for Diabetes, Digestive and Kidney Diseases (5 R01 AI090055-05 to A.P and C.M.R), National Institutes for Allergy and Infectious Diseases (2 R01 AI079031-05A1, 1 R01 AI107301-01, 1 R56 AI106005-01 to AP, 5R01 AI072613-08 to CMR) National Institutes of Health (NIH) through the NIH Roadmap for Medical Research Grant 1 R01 DK085713-01. Additional funding was provided by the Starr Foundation, the Greenberg Medical Research Institute, the Richard Salomon Family Foundation, the Ronald A. Shellow, M.D. Memorial Fund, the MGM Mirage Voice Foundation, Gregory F. Lloyd Memorial contributions, and anonymous donors. The IHC core laboratory at NYU Medical Center is funded in part by the NYU Cancer Institute. The NYU Cancer Center is supported in part by grant 5P30CA016087-32 from the National Cancer Institute. M.A.S. was supported by a National Research Service Award (F32 AI091207) from the National Institute of Allergy and Infectious Diseases. Y.P.J. was a recipient of an American Gastroenterological Association Research Scholar Award. A.P. is a recipient of the Liver Scholar Award from the American Liver Foundation. G.G. was supported by the Human Frontier Science Program (LT-000048-2009) and the German Academy of Science Leopoldina (LPDS 2009-9). M.v.S. is a recipient of a postdoctoral fellowship from the German Research Foundation. J.M.G is supported by co-funding from NIAID on iNRSA 5T32GM007388. The funding sources were not involved in the study design, collection, analysis, or interpretation of data or in the writing of the report.
List of Abbreviations
- HCV
Hepatitis C virus
- MAVS
mitochondrial antiviral signaling protein
- HIV
Human immunodeficiency virus
- TRIM5
tripartite motif containing 5
- IFN
Interferon
- IRF
Interferon response factor
- STAT
signal transducer of activated T cells
- iPSC
induced pluripotent stem cell
- Jak
Janus kinase
- OCLN
occludin
- SCARB1
scavenger receptor type B class I
- CLDN1
claudin 1
- HCVpp
HCV pseudoparticle
- PRMH
primary rhesus macaque hepatocytes
- Gluc
Gaussia luciferase
- 2’CMA
2’C methyl adenosine
- RFP
red fluorescent protein
- PAMP
pathogen associated molecular pattern
- RLR
RIG-I like helicase
- aa
amino acid
- HAV
hepatitis A virus
- kDa
kilo Dalton
- TCID
tissue culture infectious dose
- FNRG
FAH−/− NOD Rag1−/− IL2RγNULL
- FAH
fumaryl acetoacetate hydrolase
- Rag1
recombinase activating gene 1
- IL2RγNULL
Interleukin 2 receptor gamma chain
- NTBC
2-(2-nitro-4-trifluoro-methylbenzoyl)-1,3-cyclohexanedione
- ELISA
Enzyme linked immunosorbert assay
- SIV
simian immunodeficiency virus
- IPS-1
IFN-β promoter stimulator protein 1
- Cardif
CARD adaptor inducing IFN-β
- VISA
virus-induced signaling adaptor
- TRIF
Toll-IL-1 receptor domain-containing adaptor inducing IFN-β
- CypA
cyclophilin A
Footnotes
Conflict-of-interest disclosure
The authors declare the following conflicts of interest, which are managed under the Rockefeller University’s policy: C.M.R. has equity in Apath, LLC, which holds commercial licenses C.M.R. has equity in Apath, LLC, which holds commercial licenses for the Huh-7.5 cell line, HCV cell culture system, the use of OCLN to construct HCV animal models and the fluorescent cell-based reporter system to detect HCV infection.
References
- 1.Sandmann L, Ploss A. Barriers of hepatitis C virus interspecies transmission. Virology. 2013;435:70–80. doi: 10.1016/j.virol.2012.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakayama EE, Shioda T. TRIM5alpha and Species Tropism of HIV/SIV. Front Microbiol. 2012;3:13. doi: 10.3389/fmicb.2012.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 4.Newman RM, Hall L, Connole M, Chen GL, Sato S, Yuste E, Diehl W, et al. Balancing selection and the evolution of functional polymorphism in Old World monkey TRIM5alpha. Proc Natl Acad Sci U S A. 2006;103:19134–19139. doi: 10.1073/pnas.0605838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirmaier A, Wu F, Newman RM, Hall LR, Morgan JS, O’Connor S, Marx PA, et al. TRIM5 suppresses cross-species transmission of a primate immunodeficiency virus and selects for emergence of resistant variants in the new species. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McFadden G, Mohamed MR, Rahman MM, Bartee E. Cytokine determinants of viral tropism. Nat Rev Immunol. 2009;9:645–655. doi: 10.1038/nri2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dorner M, Horwitz JA, Donovan BM, Labitt RN, Budell WC, Friling T, Vogt A, et al. Completion of the entire hepatitis C virus life cycle in genetically humanized mice. Nature. 2013;501:237–241. doi: 10.1038/nature12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bitzegeio J, Bankwitz D, Hueging K, Haid S, Brohm C, Zeisel MB, Herrmann E, et al. Adaptation of hepatitis C virus to mouse CD81 permits infection of mouse cells in the absence of human entry factors. PLoS Pathog. 2010;6:e1000978. doi: 10.1371/journal.ppat.1000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sourisseau M, Goldman O, He W, Gori JL, Kiem HP, Gouon-Evans V, Evans MJ. Hepatic cells derived from induced pluripotent stem cells of pigtail macaques support hepatitis C virus infection. Gastroenterology. 2013;145:966–969 e967. doi: 10.1053/j.gastro.2013.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, et al. Complete Replication of Hepatitis C Virus in Cell Culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 11.Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci U S A. 2006;103:7408–7413. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horwitz JA, Dorner M, Friling T, Donovan BM, Vogt A, Loureiro J, Oh T, et al. Expression of heterologous proteins flanked by NS3-4A cleavage sites within the hepatitis C virus polyprotein. Virology. 2013;439:23–33. doi: 10.1016/j.virol.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones CT, Catanese MT, Law LM, Khetani SR, Syder AJ, Ploss A, Oh TS, et al. Real-time imaging of hepatitis C virus infection using a fluorescent cell-based reporter system. Nat Biotechnol. 2010;28:167–171. doi: 10.1038/nbt.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol. 2002;76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kolykhalov AA, Agapov EV, Blight KJ, Mihalik K, Feinstone SM, Rice CM. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science. 1997;277:570–574. doi: 10.1126/science.277.5325.570. [DOI] [PubMed] [Google Scholar]
- 17.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature. 2009;457:882–886. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giang E, Dorner M, Prentoe JC, Dreux M, Evans MJ, Bukh J, Rice CM, et al. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:6205–6210. doi: 10.1073/pnas.1114927109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mesa RA. Ruxolitinib, a selective JAK1 and JAK2 inhibitor for the treatment of myeloproliferative neoplasms and psoriasis. IDrugs. 2010;13:394–403. [PubMed] [Google Scholar]
- 20.de Jong YP, Dorner M, Mommersteeg MC, Xiao JW, Balazs AB, Robbins JB, Winer BY, et al. Broadly neutralizing antibodies abrogate established hepatitis C virus infection. Sci Transl Med. 2014;6:254ra129. doi: 10.1126/scitranslmed.3009512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Jong YP, Dorner M, Mommersteeg M, Balazs AB, Robbins JB, Vega K, Labitt RN, et al. Broadly neutralizing antibodies abrogate chronic hepatitis C virus infection. Science Translational Medicine. 2014 doi: 10.1126/scitranslmed.3009512. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dorner M, Horwitz JA, Robbins JB, Barry WT, Feng Q, Mu K, Jones CT, et al. A genetically humanized mouse model for hepatitis C virus infection. Nature. 2011;474:208–211. doi: 10.1038/nature10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B, Hatziioannou T, et al. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 24.Abe K, Kurata T, Teramoto Y, Shiga J, Shikata T. Lack of susceptibility of various primates and woodchucks to hepatitis C virus. J Med Primatol. 1993;22:433–434. [PubMed] [Google Scholar]
- 25.Bukh J, Apgar CL, Govindarajan S, Emerson SU, Purcell RH. Failure to infect rhesus monkeys with hepatitis C virus strains of genotypes 1a, 2a or 3a. J Viral Hepat. 2001;8:228–231. doi: 10.1046/j.1365-2893.2001.00284.x. [DOI] [PubMed] [Google Scholar]
- 26.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 27.Patel MR, Loo YM, Horner SM, Gale M, Jr, Malik HS. Convergent evolution of escape from hepaciviral antagonism in primates. PLoS Biol. 2012;10:e1001282. doi: 10.1371/journal.pbio.1001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Y, Liang Y, Qu L, Chen Z, Yi M, Li K, Lemon SM. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc Natl Acad Sci U S A. 2007;104:7253–7258. doi: 10.1073/pnas.0611506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ploss A, Khetani SR, Jones CT, Syder AJ, Trehan K, Gaysinskaya VA, Mu K, et al. Persistent hepatitis C virus infection in microscale primary human hepatocyte cultures. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:3141–3145. doi: 10.1073/pnas.0915130107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andrus L, Marukian S, Jones CT, Catanese MT, Sheahan TP, Schoggins JW, Barry WT, et al. Expression of paramyxovirus V proteins promotes replication and spread of hepatitis C virus in cultures of primary human fetal liver cells. Hepatology. 2011;54:1901–1912. doi: 10.1002/hep.24557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Letvin NL, Daniel MD, Sehgal PK, Desrosiers RC, Hunt RD, Waldron LM, MacKey JJ, et al. Induction of AIDS-like disease in macaque monkeys with T-cell tropic retrovirus STLV-III. Science. 1985;230:71–73. doi: 10.1126/science.2412295. [DOI] [PubMed] [Google Scholar]
- 32.Kestler H, Kodama T, Ringler D, Marthas M, Pedersen N, Lackner A, Regier D, et al. Induction of AIDS in rhesus monkeys by molecularly cloned simian immunodeficiency virus. Science. 1990;248:1109–1112. doi: 10.1126/science.2160735. [DOI] [PubMed] [Google Scholar]
- 33.Hatziioannou T, Ambrose Z, Chung NP, Piatak M, Jr, Yuan F, Trubey CM, Coalter V, et al. A macaque model of HIV-1 infection. Proc Natl Acad Sci U S A. 2009;106:4425–4429. doi: 10.1073/pnas.0812587106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 35.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 36.Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 37.Bellecave P, Sarasin-Filipowicz M, Donze O, Kennel A, Gouttenoire J, Meylan E, Terracciano L, et al. Cleavage of mitochondrial antiviral signaling protein in the liver of patients with chronic hepatitis C correlates with a reduced activation of the endogenous interferon system. Hepatology. 2010;51:1127–1136. doi: 10.1002/hep.23426. [DOI] [PubMed] [Google Scholar]
- 38.Marukian S, Andrus L, Sheahan TP, Jones CT, Charles ED, Ploss A, Rice CM, et al. Hepatitis C virus induces interferon-lambda and interferon-stimulated genes in primary liver cultures. Hepatology. 2011;54:1913–1923. doi: 10.1002/hep.24580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morikawa K, Lange CM, Gouttenoire J, Meylan E, Brass V, Penin F, Moradpour D. Nonstructural protein 3–4A: the Swiss army knife of hepatitis C virus. Journal of viral hepatitis. 2011;18:305–315. doi: 10.1111/j.1365-2893.2011.01451.x. [DOI] [PubMed] [Google Scholar]
- 40.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A. 2005;102:2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liao CH, Kuang YQ, Liu HL, Zheng YT, Su B. A novel fusion gene, TRIM5-Cyclophilin A in the pig-tailed macaque determines its susceptibility to HIV-1 infection. AIDS. 2007;21(Suppl 8):S19–26. doi: 10.1097/01.aids.0000304692.09143.1b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.