

Abstract
An important aspect of immune monitoring for vaccine development, clinical trials, and research is the detection, measurement, and comparison of antigen‐specific T‐cells from subject samples under different conditions. Antigen‐specific T‐cells compose a very small fraction of total T‐cells. Developments in cytometry technology over the past five years have enabled the measurement of single‐cells in a multivariate and high‐throughput manner. This growth in both dimensionality and quantity of data continues to pose a challenge for effective identification and visualization of rare cell subsets, such as antigen‐specific T‐cells. Dimension reduction and feature extraction play pivotal role in both identifying and visualizing cell populations of interest in large, multi‐dimensional cytometry datasets. However, the automated identification and visualization of rare, high‐dimensional cell subsets remains challenging. Here we demonstrate how a systematic and integrated approach combining targeted feature extraction with dimension reduction can be used to identify and visualize biological differences in rare, antigen‐specific cell populations. By using OpenCyto to perform semi‐automated gating and features extraction of flow cytometry data, followed by dimensionality reduction with t‐SNE we are able to identify polyfunctional subpopulations of antigen‐specific T‐cells and visualize treatment‐specific differences between them. © 2015 The Authors. Published by Wiley Periodicals, Inc.
Keywords: antigen‐specific T cells, automated gating, dimension reduction, intracellular cytokine staining, polyfunctionality, visualization
Introduction
Recent advancements in cytometry technologies have enabled the high‐dimensional phenotypic and functional characterization of large numbers of single cells. Current multiparameter flow cytometry experiments routinely measure 15 parameters on thousands of cells per second; it is a routine biological assay used in basic and clinical research laboratories worldwide. Thanks to recent technological development, such as mass cytometry (CyTOF; 1), we can now measure up to about 50 parameters in a single run. The resulting multidimensional and high throughput cytometry data have posed new challenges for data analysis and visualization, especially in the field of immunology.
The cellular adaptive immune response depends in part on the generation of Antigen‐specific (Ag‐specific) T‐cells. T‐cells undergo selective pressure during maturation so that those that recognize their specific antigen on antigen‐presenting cells become activated, undergo clonal expansion, transition into the blood, and eventually become a long‐lived memory population. These Ag‐specific cells are critical for antigen‐recall. T‐cells specific for a particular antigen represent a very small fraction of an individual's T‐cell repertoire. Clinicians and immunologists often rely on cytometry to distinguish and identify rare Ag‐specific T cells within heterogeneous cell samples such as blood. Polyfunctional T cells 2—subsets of Ag‐specific T cells that simultaneously produce multiple effector cytokines and other functional markers in response to activation—are believed to be of clinical relevance, and there is evidence linking their frequency to clinical outcome 3. They have been shown to be important in protective immunity and non‐progression of diseases 3. Consequently, identifying, detecting and visualizing differences amongst polyfunctional T‐cell subsets is an important goal, as they are attractive potential biomarkers for correlation with clinical outcomes.
Many computational tools have been developed for the identification of cell populations in flow cytometry. These can be broadly categorized into two groups: unsupervised approaches targeted toward cell population “discovery” and supervised or semisupervised approaches aimed at identifying known cell populations 4. Both types of approaches use a variety of statistical techniques, from model‐based clustering 5, 6, 7, 8, 9, 10, 11, to nonparametric approaches for gating, clustering, and visualization 12, 13, 14, 15.
It is generally accepted in the computational flow community that no one gating algorithm or approach will solve all specific computational problems 4, 16, and integration of these disparate tools into a coherent computational pipeline has generally been challenging, particularly for large data sets. Frameworks like GenePattern 17 and OpenCyto 18 have been developed to try and bridge the gap amongst these different techniques. OpenCyto in particular provides great flexibility for pipeline construction by simplifying data management and data representation while allowing for multiple approaches and algorithms to be used to identify different cell populations within a single analysis pipeline.
Visualization and comparison of rare cell populations across samples or treatment groups remains a challenge. The relative high dimensionality of the data together with its size poses a significant visualization problem. Traditional bivariate plots scale poorly as the numbers of markers increases, and are a poor representation of polyfunctional populations, while representation of polyfunctional profiles via pie charts can be misleading, and depending on the cell population used to normalize the data, significant differences can be obscured 19. Some effective statistical tools for quantitatively analyzing these antigen‐specific T‐cell responses to antigen stimulation following vaccinations have been developed. For example, MIMOSA (Mixture Models for Single‐cell Assays) 20 is a Bayesian model that can identify Ag‐specific changes in the proportion individual cell subsets compared to background. A multivariate extension of the model, COMPASS (COMbinatorial Polyfunctionality analysis of Antigen‐Specific T‐cell Subsets; 21) was developed to identify Ag‐specific changes across all observed T‐cell subsets simultaneously. Both of these methods present and summarize the T‐cell response profiles through heatmaps and summary scores, but only on the subject and cell subset level.
Few tools have been developed for visualization of complex polyfunctional T‐cell responses 19. The rarity of Ag‐specific subsets, (i.e., <0.1% of total T cells), renders typical dimension reduction and visualization tools ineffective 14, 15. As the number of measured parameters increases, the data will contain irrelevant features that mask the signals from rare cell subsets (such as polyfunctional Ag‐specific T‐cells). Here, we are especially interested in identifying differences in the Ag‐specific T‐cells that are correlated with a clinical outcome for a specific pathogen (e.g., HIV), as this would be a very useful first step to explore the data before conducting more sophisticated analysis in identifying relevant cell subsets for the study objective. Dimension reduction techniques such as principal component analysis (PCA) and multidimensional scaling (MDS) have proven to be useful for projecting and visualizing high‐dimensional cytometry data in lower dimensional subspaces. For example, Newell et al. 22 used PCA to visualize Ag‐specific CD8+ T‐cells measured by CyTOF. However, in this case, PCA was done on the entire T‐cell population, and only a few main phenotypic clusters were highlighted on the global PCA plot. In addition, the differences in Ag‐specific T cells across three viruses CMV, Flu, and EBV were only shown on a few selected donors, not the entire data set. Nonlinear dimensionality reduction methods such as t‐SNE 15 have also been applied in the context of flow and mass cytometry 23, 24. In particular, Amir et al. 23 have shown that t‐SNE was more robust than PCA for summarizing and visualizing high‐dimensional cytometry data. However, while these dimension reduction tools have been applied to many different studies 25, 26, 27, none of these have been used or shown to work for visualization of very rare Ag‐specific T cells across samples. Even with the help of dimension reduction techniques, informative visualization of rare cell subsets in large data sets is challenging. Visualizing many data points generates a “cluttered” display, and density estimation fails to accurately represent rare populations. The bulk of events can easily mask the presence of any signals from rare cell subsets. Hence, instead of directly applying dimension reduction techniques like previous existing approaches, we propose to first reduce the data size by gating the cell subsets of interest to extract relevant information.
In this article, we present a general automated (or semiautomated) analysis strategy for gating and visualization of rare cell subsets in cytometry data, which can be done using a consistent framework within the R environment. Using intracellular cytokine staining (ICS) assays from vaccine and immunological studies, we show how the OpenCyto framework can be used for gating and data size reduction, and integrated with dimension reduction techniques like t‐SNE in order to rapidly identify and visualize differences in rare subsets of polyfunctional antigen‐specific T‐cells.
Materials and Methods
TB Data Set
We obtained cryopreserved PBMC from an epidemiologic study of South African adolescents who were screened for the presence of latent tuberculosis infection (LTBI) using tuberculin skin testing and QuantiFERON‐TB GOLD testing of whole blood 28. This dataset includes 40 subjects, 22 mycobacterium tuberculosis (MTB)‐infected and 18 MTB‐uninfected. Subjects were classified as MTB‐infected using both TB skin testing and the Quantiferon test in‐tube gold (QFT‐gold) that measures IFNγ release in whole‐blood after stimulation with ESAT‐6, CFP‐10, and TB7.7 peptides 29. PBMCs were plated at a density of 2E5 per well and stimulated for six hours with either DMSO or peptide pools consisting of 15 mers overlapping by 12 peptides for the following mycobacterial proteins: ESAT‐6, CFP‐10, TB10.4, Ag85A, and Ag85B at a final concentration of 1 mcg/ml. All five protein‐stimulations have corresponding negative controls (i.e., the nonstimulated samples) that are used for setting cytokine gates as described below. Cells were stained using a published panel in which we replaced MIP‐1b and CD107a with IL‐17a Alexa 700 and IL‐22 PE Cy7 30, 31. Analysis of CD3+CD4+ events was performed in FlowJo (TreeStar, Ashland, OR) after first gating on single cell events, CD14‐ events, live cells, and lymphocytes. Six functions were measured at the single‐cell level in CD4+ T cells: TNFα, IFNγ, IL2, CD154, IL17A, and IL22 leading to 64 theoretical cell subsets. OpenCyto was used for automated gating of T‐cell subsets based on cytokine production, while gates for major phenotypic subsets were imported from FlowJo. It should be noted, that while we could have regated these major cell subsets using OpenCyto, we feel that this approach clearly highlights the strength of our procedure where manual and automated gating, along with statistical modeling (PCA/t‐SNE) can all be done using a consistent framework within the R environment.
HVTN 078 Data Set
HVTN 078 32 is a randomized, double blind phase 1b clinical trial (ClinicalTrials.gov registration number NCT00961883) to evaluate the safety and immunogenicity of heterologous prime/boost vaccine regimens (NYVAC‐B/rAd5 vs. rAd5/NYVAC‐B) in healthy, HIV‐1 uninfected, Ad5 seronegative adult participants. Eighty participants were enrolled into one of four groups receiving different combinations of NYVAC‐B (New York Vaccinia [NYVAC] vector containing HIV‐1BX08 gp120 and HIV‐1IIIB gag‐pol‐nef at a dose of 1x107 PFU) and rAd5 (HIV‐1 recombinant adenoviral serotype 5 [rAd5] vector vaccine VRC‐HIVADV014‐00‐VP ([HIV‐1HXB2/NL4‐3 Gag‐Pol fusion; HIV‐192RW020, HIV‐1HXB2/Bal and HIV‐197ZA012 Env]), at three increasing doses [1 × 108, 1 × 109, 1 × 1010 PFU]). We refer to the four different groups as T1, T2, T3 and T4. In the T1 group, NYVAC was the prime with rAd5 as the boost, while subjects in T2‐T4 received rAd5 as the prime and NYVAC as the boost with the three increasing doses of the prime. All four treatment groups have corresponding nonstimulated samples that are used for setting cytokine gates as described below. Here, we used a subset of the ICS data generated through the trial measuring five functions: IFNγ, IL2, IL4, TNFα, and CD154 in CD4+ T cells in the presence and absence of stimulation with ENV, GAG, and POL stimulations from T1 and T2 groups, as 21, 32 showed the difference is the most significant between these two groups.
OpenCyto
OpenCyto is a software package for the R programming language that allows for reproducible automated gating of flow cytometry data using a series of user‐defined, data‐driven gating methods. A common set of cell populations is defined across all samples in a data set, eliminating the need for cell population matching and making the results directly comparable. The position of the gates defining each cell population in each sample is data‐driven, reducing operator bias. In this study, we used OpenCyto to import manually gated FCM data and extract the CD3+CD4+ lymphocyte populations. In order to have a consistent and constant background, OpenCyto automated gating was performed for the functional markers of the CD3+CD4+ lymphocytes. The gates for the functional markers were based on the 99th percentile of the fluorescence intensity of each functional marker from the CD3+CD4+ populations in the negative control samples.
t‐SNE
t‐SNE 15 is a nonlinear dimension reduction method that projects data into a lower dimensional space. t‐SNE aims to find a mapping in low dimensional space that preserves distances between pairs of points in high dimensional space. The t‐SNE algorithm represents the distance between any two points by the probability of these two points being neighbors, and then selects the low‐dimensional space that is closet to the pairwise probabilities in the original space in terms of the Kullback‐Leibler divergence. We applied t‐SNE to compute the two‐dimensional embedding of antigen‐specific T‐cells in the two data sets presented here using the R package Rtsne. The authors of t‐SNE 15 showed that the performance of t‐SNE is fairly robust to changes in the input parameters (perplexity which is a smooth measure of the number of effective nearest neighbors and theta which controls the speed/accuracy tradeoff). In our analysis, we set perplexity to 30 and theta equals to 0.9, but found that the results were robust to different values of these parameters.
Visualizing Ag‐Specific T Cells Across Samples
Because we are interested in changes in the density of cell populations across different conditions or cohorts (e.g., TB+ and TB−), we needed to adjust for differences in the number of events in each condition. To eliminate such sample size bias before performing dimensionality reduction and visualization, events were subsampled as next described. For each of the two data sets (HVTN 078 and TB), the events from all the FCS files within each treatment group (stimulation group) were concatenated first, and subsampled such that the resulting data file had an equal number of T‐cell events in each treatment group. As such, the treatment group with the smallest number of T‐cell events will retain its entire sample. The robustness of this subsampling procedure for identifying Ag‐specific T‐cells is demonstrated below in the section: Robustness analysis.
To highlight the effect of our proposed strategy in excluding non‐informative data before identifying rare Ag‐specific T cells, we also ran t‐SNE simply on the total T cells. However, due to run‐time and memory limitations, we had to run t‐SNE on a randomly selected sample from each treatment group for both data sets.
Result
Flow Data Analysis and Visualization Pipeline
We gated the FCS data using OpenCyto as described in the Methods before applying t‐SNE in order to remove events and exclude dimensions (noncytokine markers) that are non informative for the identification of polyfunctional T‐cells. OpenCyto identified 2,597,443 CD4+ T cells in the HVTN data set, of which 22,881 express at least one functional marker, and 5,614 are polyfunctional (>2 markers). From the tuberculosis study, 3,395,456 CD4+ T cells were identified, of which 18,324 express at least one functional marker, and 4,921 are polyfunctional. Dotplots showing gated FCS files from each data set are shown in Supporting Information Figures S1 and S2. An overview of our data analysis pipeline is shown in Figure 1.
Figure 1.
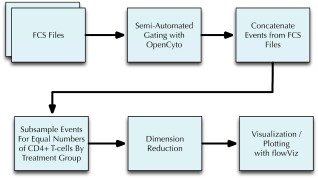
Pipeline for visualizing t‐SNE projected T‐cell subsets. Using OpenCyto FCS files are first gated to extract major T‐cell populations (e.g., CD4 +) and their different cytokine producing subsets. The samples within each group are concatenated and subsampled such that each concatenated sample contains the same number of T‐cell events. t‐SNE is then used to project and visualize individual cell events from these concatenated samples into a two dimensional space.
Distinct Ag‐Specific T‐Cell Signatures for MTB Infection
Applying t‐SNE mapping to the gated TB CD4+ T cells, we obtained 2D scatter plots in which each data point represents an individual cell in the original data and the data points are colored according to selected cell features (e.g., degree of functionality or marker expression). Figure 2 shows two‐dimensional scatter plots of cells stratified by two selected peptide stimulations (Ag85B and ESAT‐6) and MTB infection status. The ESAT‐6 peptide stimulation can be classified as “MTB‐specific” because these proteins are known to be absent in Bacillus Calmette‐Guérin (BCG) and many environmental mycobacteria. By extension, Ag85B can be defined as “MTB‐nonspecific” because these proteins are present in MTB as well as BCG and many environmental mycobacteria.
Figure 2.
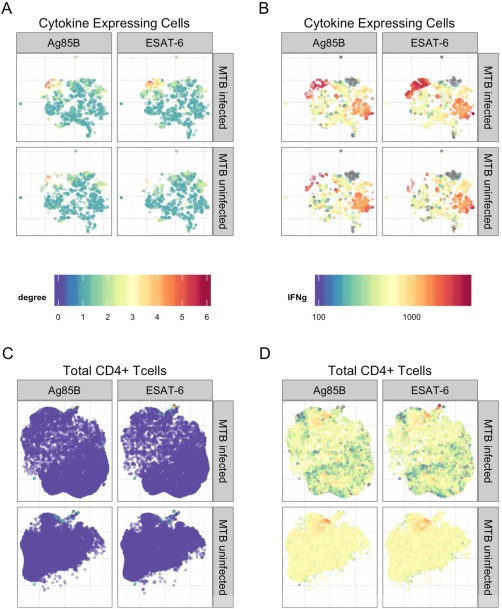
Application of t‐SNE to the TB data set. A: t‐SNE plots for the Ag‐specific T cells on the two selected peptide stimulations (Ag85B: MTB nonspecific, and ESAT‐6: MTB specific) with subjects stratified according to their MTB infection status. Colors of the points indicate the number of cytokines expressed by each single cell (degree of functionality). B: Similar to (A), but the colors indicate the fluorescent intensities of cytokine IFNγ at the single‐cell level. C: Similar to (A), but t‐SNE was applied to total CD4+ T‐cells. D: Similar to (B), but t‐SNE was applied to total CD4+ T‐cells.
Figures 2A and 2B show clusters of polyfunctional T‐cells with IFNγ expression in both stimulations (colored in red which are of higher intensity in IFNγ). However, in the MTB‐specific stimulation, the cluster has higher density and contains more polyfunctional T‐cells, indicating that MTB‐specific stimulation induced a greater polyfunctional T‐cell response than MTB‐non‐specific stimulation. TB‐infected subjects also tend to a have much higher density of polyfunctional cells (Fig. 2A). These cells are predominantly polyfunctional cell subsets with degree of functionality [mt]2, and they all expressed IFNγ marginally (Fig. 2B), in line with the QFT‐gold test. Supporting Information Figures S3–S8 show these clusters of cells for MTB‐specific and non‐specific groups colored by the expression of each cytokine across all peptide stimulations. Figures 2C and 2D show the disadvantage in simply applying dimension reduction to all CD4+ T‐cells in the manner that has been previously described 22. Nonspecific T‐cells easily mask the useful information presented in Figures 2A and 2B. It should be noted that it is possible to group cells in the t‐SNE projected 2‐dimensional space and map the selected cells into their original space. Supporting Information Figure S9 shows the cytokine expressions for the group of cells colored in red (with IFNγ expression [mt]1500 and horizontal axis <−5, vertical axis [mt]5, which are of higher intensity in IFNγ in Figure 2B for MTB infected and ESAT‐6 stimulated group. The mapping to the original cytokine space reveals heterogeneities among the selected group of cells: IL2 expression varies the most among these cells.
We also show the t‐SNE mapping color‐coded by polyfunctional and marginal cytokine expression in Figure 3. For more effective visualization, we only show a subset of polyfunctional cell populations (those with [mt]100 cells, Figure 3A, and differences between peptide stimulations are clearly visible. For cells of degree 1 (cells expressing a single cytokine, Figure 3B, there is no obvious difference across MTB infection status and peptide stimulations. This highlights the importance of joint analysis of marker expression, that is, the study of multifunctional cell subsets is necessary to detect biological changes. Supporting Information Figures S10–S12 show t‐SNE plots for degree of functionality, polyfunctional and marginal cytokine expression for all five stimulations, respectively. Differences between MTB specific and nonspecific peptides are evident when visualizing both degree of functionality and polyfunctional cytokine expression, but not for cells of degree‐1.
Figure 3.
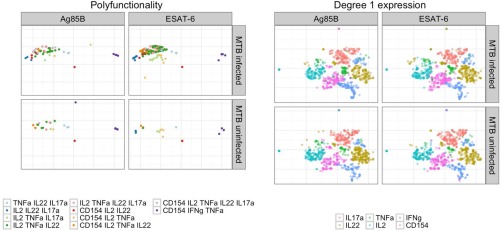
Application of t‐SNE to the Ag‐specific T cells for TB data set. Left panel: colors indicate different polyfunctional cell subsets (degree > 1). Condition‐specific differences are visible. Right panel: colors indicate degree 1 (single‐marker) expression of different cytokines in single cells. No condition‐specific differences are visible.
t‐SNE Reveals Differences in Ag‐Specific T‐Cells Induced by Different Vaccine Regimens
t‐SNE was also applied to the HVTN078 data set on CD4+ T cells. Figure 4 revealed differences among cells between the two treatment groups (T1: NYVAC prime + rAd5 boost, T2: rAd5(1 × 108) prime + NYVAX boost). In order to compare difference in vaccine regimens and stimulation, we fixed the total T‐cell counts to be the same for the six different conditions (two treatment groups each with three stimulation conditions). Figures 4 and 5 show the results for the ENV stimulation group, where it can be seen that there is a clear difference between treatment groups induced by cell‐subsets with higher degree (≥ 3) cytokine expression, especially the four‐degree subset simultaneously expressing CD154, IFNγ, TNFα, and IL4, and the five‐degree subset simultaneously expressing all the markers. Supporting Information Figures S13–S20 also show that this observation is consistent across stimulation conditions. However, Env stimulation activated more Ag‐specific T cells than the other two stimulations. No differences were evident between treatments for lower degree functional cell subsets (e.g., degree functionality less than or equal to 3, Fig. 5). This suggested that priming with rAd5 followed by NYVAC boost (T2) induced an increased percentage of Ag‐specific T cells producing CD154 or IFNγ or TNFα or IL4, while targeting on specific polyfunctional cell subsets. This finding revealed by t‐SNE is consistent with COMPASS analysis in 21 where the authors identified vaccine‐induced differences in polyfunctional subsets supporting the validity of t‐SNE as a visualization tool for Ag‐specific T cells.
Figure 4.
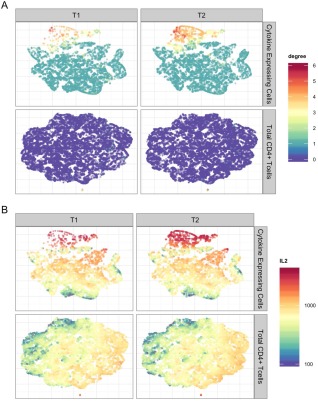
Application of t‐SNE to the HVTN078 data set. A: t‐SNE plots for Ag‐specific T cells (upper half of panel) and total CD4+ T cells (lower half of panel) for ENV stimulated samples from two different vaccine treatment groups (T1 and T2). Colors of the points indicate the number of cytokines expressed by each single cell (degree of functionality). B: Similar to (a), but the colors indicate the fluorescent intensities of cytokine IL2 at the single‐cell level.
Figure 5.
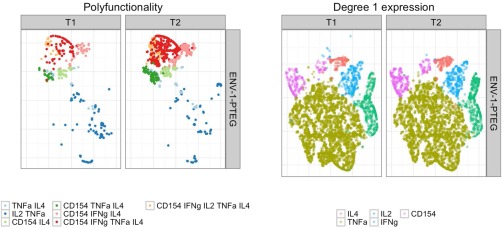
Application of t‐SNE to the Ag‐specific T cells for HVTN078 data set. Left panel: colors indicate cell subsets of differing cytokine polyfunctionality (degree > 1). Condition‐specific differences are visible. Right panel: colors indicate degree 1 (single‐marker) expression of different cytokines in single cells. No condition‐specific differences are visible.
Robustness Analysis
We also conducted robustness analysis to assess the effect of sampling variation on visualization results. Supporting Information Figures S21 and S22 show the three t‐SNE plots on three different randomly sampled subsets of TB and HVTN 078 data, respectively. Each plot can clearly show the difference between TB specific and TB non‐specific stimulation, as well as T1 and T2 treatment groups. While the geometry of the points is different (due to invariant rotation in t‐SNE), the qualitative aspects (i.e., differences between treatment groups) agree with Figures 2 and 3. Stated more simply, sampling variation will alter the global shape of the point cloud, but it is not the absolute position of cell clusters that is important, but rather their relative position compared to each other within a run and between treatment groups that is the salient feature of this approach. This indicates that the results are robust to the subsampling scheme.
PCA Analysis
To compare t‐SNE with PCA, we have also performed PCA analysis instead of t‐SNE on the two data sets described above. Supporting Information Figures S23 and S24 show the PCA projections for the TB and HVTN078 data sets, which correspond to Figure 2 and 4, respectively. It can be seen that first PCA is more sensitive to outliers. Second, while PCA can show some differences between subjects group, t‐SNE leads to better visualization of differences in polyfunctional cell subsets between subject groups. This supports our choice of using t‐SNE for visualization of Ag‐specific cells instead of PCA.
Discussion
We demonstrate a new, exploratory approach to identify and visualize changes in very rare, multifunctional, Ag‐specific T cells across biological conditions in flow assay data. This framework can be a useful component of an exploratory data analysis pipeline meant to visualize differences before applying more complex and formal statistical modeling or statistical testing frameworks such as MIMOSA or COMPASS 20, 21. While tools like COMPASS can quantify changes in antigen‐specific polyfunctional cell subsets, they can be time‐consuming to run and require large sample sizes (i.e., many subjects). Furthermore, results are summarized at the cell‐subset level or per‐subject. In contrast, visualization of specific cell‐level differences between treatment groups using the pipeline presented here can provide a rapid, qualitative, but informative “at‐a‐glance” summary of an experiment. Gating the FCM data to include only the relevant cell populations is critical, as we show, since including irrelevant events leads to crowding and over plotting of points, as well as technical limitations on the number of events that can be processed by the t‐SNE algorithm within a reasonable amount of time and memory. We have also showed that extracting Ag‐specific T cells helps t‐SNE for visualizing rare cell subsets. OpenCyto provides a convenient and unified platform for gating relevant events, and performing subsequent dimension reduction and visualization. Controlling for constant background in non‐stimulated samples is critical for the subsequent visualization to be interpretable. OpenCyto makes this gating very simple. Dimension reduction with t‐SNE provides a complementary visualization tool to current analytic methods 20, 21. The two examples shown here highlight t‐SNE's ability to effectively reveal the heterogeneity in antigen‐specific T‐cell subsets at the single‐cell level. Our approach provides a global view of rare cell populations, with colors highlighting the heterogeneity of these populations across treatments. This visualization provides a more comprehensive understanding of the data.
Supporting information
Supporting Information
Acknowledgment
The authors are grateful to Tom Scriba, Hassan Mahomed, and Willem Hanekom for providing PBMC from South African adolescents used in this study. They also acknowledge Glenna Peterson for technical assistance with all assays related to the TB dataset.
Literature Cited
- 1. Bandura DR, Baranov VI, Ornatsky OI, Antonov A, Kinach R, Lou X, Pavlov S, Vorobiev S, Dick JE, Tanner SD. Mass cytometry: Technique for real time single cell multitarget immunoassay based on inductively coupled plasma time‐of‐flight mass spectrometry. Anal Chem 2009;81: 6813−6822. [DOI] [PubMed] [Google Scholar]
- 2. De Rosa SC, Lu FX, Yu J, Perfetto SP, Falloon J, Moser S, Evans TG, Koup R, Miller CJ, Roederer M. Vaccination in humans generates broad T cell cytokine responses. J Immunol 2004;173: 5372–5380. [DOI] [PubMed] [Google Scholar]
- 3. Seder RA, Darrah PA, Roederer M. T‐cell quality in memory and protection: Implications for vaccine design. Nat Rev Immunol 2008;8: 247–258. [DOI] [PubMed] [Google Scholar]
- 4. O'Neill K, Aghaeepour N, Spidlen J, Brinkman R. Flow cytometry bioinformatics. PLoS Comput Biol 2013;12:e1003365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chan C, Lin L, Frelinger J, Hérbert V, Gagnon D, Landry C, Sékaly R‐P, Enzor J, Staats J, Weinhold KJ, Jaimes M, West M. Optimization of a highly standardized carboxyfluorescein succinimidyl ester flow cytometry panel and gating strategy design using discriminative information measure evaluation. Cytometry Part A 2010;77A: 1126–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lo K, Brinkman RR, Gottardo R. Automated gating of flow cytometry data via robust model‐based clustering. Cytometry Part A 2008:73A: 321–332. [DOI] [PubMed] [Google Scholar]
- 7. Lo K, Hahne F, Brinkman RR, Gottardo R. flowClust: A Bioconductor package for automated gating of flow cytometry data. BMC Bioinformatics 2009;10:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Finak G, Bashashati A, Brinkman R, Gottardo R. Merging mixture components for cell population identification in flow cytometry. Adv. Bioinformatics 2009;247646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin L, Chan C, Hadrup SR, Froesig TM, Wang Q, West M. Hierarchical Bayesian mixture modelling for antigen‐specific T‐cell subtyping in combinatorially encoded flow cytometry studies. Stat Appl Genet Mol Biol 2013;12:309–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cron A, Gouttefangeas C, Frelinger J, Lin L, Singh SK, Britten CM, Welters MJP, van der Burg SH, West M, Chan C. Hierarchical modeling for rare event detection and cell subset alignment across flow cytometry samples. PLoS Comput Biol 2013;9:e1003130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mosmann TR, Naim I, Rebhahn J, Datta S, Cavenaugh JS, Weaver JM, Sharma G. SWIFT‐scalable clustering for automated identification of rare cell populations in large, high‐dimensional flow cytometry datasets, part 2: Biological evaluation. Cytometry A 2014;85:422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rogers WT, Holyst HA. FlowFP: A Bioconductor package for fingerprinting flow cytometric data. Adv Bioinformatics 2009;193947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Applegate D, Dasu T, Krishnan S, Urbanek S. Unsupervised clustering of multidimensional distributions using earth mover distance. Proceedings of the 17th ACM SIGKDD international conference on Knowledge discovery and data mining ‐ KDD '11, San Diego, CA, 2011. pp. 636. [Google Scholar]
- 14. Qiu P, Simonds EF, Bendall SC, Gibbs KD, Bruggner RV, Linderman MD, Sachs K, Nolan GP, Plevritis SK. Extracting a cellular hierarchy from high‐dimensional cytometry data with SPADE. Nat Biotechnol 2011;29:886–891, Oct. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. van der Maaten L, Hinton G. Visualizing Data using t‐SNE. J Mach Learn Res 2008;9:2579 – 2605. [Google Scholar]
- 16. Aghaeepour N, Finak G, Hoos H, Mosmann TR, Brinkman R, Gottardo R, Scheuermann RH. Critical assessment of automated flow cytometry data analysis techniques. Nat Methods 2013;10:228–238, Mar. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Spidlen J, Barsky A, Breuer K, Carr P, Nazaire M‐D, Hill BA, Qian Y, Liefeld T, Reich M, Mesirov JP, Wilkinson P, Scheuermann RH, Sekaly R‐P, Brinkman RR. GenePattern flow cytometry suite. Source Code Biol Med 2013;8:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Finak G, Frelinger J, Jiang W, Newell EW, Ramey J, Davis MM, Kalams SA, De Rosa SC, Gottardo R. OpenCyto: An open source infrastructure for scalable, robust, reproducible, and automated, end‐to‐end flow cytometry data analysis. PLoS Comput Biol 2014;10:e1003806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roederer M, Nozzi JL, Nason MC. SPICE: Exploration and analysis of post‐cytometric complex multivariate datasets. Cytometry A 2011;79: 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Finak G, McDavid A, Chattopadhyay P, Dominguez M, De Rosa S, Roederer M, Gottardo R. Mixture models for single‐cell assays with applications to vaccine studies. Biostatistics 2013:1–15. doi:10.1093/biostatistics/kxt024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lin L, Finak G, Ushey K, Seshadri C, Hawn T, Frahm N, Scriba T, Mahomed H, Hanekom W, Bart P‐A, Pantaleo G, Tomaras GD, Rerks‐Ngarm S, Kaewkungwal J, Nitayaphan S, Pitisuttithum P, Michael N, Kim JH, Robb ML, O'Connell R, Karasawas N, Gilbert P, DeRosa SC, McElrath MJ, Gottardo R. Combinatorial polyfunctionality analysis of antigen‐specific T‐cell subsets identifies novel cellular subsets correlated with clinical outcomes. Nature Biotechnology 2015; in press. [DOI] [PMC free article] [PubMed]
- 22. Newell EW, Sigal N, Bendall SC, Nolan GP, Davis MM. Cytometry by time‐of‐flight shows combinatorial cytokine expression and virus‐specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity 2012;36:142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Amir ED, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, Pe'er D. viSNE enables visualization of high dimensional single‐cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol 2013;31:545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shekhar K, Brodin P, Davis MM, Chakraborty AK. Automatic classification of cellular expression by nonlinear stochastic embedding (ACCENSE). Proc Natl Acad Sci USA 2014;111:202–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Platzer A. Visualization of SNPs with t‐SNE. PLoS One 2013;8:e56883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arsenio J, Kakaradov B, Metz PJ, Kim SH, Yeo GW, Chang JT. Early specification of CD8+ T lymphocyte fates during adaptive immunity revealed by single‐cell gene‐expression analyses. Nat Immunol 2014;15:365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bushati N, Smith J, Briscoe J, Watkins C. An intuitive graphical visualization technique for the interrogation of transcriptome data. Nucleic Acids Res 2011;39:7380–7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mahomed H, Hawkridge T, Verver S, Geiter L, Hatherill M, Abrahams D‐A, Ehrlich R, Hanekom WA, Hussey GD. Predictive factors for latent tuberculosis infection among adolescents in a high‐burden area in South Africa. Int J Tuberc Lung Dis 2011;15:331 −336. [PubMed] [Google Scholar]
- 29. Pai M, Denkinger CM, Kik SV, Rangaka MX, Zwerling A, Oxlade O, Metcalfe JZ, Cattamanchi A, Dowdy DW, Dheda K, Banaei N. Gamma interferon release assays for detection of mycobacterium tuberculosis infection. Clin Microbiol Rev 2014;27: 3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Horton H, Thomas EPE, Stucky JA, Frank I, Moodie Z, Huang Y, Chiu YLL, McElrath MJ, De Rosa SC. optimization and validation of an 8‐color intracellular cytokine staining (ICS) assay to quantify antigen‐specific T cells induced by vaccination. J Immunol Methods 2007;323: 39–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. De Rosa SC, Carter DK, McElrath MJ. OMIP‐014: validated multifunctional characterization of antigen‐specific human T cells by intracellular cytokine staining. Cytometry A 2012;81A: 1019–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bart P, Huang Y, Frahm N, Karuna S, Allen M, Kochar N, Chappuis S, Gaillard J, Graham B, Pantaleo G. rAd5 prime/NYVAC‐B boost regimen is superior to NYVAC‐B prime/rAd5 boost regimen for both response rates and magnitude of CD4 and CD8 T‐cell responses. Retrovirology 2012;9:O72. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information