

Abstract
Independent evidence associates β-amyloid pathology with both NREM sleep disruption and memory impairment in older adults. However, whether the influence of β-amyloid pathology on hippocampus-dependent memory is, in part, driven by impairments of NREM slow wave activity (SWA) and associated overnight memory consolidation is unknown. Here, we show that β-amyloid burden within medial prefrontal cortex (mPFC) is significantly correlated with the severity of impairment in NREM SWA generation. Moreover, reduced NREM SWA generation was further associated with impaired overnight memory consolidation and impoverished hippocampal-neocortical memory transformation. Furthermore, structural equation models revealed that the association between mPFC β-amyloid pathology and impaired hippocampus-dependent memory consolidation is not direct, but instead, statistically depends on the intermediary factor of diminished NREM SWA. By linking β-amyloid pathology with impaired NREM SWA, these data implicate sleep disruption as a novel mechanistic pathway through which β-amyloid pathology may contribute to hippocampus-dependent cognitive decline in the elderly.
Introduction
Cognitive decline is a problematic and disabling consequence of aging, with impairments in hippocampus-dependent memory being one of the most debilitating symptoms1–4. The accumulation of cortical β-amyloid (Aβ) and subcortical tau proteins within the brain are leading candidate mechanisms underlying hippocampus-dependent memory impairment in aging and Alzheimer’s disease (AD)2–5. While tau pathology predominantly accumulates initially within medial temporal lobe structures, including the hippocampus3, 5, β-amyloid pathology predominates in cortex, with the earliest deposition including cortical regions such as the medial prefrontal cortex (mPFC)2, 4. Evidence implicates both pathologies in memory failure in even healthy older adults3, 5, 6. Though tau pathology appears to exert its influence on memory through direct degeneration of hippocampal synapses5, the mechanisms through which Aβ compromises hippocampus-dependent memory remain unclear. Aβ pathology does not aggregate substantively within the hippocampus2, 3, but has been associated with memory through its effects on hippocampal-neocortical network structure, function, and connectivity6, 7, and through its association with hippocampal tau pathology5. However, since the direct influence of Aβ and tau neuropathology explains only a moderate proportion of the variance in age-related cognitive decline1, it remains possible that Aβ pathology also impacts hippocampus-dependent memory indirectly, through other pathways that impact memory-relevant hippocampal-neocortical functioning.
One novelpathway through which cortical Aβ may trigger hippocampus-dependent memory deficits is through its disruption of non-rapid eye movement (NREM) sleep and associated slow wave activity (SWA)8–11. Several independent lines of evidence support this hypothesis. First, older adults exhibit marked reductions in NREM SWA, with these reductions being associated with the degree of memory impairment observed8–13. Second, the degree of disrupted prefrontal NREM SWA in older adults is associated not only with the degree of impaired overnight memory retention11, but is further associated with persistent retrieval-related hippocampal activation that reflects impoverished hippocampal-neocortical memory transformation11. Third, experimentally increasing NREM SWA, particularly in the slow <1Hz frequency range, causally enhances subsequent consolidation and thus long-term memory retention in young adults14. Fourth, there is strong homology between source generators of NREM slow wave oscillations, which predominate in medial prefrontal cortex (mPFC)15, and the cortical regions where Aβ preferentially aggregates in both cognitively normal older adults and Alzheimer’s disease patients2, 15. Fifth, age-related NREM slow wave sleep disruption is exacerbated early in the course of Alzheimer’s disease and mild cognitive impairment; groups known to have elevated Aβ burden13, 16, 17, with these NREM sleep disruptions predicting the severity of observed memory impairment13, 18. Finally, recent studies in humans and rodents demonstrate that interstitial Aβ levels rise and fall with the brain states of wake and NREM sleep, respectively. Indeed, shorter NREM sleep duration and greater NREM sleep fragmentation has been reported in mice over-expressing Aβ proteins19, 20, while human subjective reports of reduced sleep duration and diminished sleep quality correlate with cortical Aβ burden in healthy older adults21. Moreover, direct manipulations of sleep and Aβ production in rodent models of AD have established bidirectional relationships between both factors19, 20, 22.
Together, these data generate the untested hypothesis that the severity of local Aβ accumulation within mPFC is significantly associated with diminished NREM slow wave activity that, in turn, further correlates with the extent of impaired overnight hippocampus-dependent memory consolidation in older adults. Here, we test this hypothesis by combining [11C]PIB PET scanning, offering in vivo estimates of regional Aβ burden, together with an experimental night of sleep electroencephalography (EEG) recording, and a behavioral and functional magnetic resonance imaging (fMRI) test of sleep-dependent memory consolidation. Utilizing these methods, we hypothesized that the accumulation of Aβ within the mPFC would be associated with disrupted memory retention through its association with NREM slow wave activity. Specifically, we predicted that mPFC Aβ burden would significantly correlate with severity of disrupted NREM slow wave activity, particularly in the 0.6–1Hz range known to promote memory consolidation14, 23. Second, we hypothesized that such disruption in NREM SWA would correlate with the degree of impaired overnight memory retention and persistent reliance (rather than progressive independence) of post-sleep retrieval on hippocampal activity. Third, bridging these relationships, we posited that the association between mPFC β-amyloid pathology and impaired hippocampus-dependent memory was not direct (i.e., independent of sleep), but instead, significantly accounted for by the intermediary factor of diminished NREM SWA.
Results
In short (see Methods), twenty-six cognitively normal older adults (Table 1) received a PIB-PET scan, and performed a sleep-dependent episodic associative (word-pair) task before and after a night of polysomnographically (PSG) recorded sleep, with next-day retrieval-related brain activity measured using functional MRI (fMRI). For the memory test, all participants were initially trained to criterion on a set of word pairs in the evening, pre-sleep, followed by two separate recognition memory tests. The first (“short delay”) recognition memory test occurred 10 minutes after the initial study session, where a subset of the studied word pairs were tested.
Table 1.
Demographic and Neuropsychological Measures (mean±s.d.)
| Variable | Subjects (n = 26) |
|---|---|
| Age (yr) | 75.1±3.5 |
| Gender | 18 Female |
| Education (yr) | 16.5±2.2 |
| MMSE | 29.5±1.0 |
| PIB Index | 1.13±0.21 |
| mPFC PIB | 1.16±0.25 |
| Occipital Lobe PIB | 1.10±0.08 |
| Temporal Lobe PIB | 1.07±0.18 |
| Parietal Lobe PIB | 1.15±0.19 |
| DLPFC PIB | 1.15±0.26 |
| Mean bed time | 22:55±1:20 |
| Mean wake time | 7:20±1:15 |
| Mean prestudy time in bed (hr) | 8.42±0.73 |
| Mean prestudy sleep time (hr) | 7.24±0.91 |
| Mean prestudy sleep latency (min) | 38.5±45.3 |
| Mean prestudy sleep efficiency (%) | 86.2±10.1 |
| Short delay recognition (HR–FAR–LR) | 0.14±0.25 |
| Long delay recognition (HR–FAR–LR) | −0.49±0.32 |
| Memory Change (long–short delay) | −0.64±0.29 |
| Neuropsychological Measures | |
| CVLT (long delay, # free recalled) | 10.6±3.0 |
| WMS (visual reproduction %) | 71.3±15.9 |
| Trailmaking B (seconds) | 76.0±35.7 |
| Stroop (# correct in 60 seconds) | 47.6±13.6 |
Following the short delay recognition test, participants were given the in laboratory 8 hour sleep recording period in accordance with habitual sleep-wake habits. The next morning, participants performed the second (“long delay”) post-sleep recognition test during an fMRI scanning session, where the remaining subset of originally studied word pairs were tested. Functional MRI scanning was employed to assess post-sleep retrieval-related activity, focused a priori on the hippocampus24. The measure of overnight memory retention was calculated by subtracting short delay recognition performance from long delay recognition performance8, 11.
β-amyloid and NREM SWA
Given our hypothesized associations between Aβ pathology, NREM slow wave sleep and episodic memory retention, we first examined associations between mPFC NREM SWA (mean at FZ and CZ derivations) and Aβ pathology (the latter indexed using PIB-PET distribution volume ratios; DVRs) (Fig. 1a–d). Because of the causal role of lower frequency (<1Hz) NREM SWA in memory consolidation14, 23, we first examined the impact of Aβ on NREM SWA by frequency. A two-way, repeated measures ANCOVA, with frequency [0.6–1Hz/1–4Hz] as a within subjects factor and mPFC PIB as a between subjects covariate, revealed a significant frequency×mPFC PIB interaction (P=0.032; Fig. 2a). Specifically, greater mPFC PIB DVR was associated with lower NREM SWA in the 0.6–1Hz range (parameter estimate t=−2.29, P=0.031), but not within the faster frequency range NREM SWA (1–4Hz: parameter estimate t=1.85, P=0.076) over prefrontal cortex. The proportion of mPFC SWA 0.6–1Hz was also negatively associated with mPFC PIB (r=−0.45, P=0.020; Fig. 2b). Moreover, the relationship between mPFC PIB and proportion of mPFC NREM SWA 0.6–1Hz persisted when accounting for the non-normal distribution of PIB DVRs by utilizing nonparametric analysis (Kendall’s τ=−0.30, P=0.035). Indicating specificity, no other significant NREM sleep associations were detected (Supplementary Table 1).
Figure 1. Aβ, NREM SWA, and memory retention measures in three sample subjects.

[11C]PIB-PET DVR images demonstrating Aβ deposition (a), NREM SWA and associated localized slow wave source (b), proportion of NREM SWA 0.6–1Hz at FZ&CZ derivations (c), and overnight memory retention (long delay recognition testing – short delay recognition testing; d) measures in three sample participants with low mPFC PIB DVR (left column), intermediate mPFC PIB DVR (middle column), and high mPFC PIB DVR (right column). PIB-PET mPFC region of interest (ROI) is outlined in white (a) and the mPFC EEG derivations are outlined in black (b), with accompanying source analysis (thresholded at ±7) verifying mPFC overlap across PIB-PET and EEG ROIs (see Methods and Supplementary Fig. 2). Prop. denotes proportion, PIB denotes Pittsburgh compound B, PET denotes positron emission tomography, DVR denotes distribution volume ratio referenced against the whole cerebellum, mPFC denotes medial prefrontal cortex, and [HR–FAR–LR] denotes [hit rate to originally studied word pairs – false alarm rate to new, unstudied words – false alarm rate to originally studied word pairs].
Figure 2. Associations between Aβ, NREM SWA, and memory retention measures.

Associations between LN transformed [11C]PIB-PET DVR measured mPFC Aβ deposition, mPFC relative SWA, mPFC SW density, and overnight memory retention. Interaction plots of two-way, repeated measures ANCOVAs, which revealed that Aβ burden was associated with lower relative mPFC NREM SWA and SW density 0.6–1Hz and higher mPFC NREM SWA and SW density at 1–4Hz (parameter estimates for each frequency bin plotted in a) for SWA and c) for SW density. mPFC Aβ burden was also negatively associated with proportion of mPFC NREM SWA 0.6–1Hz (b). mPFC NREM SWA 0.6–1Hz, in turn, positively predicted overnight memory retention (d). %PTOT denotes percentage of total spectral power (0.6–50Hz), Prop. denotes proportion, PIB denotes Pittsburgh compound B, PET denotes positron emission tomography, DVR denotes distribution volume ratio referenced against the cerebellum, mPFC denotes medial prefrontal cortex, and [HR–FAR–LR] denotes [hit rate to originally studied word pairs – false alarm rate to new, unstudied words – false alarm rate to originally studied word pairs].
These Aβ pathology associations remained significant when accounting for the factors of age, mPFC grey matter volume (optimized voxel based morphometry measure), and gender within the same statistical model (P=0.026 for mPFC PIB, yet P=0.753 for age, P=0.781 for grey matter, and P=0.660 for gender). While an exploration of gender was not part of the primary hypotheses, the latter non-significant effect should be appreciated cautiously, since the design and power of the study is not adequate to discount potential gender interactions.
To address the mPFC specificity of our PIB DVR and NREM SWA 0.6–1Hz associations, two-way, repeated measures ANCOVA models were employed. In the first model we examine whether SWA at different derivations was associated with mPFC PIB DVRs. In this model, mPFC PIB was included as a between subjects covariate with location of proportion of NREM SWA 0.6–1Hz included as a within subjects factor [mPFC/dlPFC/Parietal/Temporal/Occipital]. In this model, Location (F=24.002, P<0.001) and Location×mPFC PIB DVR (F=2.568, P=0.043) were significant whereas mPFC PIB DVR was not (F=3.871, P=0.061). Parameter estimates from this model suggested that only frontal ‘locations’ were significantly associated with mPFC PIB DVR, with peak significance being detected over mPFC (for mPFC P=0.020; for dlPFC P=0.027; with non-significant associations for Parietal P=0.118, Temporal P=0.105 and Occipital P=0.162 locations). In the second ANCOVA model, the proportion of mPFC SWA 0.6–1Hz was included as a between subjects covariate with location of PIB DVR included as a within subjects factor [mPFC/dlPFC/Parietal/Temporal/Occipital]. In this model, Location (F=8.331, P<0.001) and Location×Proportion of mPFC SWA 0.6–1Hz (F=6.219, P<0.001) were significant, whereas the Proportion of mPFC SWA 0.6–1Hz was a trend (F=4.084, P=0.055). Thus, parameter estimates from this model suggested that only frontal regions were significantly associated with proportion of mPFC SWA 0.6–1Hz, with peak significance being detected over mPFC (for mPFC P=0.020; for dlPFC P=0.034; and trends or non-significant associations for Parietal P=0.072, Temporal P=0.162, and Occipital P=0.217). Finally, mPFC PIB was not associated with the proportion of REM delta power 0.6–1Hz over prefrontal cortex (r=−0.31, P=0.123; Kendall’s τ=−0.16, P=0.269), demonstrating that the association between mPFC PIB and SWA 0.6–1Hz was specific to NREM sleep. Further, no significant association between mPFC PIB and NREM spectral power was detected beyond the SWA range (Supplementary Fig. 3). Together, these data indicate that mPFC Aβ aggregation significantly predicts the degree of impoverished NREM SWA expressed over mPFC in the memory-relevant 0.6–1Hz range.
To determine whether the association between β-amyloid pathology and NREM SWA 0.6–1Hz was driven by a reduction in the number of slow waves generated or a disruption in slow wave morphology, slow waves (SW) were detected and examined using an established algorithm25. Similar to mPFC NREM SWA, a two-way, repeated measures ANCOVA, with frequency [0.6–1Hz/1–4Hz] as a within subjects factor and mPFC PIB as a between subjects covariate, revealed a significant frequency×mPFC PIB interaction predicting mPFC slow wave density (P=0.020; Fig. 2c) but not mean slow wave period (P=0.257), amplitude (P=0.685) or slope (P=0.535). Congruent with the measure of mPFC NREM SWA, mPFC PIB was associated with lower mPFC SW density 0.6–1Hz (parameter estimate t=−2.623, P=0.015) and higher SW density at 1–4Hz (parameter estimate t=2.416, P=0.024). These data suggest that the association between NREM SWA 0.6–1Hz and β-amyloid pathology is statistically accounted for by the reduction in the incidence of 0.6–1Hz slow waves, rather than morphological changes in SW slope, amplitude, or period.
That mPFC PIB was associated with reduced SW generation within the mPFC was explored by performing source analysis, using the validated sLORETA method26, time-locking to the negative slow wave peak. Source analysis revealed that the peak current density associated with the negative peak of slow waves 0.6–1Hz detected at CZ and FZ was localized within the mPFC (Fig. 1b and Supplementary Fig. 1). These data support the conclusion that mPFC Aβ deposition is associated with fewer slow waves 0.6–1Hz generated within the mPFC.
NREM SWA and hippocampus-dependent memory
Next, we sought to determine whether reduced mPFC NREM SWA 0.6–1Hz, associated with higher mPFC β-amyloid burden, predicted impaired long-term memory retention in cognitively healthy older adults. The proportion of mPFC NREM SWA 0.6–1Hz (r=0.50, P=0.019; Fig. 2d) positively predicted memory retention, and remained a significant predictor when controlling for age and gender (P=0.022 for mPFC NREM SWA 0.6–1Hz, P=0.980 for age, P=0.494 for gender). Therefore, reductions in mPFC NREM SWA 0.6–1Hz predicted worse overnight memory retention.
At the neural level, and consistent with a NREM sleep-dependent hippocampal-neocortical model of memory consolidation27, 28, the severity of impairment in NREM SWA 0.6–1Hz was further associated with greater persistence (rather than progressive independence11, 29–31) of post-sleep retrieval-related hippocampal activation (r=−0.59, P=0.004; Fig. 3a). This association also remained significant when controlling for age and gender (P=0.006 for mPFC NREM SWA 0.6–1Hz, P=0.897 for age, P=0.657 for gender). Though this association was maximal in the left hippocampus, it was present bilaterally (Supplementary Fig. 2). No significant associations between retrieval-related HC activation and NREM spectral power were detected beyond the SWA range (Supplementary Fig. 3). Implicating diminished memory consolidation in this association between NREM SWA disruption and persistent hippocampal activity, post-sleep retrieval-related activation within the hippocampus significantly predicted worse overnight memory retention (r=−0.50, P=0.017) (Fig. 3b). Taken together, these data are consistent with the prediction that the severity of impairment in NREM SWA 0.6–1Hz detected over mPFC, in older adults, is significantly associated with worse overnight memory retention and persistent reliance on the hippocampus during next day retrieval.
Figure 3. Associations between NREM SWA, retrieval-related hippocampus activation, and memory retention.
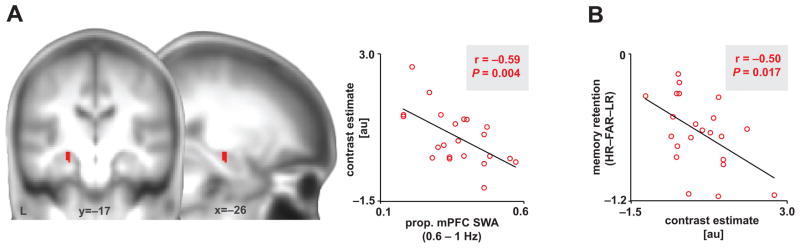
(a) Negative association between proportion of mPFC SWA 0.6–1Hz and left hippocampal activation greater during successful associative episodic retrieval than correct rejection of novel words (Hits-Correct Rejections); 8mm-sphere ROI: [x=−22, y=−14, z=−12; x=−23, y=−15, z=−16 in mni coordinates]24. Activations were inclusively masked by hippocampal anatomy and displayed and considered significant at the voxel level of P<0.05 family-wise error (FWE) corrected for multiple comparisons within the a priori hippocampal region of interest. Peak effects were detected at [x=−24, y=−16, z=−14]. Hot colors represent the extent of the negative association between hippocampal activation and proportion of SWA 0.6–1Hz. (b) Negative association between overnight memory retention and the average contrast estimate of significant hippocampal voxels, extracted using marsbar46. au denotes arbitrary units, prop. denotes proportion, and [HR–FAR–LR] denotes [hit rate to originally studied word pairs – false alarm rate to new, unstudied words – false alarm rate to originally studied word pairs].
Aβ, SWA, and hippocampus-dependent memory consolidation
Having characterized the separate associations between mPFC Aβ pathology, NREM SWA deficits, and hippocampus-dependent memory impairment, we finally sought to determine the interactions between factors using path analysis32. Specifically, we tested the hypothesis that mPFC Aβ pathology exerted an influence on memory not directly, but instead, indirectly, through its association with impaired NREM SWA, thus compromising sleep-dependent consolidation of hippocampus-dependent memory. Three models were constructed (Fig. 4a–c) and compared to each other and standard saturation and independence control models to determine the nature of these interactions. The standardized metrics used to determine these interactions were: (1) root mean square residual (RMR), (2) goodness of fit index (GFI), and (3) Bayesian information criterion (BIC; see Methods)33–35. In short, RMR values near 0 and GFI values above 0.9 are considered evidence for sufficient model fit34. Lower BIC values suggest better model fits, with a difference in BIC of over 10 suggesting marked differences between the models, a difference of 6–10 suggesting a strong difference, and a difference of 2–6 suggesting marginal difference is present35. In the first model (Fig. 4a), mPFC Aβ pathology was allowed to directly predict deficits in memory retention independent of NREM SWA (proportion of mPFC NREM SWA 0.6–1Hz) and retrieval-related hippocampal activation. In the second model (Fig. 4b), mPFC Aβ pathology was associated with diminished memory retention independent of NREM SWA, instead, being directly associated through its impact on retrieval-related hippocampal activation. In the third, sleep-dependent, model (Fig. 4c), the associated influence of mPFC Aβ pathology on impaired memory retention was not direct. Instead, the influence of mPFC Aβ pathology was indirect, through its effects on diminished NREM SWA, which consequentially predicted deficits in overnight memory retention and hippocampus-dependent memory transformation. Of the three, the third, sleep-dependent, model provided the superior statistical fit (Fig. 4c). Specifically, this sleep-dependent model provided (i) the lowest RMR (RMR=0.006, compared to 0.021 for Model 1 and 0.021 for Model 2), (ii) the only GFI above 0.9 (GFI=0.931, compared to 0.858 for Model 1 and 0.873 for Model 2), and (iii) the lowest BIC value (BIC=24.676, compared to 29.640 for Model 1 and 29.131 for Model 2). Moreover, only the sleep-dependent model outperformed both the saturation (RMR=0.000, GFI=1.000, BIC: 30.910) and independence control models (RMR=0.046, GFI=0.617, BIC: 30.747). Critically, however, while all three models demonstrated significant associations between (i) NREM SWA 0.6–1Hz and post-sleep retrieval-related hippocampal activation (all P<0.005; Fig. 4a–c), and (ii) post-sleep retrieval-related hippocampal activation and overnight memory retention (all P<0.010; Fig. 4a–c), the only significant path linking mPFC Aβ pathology to impaired memory retention was the sleep-dependent model (Model 3), by way of the influence of mPFC Aβ on NREM SWA (P=0.017; Fig. 4c). These results indicate that the association between mPFC Aβ pathology and diminished memory consolidation is significantly accounted for by the impairing influence of mPFC Aβ pathology on NREM SWA, resulting in a profile of greater overnight forgetting and persistent reliance on the hippocampus during next day retrieval.
Figure 4. Path models linking Aβ, NREM SWA, retrieval-related hippocampus activation, and memory retention.

Path analysis models examining the relative contributions of [11C]PIB-PET DVR measured mPFC β-amyloid (Aβ) deposition, proportion of mPFC NREM SWA 0.6–1Hz, and retrieval-related hippocampal (HC) activation to overnight memory retention (long delay recognition testing — short delay recognition testing) in three hypothesized models (a–c). Values represent standardized regression weights. Models were estimated and model fit for the sleep and HC-independent model (a, BIC = 29.640; RMR = 0.021; GFI = 0.858), the sleep-independent and HC-dependent model (b, BIC = 29.131; RMR = 0.021; GFI = 0.873), and the sleep-dependent model (c, BIC = 24.676; RMR = 0.006; GFI = 0.931) were compared against a saturated model (BIC = 30.910; RMR = 0.000; GFI = 1.000), and an independence model (BIC = 30.747; RMR = 0.046; GFI = 0.617). * denotes path significance at P<0.05.
Discussion
To the best of our knowledge, the current findings provide the first evidence that cortical Aβ pathology is associated with impaired generation of NREM slow wave oscillations that, in turn, predict the failure in long-term hippocampus-dependent memory consolidation. While it is important to recognize that the current findings are cross-sectional and correlational, limiting causal claims, they nevertheless establish that the factors of Aβ and NREM sleep physiology and hippocampus-dependent memory are significantly and directionally inter-related. Thus, in addition to already established pathways associated with diminished cognitive function2, 3, 5, 6, Aβ may additionally impair hippocampus-dependent memory in older adults through its impact on NREM SWA. Moreover, since sleep is a potentially modifiable factor, such findings raise the possibility that therapeutic sleep intervention may aid in minimizing the degree of cognitive decline associated with β-amyloid pathology in old age.
To date, age-related NREM sleep disruption has been described in older adult, MCI, and AD cohorts13, 16–18. Moreover, subjective reports of poor quality sleep are associated with high Aβ burden in healthy older individuals21, with reductions in SWS and REM sleep time associated with CSF Aβ and tau protein levels in AD patients18. These findings are supported by animal studies linking Aβ pathology to NREM sleep fragmentation20. The current study extends these reports by demonstrating that regionally specific aggregation of Aβ within mPFC is associated with the selective electrophysiological impairment of NREM slow wave activity, and that this sleep disruption subsequently correlates with impaired hippocampal-neocortical memory transformation and related overnight memory retention.
Our findings further highlight specificity within this pathological interaction at two levels: anatomical and electrophysiological. Anatomically, the selective association between mPFC Aβ pathology (and not other common Aβ accumulating regions) and diminished slow waves suggests that this region may be especially critical to the generation of such NREM sleep oscillations. Indeed, source localization analyses in healthy young adults has revealed slow wave generators within the same mPFC regions that commonly suffer early and extensive Aβ burden2, 4, 15. Electrophysiologically, the Aβ association with NREM SWA was additionally specific to the low frequency range of SWA between 0.6–1Hz. This is of particular relevance considering the recognition of the two mechanistically distinct forms of NREM slow waves: the <1Hz slow oscillation and the delta wave (1–4 Hz)36, 37. While the mechanisms underlying the mPFC Aβ frequency-specific association identified in the current study (0.6–1Hz) remains unknown, it is plausible that β-amyloid pathology impacts the generation/expression of slow oscillations through an impact on coordinated cortico-thalamic hyperpolarized down states and depolarized up states36, 37. This may include the recognized reduction in synaptic NMDAR functioning by Aβ38, 39; receptors that are also necessary to generate NREM slow oscillations (and not delta waves)36–39. In addition, or alternatively, Aβ may exacerbate age-related prefrontal atrophy due to the neurotoxic effects of Aβ2, 3, 38, 39 or Aβ-coordinated spread of tau pathology through hippocampal-thalamic loops that interact with the reticular nucleus of the thalamus and the cortico-thalamic loops that generate NREM slow oscillations3, 5, 40. Importantly, all these hypotheses offer clear, testable predictions for future exploration within varied clinical and animal model systems.
In addition to Aβ being associated with diminished NREM sleep, a growing body of evidence suggests that NREM sleep disruption reciprocally promotes the build-up of Aβ. Interstitial Aβ levels in both humans and rodents rise during periods of wakefulness and fall during sleep19. Moreover, sleep deprivation increases Aβ plaque formation in rodent cortex19 and alters CSF Aβ levels in humans41, yet the presence of NREM sleep facilitates Aβ clearance19, 22. These data, combined with evidence linking Aβ pathology to NREM sleep disruption in rodents20 and reduced NREM SWA between 0.6–1Hz in the current study, supports the interpretation of a bidirectional relationship between sleep and Aβ pathology. While remaining speculative, such an interpretation supports the proposal of a self-perpetuating cycle in which the initial emergence of Aβ impairs the generation of NREM sleep oscillations, which, in turn, consequently results in wake-dependent increases in Aβ while diminishing the sleep-dependent clearance of Aβ42. As a result, Aβ buildup would accelerate, exacerbating the pathological cascade leading to AD3.
Beyond the association between Aβ burden and impaired NREM SWA, the current findings additionally characterize a functional consequence of this association: impaired overnight consolidation of long-term memory. While prior evidence suggests that the strength of association between Aβ burden and memory retention in healthy older adults is only modest when memory is assessed immediately after encoding1, 3, 6, the current findings indicate that this association becomes significant when the retention interval is delayed, and thus involves sleep-dependent memory processes. Consequentially, our data suggest that one additional novel pathway linking cortical Aβ pathology to hippocampus-dependent long-term memory functioning is through the association between Aβ aggregation and disrupted NREM slow oscillations. Specifically, the data support a model in which the severity of Aβ aggregation in mPFC regions that generate NREM slow waves15 predicts reductions in NREM slow waves 0.6–1Hz. This reduction, in turn, is associated with diminished sleep-dependent memory consolidation and the persistence (rather than the typical progressive independence27, 28, 11, 30) of next day memory retrieval upon hippocampal activity. Indeed, results from the path analyses support the hypothesis that the influence of mPFC Aβ on hippocampus-dependent memory consolidation was not direct, but through its impact on NREM slow waves 0.6–1Hz. These associations remained robust when adjusting for age, gender, and atrophy.
Importantly, it should be noted that these data in no way preclude the possibility that Aβ can influence memory independent of NREM slow waves, or that other factors, such as atrophy or tau pathology may influence memory independent of or dependent on associations with NREM slow waves. While the SEM models did demonstrate significant inter-relatedness between Aβ pathology, NREM sleep, and hippocampus-dependent memory, it does not explain the influence of other, unmeasured factors, such as tau pathology, which may explain additional variance in age-related, sleep-dependent memory impairment. It is therefore necessary for future studies to employ models that examine multiple factors associated with age-related cognitive decline, to develop a more comprehensive account of how these factors interact with sleep and impact sleep-dependent memory. However, these findings do establish that one influence of Aβ pathology on hippocampus-dependent memory includes an impact on the cortical generation of NREM slow waves and the associated consolidation of sleep-dependent memory.
Building on this model, and more generally, our findings offer several clinical and public health considerations. First, should these associations prove to be causal in cognitively normal older adults and AD cohorts, screening for and treating NREM slow wave sleep abnormalities in older adult populations may aid in reducing both the risk for developing AD and the rate at which AD progresses. Indeed, disordered sleep is recognized to carry an increased risk for cognitive decline and AD43, 44, while superior sleep quality is associated with resilience to cognitive decline and a reduced risk of developing AD45. Second, since associations between Aβ pathology and NREM sleep physiology are observed in preclinical older adults as well as in MCI and AD cohorts13, 18, it is possible that sleep disruption, specifically in the electrophysiological index of NREM slow oscillatory activity (<1Hz), may represent an additional preclinical AD biomarker. Finally, these data offer the empirical foundations on which future work may determine whether Aβ-related sleep disruption plays a causal role in the progression of cognitive decline in neurodegenerative dementias. They further warrant the exploration of whether interventions that promote NREM SWA (in the 0.6–1Hz frequency range) minimize the progression of neurodegeneration and the cognitive dysfunction associated with Aβ pathology.
Methods
Thirty healthy older adult participants were recruited, with twenty-six participants completing the study (18 females, mean±s.d., 75.1±3.5 years; Table 1). No statistical methods were used to pre-determine sample sizes, but our sample sizes are similar to those reported in previous publications6–8, 11, 47. Data from ten of these participants were included in a previous publication11. These ten participants were selected for the present study, as they were the only participants with concurrently acquired PIB-PET data. The study was approved by the local human studies committee, with all participants providing written informed consent. Exclusion criteria included presence of neurologic, psychiatric or sleep disorders, current use of antidepressant or hypnotic medications, or being left handed. Participants were free of depressive symptoms48, and all scored >25 on the mini mental state exam49. Further, and in addition to neuroradiological assessments and medical interviews (cf.11, 47; obtained within one year of study entry), participants performed within 1.5 standard deviations of their age, gender, and education-matched control group on tests of 1) episodic memory50, 51 and 2) frontal function52, 53 (Table 1). Episodic memory task data were specifically excluded when below two standard deviations of the mean across participants, or when performing at chance levels. PIB DVR does not follow a normal distribution, and, unlike behavioral assessment, there are no numerical boundaries of the PIB DVR measure that render this metric without scientific or clinical relevance. Consequently, there was no PIB DVR exclusion threshold (within biological limits) employed in the current study. Prior to study entry, participants underwent sleep disorders screening with a polysomnography (PSG) recording night (described below) reviewed by a board certified sleep medicine specialist (author B.L.). Participants were excluded if they displayed evidence of a parasomnia or an Apnea/Hypopnea Index ≥1554, with four participants being excluded due to evidence of sleep apnea). All participants abstained from caffeine, alcohol, and daytime naps for the 48 hr before and during the study. Participants kept normal, habitual sleep-wake rhythms and averaged 7–9 hr of reported time in bed per night prior to study participation, verified by sleep logs (Table 1). The recording of sleep in the laboratory environment, as in the current study, is advantageous for a number of data acquisition and quality control reasons. However, it represents an important limitation considering that sleep amounts and efficiency are often greater in the home setting. While total sleep time and NREM SWS time often differ across these two contexts, it is of note that the measure of NREM sleep spectral EEG power is highly consistent across nights within an individual in a variety of contexts, such that within-subject night-to-night variability is much smaller than between subjects variability in NREM SWA55, 56. This is of potential relevance to the current findings, since it was spectral NREM SWA that demonstrated associations with PIB and memory measures rather than any sleep stage metrics. Nevertheless, home PSG assessments will be necessary to provide a more ecologically valid exploration of the interaction between β-amyloid pathology, sleep and memory.
General Experimental Design
All participants underwent positron emission tomography (PET) scanning following [11C]PIB injection. Within one year of PIB-PET scanning, participants then entered the lab in the evening and trained to criterion on a sleep-dependent episodic memory task (describe below), followed by a short delay (10 min) recognition test. Participants were then given an 8 hour sleep opportunity, measured with PSG, starting at their habitual bed time (Table 1). Approximately two hours post-awakening, participants performed an event-related functional MRI (fMRI) scanning session while performing a long delay (10 hr) recognition test. PIB-PET data were acquired and analyzed separately (authors S.M. and J.V.) from all other data analyzed (author B.M.), thus ensuring PSG, fMRI, and memory data acquisition, preprocessing, and analysis were conducted blind to participant Aβ status.
Episodic Memory Task
The word-pairs task11 had an intentional encoding phase immediately followed by a training to criterion phase, which was then followed by a short delay recognition test (10 min; 30 studied trials and 15 foil trials) and a long delay recognition test (10 hr, occurring 2 hours post-awakening within the MRI scanner; 90 studied trials and 45 foil trials).
As described previously11, associative recognition memory was calculated by subtracting both the ‘False alarm rate’ (FAR; proportion of foil words endorsed as “previously studied”) and the ‘Lure rate’ (LR; proportion of previously studied words erroneously paired with the lure) from the ‘Hit rate’ (HR; proportion of previously studied words paired with the correct nonsense word).11 Episodic memory retention was subsequently calculated as the difference in short and long delay recognition memory performance [long delay – short delay]11, 57. Two participants were excluded from analysis as outliers (memory performance more than 2 standard deviations from the mean), and two participants had memory and fMRI data lost due to computer theft.
PET scanning and analysis
PIB-PET scans were collected within one year of sleep and memory assessment, as PIB distribution volume ratio (DVR) values change minimally within this duration58, 59. Scanning was performed on 23 participants using a Siemens ECAT EXACT HR PET scanner and on 3 participants using a Siemens Biograph 6 PET/CT scanner in 3D acquisition mode post [11C]PIB injection (approximately 15 mCi) into the antecubital vein. PIB DVR values have been shown to be highly comparable across these two scanners, having no effect on the global PIB measure60. Dynamic acquisition frames were obtained over 90 minutes, as reported6, 61, following transmission or CT scans for attenuation correction. PIB-PET data were reconstructed using an ordered subset expectation maximization algorithm with weighted attenuation, and images were smoothed using a 4 mm Gaussian kernel with scatter correction. Each image is evaluated for excessive motion and adequacy of statistical counts. PET image processing and analysis were performed using SPM8 to realign frames. Realigned PIB frames from the first 20 minutes of acquisition were averaged and used to guide coregistration of each individual’s PIB-PET scan to their structural MRI scan. Logan graphical analysis was used to calculate voxel-wise distribution volume ratios (DVRs) with a cerebellar grey matter region of interest (ROI) used as a reference region, as described previously6, 61. This analysis yielded a voxelwise DVR image for each participant. Targeting our mPFC hypothesis, the following Desikan-Killiany Atlas-derived62 regions were used to construct our mPFC ROI: left and right hemisphere superior frontal, rostral and caudal anterior cingulate, and medial orbitofrontal regions (Fig. 1a). In addition, occipital cortex (right and left hemisphere cuneus, lingual, pericalcarine, and lateral occipital regions), temporal cortex (right and left hemisphere middle and superior temporal regions), parietal cortex (right and left hemisphere inferior and superior parietal, supramarginal gyrus, and precuneus regions), and dorsolateral prefrontal cortex (right and left hemisphere rostral and caudal middle frontal, pars opercularis, and pars triangularis regions; DLPFC) ROIs were used as control measures to determine specificity of mPFC Aβ effects. ROI DVR values were derived by calculating the mean of all voxelwise DVR values within each ROI. To account for the non-normal distribution of Aβ in the population, DVR measures were normalized using the natural logarithm, as described previously63–65.
MRI scanning
Scanning was performed on a Siemens Trio 3 Tesla scanner equipped with a 32-channel head coil. Functional scans were acquired using a susceptibility-weighted, single-shot echo-planar imaging (EPI) method to image the regional distribution of the blood oxygenation level-dependent signal [time repetition (TR)/time echo (TE) 2000/23 ms; flip angle 90°; FatSat, FOV 224 mm; matrix 64×64; 37 3mm slices with 0.3mm slice gap, descending sequential acquisition], and using parallel imaging reconstruction (GRAPPA) with acceleration factor 2. Three functional runs were acquired (159 volumes, 5.3 minutes). Following functional scanning, two high-resolution T1-weighted anatomical images were acquired using a 3D MPRAGE protocol with the following parameters: repetition time (TR), 1900 ms; echo time (TE), 2.52 ms; flip angle, 9°; field of view (FOV), 256 mm; matrix, 256 × 256; slice thickness, 1.0 mm; and 176 slices. Optimized voxel-based morphometry (VBM) was performed on coregistered mean MPRAGE images to examine grey matter volume within the same mPFC ROI used to extract mPFC PIB DVR values; VBM methods described in detail in11.
fMRI analysis
Functional MRI data were analyzed using SPM8 (Wellcome Department of Imaging Neuroscience; http://www.fil.ion.ucl.ac.uk/spm/software/) beginning with standardized preprocessing (realignment, slice timing correction, and coregistration), and with normalization accomplished using a template containing elderly brains, as described previously11, 47.
Following preprocessing, retrieval trials were sorted into “Hits” (correct word-nonsense word recognition), “Lures” (selection of the incorrect, previously studied, nonsense word), “Misses” (incorrect selection of never studied nonsense word or endorsement of word as “new”), “Correct Rejections” (novel words correctly endorsed as “new”), “False Alarms” (novel words incorrectly endorsed as “studied”), and “Omissions” (trials with no subject response)11, with each trial modeled using a canonical hemodynamic response function. To generate a validated contrast for retrieval-related activity, Hit events were contrasted with Correct Rejection events [Hits – Correct Rejections]11. Individual activation maps were then taken to a second-level random effects analysis to examine retrieval-related activation negatively associated with NREM SWA and overnight memory retention measures. Activations were assessed at the voxel level of p<0.05 family wise error (FWE)66 corrected for multiple comparisons within an a priori hippocampal region of interest (ROI; 8 mm sphere [x=−22, y=−14, z=−12]24 in Talairach space and [x=−23, y=−15, z=−16] after MNI conversion67), further inclusively masked using an anatomical hippocampus ROI. To determine associations between hippocampus activation and other variables of interest, the cluster average of significant voxels was extracted using Marsbar46.
Sleep monitoring and EEG analysis
PSG on the experimental night was recorded using a Grass Technologies Comet XL system (Astro-Med, inc., West Warwick, RI), including 19-channel electroencephalography (EEG) placed using the 10–20 system, electrooculography (EOG) recorded at the right and left outer canthi (right superior; left inferior), and electromyography (EMG). Reference electrodes were recorded at both the left and right mastoid (A1, A2). Data were digitized at 400Hz, and stored unfiltered (recovered frequency range of 0.1–100 Hz), except for a 60-Hz notch filter. Sleep was scored using standard criteria68. Sleep monitoring on the screening night was recorded using a Grass Technologies AURA PSG Ambulatory system (Astro-Med, inc., West Warwick, RI), and additionally included nasal/oral airflow, abdominal and chest belts, and pulse oximetry.
EEG data from the experimental night were imported into EEGLAB (http://sccn.ucsd.edu/eeglab/) and epoched into 5 s bins. Epochs containing artifacts were manually rejected by a trained scorer (author B.A.M.), and the remaining epochs were filtered between 0.4–50Hz (645±80 epochs per participant with 4.6%±2.1% of epochs rejected). A fast Fourier transform (FFT) was then applied to the filtered EEG signal at 5-second intervals with 50% overlap and employing hanning windowing. Analyses in the current report focused, a priori, on slow wave activity (SWA), defined as relative spectral power between 0.6–4.6Hz during slow wave sleep (NREM stages 3&4)10, 11. Spectral power was subdivided into two bins for analysis (0.6–1Hz/1–4Hz), to examine the impact of β-amyloid on SWA frequencies particularly relevant to memory functions14, 23. A single summary proportional measure was also derived by dividing the spectral power between 0.6–1Hz by the sum of spectral power between 0.6–4Hz, to determine the relative dominance of memory-relevant slow waves. Furthermore, due to our a priori focus on mPFC, SWA measures at FZ and CZ derivations were averaged and used as a measure of mPFC SWA (Fig. 1b). To ascertain topographic specificity of effects, SWA measures at F3, F4, F7, and F8 derivations were averaged and used as a measure of dlPFC SWA, SWA measures at P3, P4, and PZ derivations were averaged and used as a measure of Parietal SWA, SWA measures at T3, T4, T5, and T6 derivations were averaged and used as a measure of Temporal SWA, and SWA measures at O1 and O2 derivations were averaged and used as a measure of Occipital SWA.
Slow wave detection and source analysis were performed to (1) calculate the impact of mPFC Aβ on slow wave density, and (2) determine whether memory-relevant FZ and CZ measured slow waves (0.6–1Hz) have an mPFC source (Fig. 1b and Supplementary Fig. 1). EEG data were filtered between 0.5–4Hz, and individual slow waves were detected using a validated algorithm25. Standardized low resolution brain electromagnetic tomography (sLORETA) was employed26 as previously described69, 70. In short, this method calculates current density sources using a discrete, three-dimensionally distributed, linear minimum norm solution to the forward problem. Computations are made using a head model based on the MNI152 template71. Prior to sLORETA analysis, EEG preprocessing was conducted in MATLAB using the EEGLAB toolbox. For each participant, filtered (0.5Hz–4Hz), artifact-rejected EEG was event-marked separately for detected slow wave (0.6–1Hz) midpoints in the FZ and CZ derivations. EEG was then epoched around each detected slow wave midpoint (±100 ms). Slow wave epochs were then averaged and exported separately for CZ and FZ detected slow waves. sLORETA analyses of slow wave epochs were carried out using the freeware sLORETA utilities (http://www.uzh.ch/keyinst/loreta.htm), consistent with previous source analysis examinations69, 70. Prior to current density source calculation, all electrode derivations were registered and transformed into 3D MNI space, yielding a spatial transformation matrix. Current density source maps were then derived for each participant separately for CZ and FZ time-locked EEG averages. CZ and FZ source maps were then averaged within each participant, with CZ-FZ averaged source maps then averaged across participants to generate a grand mean average source image for memory-relevant CZ and FZ slow waves (Supplementary Fig. 1).
Statistical Analysis
Two-way repeated measures ANCOVA models were used to determine the influence of PIB-PET measures on NREM slow wave measures, with PIB-PET DVR measures as a between subjects covariate and frequency (0.6–1Hz/1–4hz) as a within-subjects factor. Associations between PIB DVR measures, sleep measures, hippocampal activation, and episodic memory retention were assessed using regression models. Normality was formally tested, and all variables exhibited the skewness and kurtosis of a normal distribution except PIB-PET DVR measures, which exhibited a normal kurtosis but a right skewed distribution. Since PIB-PET DVR measures followed a right skewed non-normal distribution, PIB DVR values were natural logarithm transformed before analysis and regressions were further affirmed with follow-up nonparametric Kendall’s Tau correlations. Analyses were completed using SPSS version 22.0 (SPSS, Inc., Chicago, IL).
To determine whether mPFC Aβ statistically influenced hippocampal-dependent episodic memory retention through mPFC NREM SWA, path analyses were performed using a structural equation modeling framework32, 34 in Amos version 22 (IBM Corp., Armonk, NY). This multivariate modeling technique calculates the path coefficients, i.e. coupling between model variables, given a specified model. Path coefficients reflect the direct and proportional influence of one variable on another while controlling for other variables in the model. Three hypothesized models were specified with an equal number of paths. These models were then compared to each other and saturation and independence models. The first model allowed Aβ to directly impact memory retention independently of NREM SWA. The second model allowed Aβ to impact memory retention independently of NREM SWA, but this time indirectly through its effects on hippocampal activation. Finally, model three instead required Aβ to impact memory retention solely through its influence on NREM SWA. Three validated metrics were used to compare model fits: BIC (Bayesian information criterion), RMR (root mean square residual), and GFI (goodness of fit index)33–35. In short, models with RMR near 0 and GFI above 0.9 were considered sufficient model fits34. The model with the lowest BIC value was considered the superior model, with a difference of 10+ suggesting large model differences, a difference of 6–10 suggesting medium model differences, and a difference of 2–6 suggesting small model differences are present35. Within the superior model, individual path coefficients were then examined for significance.
A supplementary methods checklist is available.
Supplementary Material
Acknowledgments
We thank David Baquirin, Maggie Belshe, Meghna Bhatter, Michelle Binod, Sam Bowditch, Catherine Dang, Jay Gupta, Amynta Hayenga, Danny Holzman, April Horn, Emily Hur, Jonathan Jeng, Samika Kumar, Candace Markeley, Elizabeth Mormino, Molly Nicholas, Sina Rashidi, Matthew Shonman, Lily Zhang, and Alyssa Zhu for their assistance; Anthony Mander for his aid in task design; and Michael Rubens and Adam Gazzaley for use of their aging template brain. This work was supported by awards R01–AG031164(MPW), R01–AG034570(WJ) and F32–AG039170(BAM), from the National Institutes of Health.
Footnotes
Author contributions
B.A.M. designed the study, conducted the experiments, analyzed the data, and wrote the manuscript. S.M. aided in data analysis and manuscript preparation. J.V. aided in data collection, analysis, and manuscript preparation. V.R. aided in data analysis and manuscript preparation. B.L. aided in study screening procedures and manuscript preparation, J.M.S. provided data analytic tools, aided in data analysis, and manuscript preparation. S.A.I aided in study design and manuscript preparation. W.J. provided the subject pool and data analytic tools, aided in study design, PET data analysis, and manuscript preparation. M.P.W. designed the study, aided data analysis, and wrote the manuscript.
References
- 1.Boyle PA, et al. Much of late life cognitive decline is not due to common neurodegenerative pathologies. Ann Neurol. 2013 doi: 10.1002/ana.23964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buckner RL, et al. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jack CR, Jr, et al. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sepulcre J, Sabuncu MR, Becker A, Sperling R, Johnson KA. In vivo characterization of the early states of the amyloid-beta network. Brain. 2013;136:2239–2252. doi: 10.1093/brain/awt146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spires-Jones TL, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron. 2014;82:756–771. doi: 10.1016/j.neuron.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mormino EC, et al. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain. 2009;132:1310–1323. doi: 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oh H, Jagust WJ. Frontotemporal network connectivity during memory encoding is increased with aging and disrupted by beta-amyloid. J Neurosci. 2013;33:18425–18437. doi: 10.1523/JNEUROSCI.2775-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Backhaus J, et al. Midlife decline in declarative memory consolidation is correlated with a decline in slow wave sleep. Learn Mem. 2007;14:336–341. doi: 10.1101/lm.470507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carrier J, et al. Sleep slow wave changes during the middle years of life. Eur J Neurosci. 2011;33:758–766. doi: 10.1111/j.1460-9568.2010.07543.x. [DOI] [PubMed] [Google Scholar]
- 10.Dijk DJ, Beersma DG, van den Hoofdakker RH. All night spectral analysis of EEG sleep in young adult and middle-aged male subjects. Neurobiol Aging. 1989;10:677–682. doi: 10.1016/0197-4580(89)90004-3. [DOI] [PubMed] [Google Scholar]
- 11.Mander BA, et al. Prefrontal atrophy, disrupted NREM slow waves, and impaired hippocampal-dependent memory in aging. Nature Neuroscience. 2013;16:357–364. doi: 10.1038/nn.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Cauter E, Leproult R, Plat L. Age-related changes in slow wave sleep and REM sleep and relationship with growth hormone and cortisol levels in healthy men. Jama. 2000;284:861–868. doi: 10.1001/jama.284.7.861. [DOI] [PubMed] [Google Scholar]
- 13.Westerberg CE, et al. Concurrent Impairments in Sleep and Memory in Amnestic Mild Cognitive Impairment. J Int Neuropsychol Soc. 2012:1–11. doi: 10.1017/S135561771200001X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marshall L, Helgadottir H, Molle M, Born J. Boosting slow oscillations during sleep potentiates memory. Nature. 2006;444:610–613. doi: 10.1038/nature05278. [DOI] [PubMed] [Google Scholar]
- 15.Murphy M, et al. Source modeling sleep slow waves. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:1608–1613. doi: 10.1073/pnas.0807933106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hita-Yanez E, Atienza M, Cantero JL. Polysomnographic and subjective sleep markers of mild cognitive impairment. Sleep. 2013;36:1327–1334. doi: 10.5665/sleep.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prinz PN, et al. Sleep, EEG and mental function changes in senile dementia of the Alzheimer’s type. Neurobiol Aging. 1982;3:361–370. doi: 10.1016/0197-4580(82)90024-0. [DOI] [PubMed] [Google Scholar]
- 18.Liguori C, et al. Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA Neurol. 2014;71:1498–1505. doi: 10.1001/jamaneurol.2014.2510. [DOI] [PubMed] [Google Scholar]
- 19.Kang JE, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roh JH, et al. Disruption of the sleep-wake cycle and diurnal fluctuation of beta-amyloid in mice with Alzheimer’s disease pathology. Sci Transl Med. 2012;4:150ra122. doi: 10.1126/scitranslmed.3004291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spira AP, et al. Self-reported Sleep and beta-Amyloid Deposition in Community-Dwelling Older Adults. JAMA Neurol. 2013 doi: 10.1001/jamaneurol.2013.4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie L, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–377. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chauvette S, Seigneur J, Timofeev I. Sleep oscillations in the thalamocortical system induce long-term neuronal plasticity. Neuron. 2012;75:1105–1113. doi: 10.1016/j.neuron.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim H. Neural activity that predicts subsequent memory and forgetting: a meta-analysis of 74 fMRI studies. Neuroimage. 2011;54:2446–2461. doi: 10.1016/j.neuroimage.2010.09.045. [DOI] [PubMed] [Google Scholar]
- 25.Riedner BA, et al. Sleep homeostasis and cortical synchronization: III. A high-density EEG study of sleep slow waves in humans. Sleep. 2007;30:1643–1657. doi: 10.1093/sleep/30.12.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pascual-Marqui RD. Standardized low-resolution brain electromagnetic tomography (sLORETA): technical details. Methods Find Exp Clin Pharmacol. 2002;24(Suppl D):5–12. [PubMed] [Google Scholar]
- 27.Buzsaki G. The hippocampo-neocortical dialogue. Cerebral Cortex. 1996;6:81–92. doi: 10.1093/cercor/6.2.81. [DOI] [PubMed] [Google Scholar]
- 28.Frankland PW, Bontempi B. The organization of recent and remote memories. Nat Rev Neurosci. 2005;6:119–130. doi: 10.1038/nrn1607. [DOI] [PubMed] [Google Scholar]
- 29.Diekelmann S, Born J. The memory function of sleep. Nature Reviews Neuroscience. 2010;11:114–126. doi: 10.1038/nrn2762. [DOI] [PubMed] [Google Scholar]
- 30.Takashima A, et al. Declarative memory consolidation in humans: a prospective functional magnetic resonance imaging study. Proc Natl Acad Sci U S A. 2006;103:756–761. doi: 10.1073/pnas.0507774103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walker MP. The role of sleep in cognition and emotion. Annals of the New York Academy of Sciences. 2009;1156:168–197. doi: 10.1111/j.1749-6632.2009.04416.x. [DOI] [PubMed] [Google Scholar]
- 32.Stage FK, Carter HC, Nora A. Path analysis: An introduction and analysis of a decade of research. Journal of Educational Research. 2004;98:5–12. [Google Scholar]
- 33.Bozdogan H. Model Selection and Akaike Information Criterion (Aic) - the General-Theory and Its Analytical Extensions. Psychometrika. 1987;52:345–370. [Google Scholar]
- 34.Kline RB. Principles and Practice of Structural Equation Modeling. The Guilford Press; New York, N.Y: 2011. [Google Scholar]
- 35.Raftery AE. Bayesian model selection in social research. Sociological Methodology 1995. 1995;25:111–163. [Google Scholar]
- 36.Steriade M, Contreras D, Curro Dossi R, Nunez A. The slow (< 1 Hz) oscillation in reticular thalamic and thalamocortical neurons: scenario of sleep rhythm generation in interacting thalamic and neocortical networks. J Neurosci. 1993;13:3284–3299. doi: 10.1523/JNEUROSCI.13-08-03284.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steriade M, Nunez A, Amzica F. A novel slow (< 1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993;13:3252–3265. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurup P, et al. Abeta-mediated NMDA receptor endocytosis in Alzheimer’s disease involves ubiquitination of the tyrosine phosphatase STEP61. J Neurosci. 2010;30:5948–5957. doi: 10.1523/JNEUROSCI.0157-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Snyder EM, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 40.Cavdar S, et al. The pathways connecting the hippocampal formation, the thalamic reuniens nucleus and the thalamic reticular nucleus in the rat. J Anat. 2008;212:249–256. doi: 10.1111/j.1469-7580.2008.00858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ooms S, et al. Effect of 1 Night of Total Sleep Deprivation on Cerebrospinal Fluid beta-Amyloid 42 in Healthy Middle-Aged Men: A Randomized Clinical Trial. JAMA Neurol. 2014 doi: 10.1001/jamaneurol.2014.1173. [DOI] [PubMed] [Google Scholar]
- 42.Ju YE, Lucey BP, Holtzman DM. Sleep and Alzheimer disease pathology--a bidirectional relationship. Nat Rev Neurol. 2014;10:115–119. doi: 10.1038/nrneurol.2013.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cricco M, Simonsick EM, Foley DJ. The impact of insomnia on cognitive functioning in older adults. J Am Geriatr Soc. 2001;49:1185–1189. doi: 10.1046/j.1532-5415.2001.49235.x. [DOI] [PubMed] [Google Scholar]
- 44.Yaffe K, et al. Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA. 2011;306:613–619. doi: 10.1001/jama.2011.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lim AS, et al. Modification of the relationship of the apolipoprotein E epsilon4 allele to the risk of Alzheimer disease and neurofibrillary tangle density by sleep. JAMA Neurol. 2013;70:1544–1551. doi: 10.1001/jamaneurol.2013.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brett M, Anton JL, Valabregue R, Poline JB. Region of interest analysis using an SPM toolbox [abstract]. NeuroImage; 8th International Conference on Functional Mapping of the Human Brain; Sendai, Japan. 2002. [Google Scholar]
- 47.Mander BA, et al. Impaired Prefrontal Sleep Spindle Regulation of Hippocampal-Dependent Learning in Older Adults. Cereb Cortex. 2014;24:3301–3309. doi: 10.1093/cercor/bht188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yesavage JA, et al. Development and validation of a geriatric depression screening scale: a preliminary report. J Psychiatr Res. 1982;17:37–49. doi: 10.1016/0022-3956(82)90033-4. [DOI] [PubMed] [Google Scholar]
- 49.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. Journal of Psychiatric Research. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 50.Delis D, Kramer J, Kaplan E, Ober B. California verbal learning test. The Psychological Corporation; San Antonio, TX: 2000. [Google Scholar]
- 51.Wechsler D. Wechsler memory scale-revised. The Psychological Corporation; San Antonio, TX: 1987. [Google Scholar]
- 52.Reitan RM. Validity of the trail-making test as an indication of organic brain damage. Percept Mot Skills. 1958;8:271–276. [Google Scholar]
- 53.Zec RF. The stroop color-word test: A paradigm for procedural learning. Arch Clin Neuropsychol. 1986;1:274–275. [Google Scholar]
- 54.Young T, Peppard PE, Gottlieb DJ. Epidemiology of obstructive sleep apnea: a population health perspective. Am J Respir Crit Care Med. 2002;165:1217–1239. doi: 10.1164/rccm.2109080. [DOI] [PubMed] [Google Scholar]
- 55.Tan X, Campbell IG, Feinberg I. Internight reliability and benchmark values for computer analyses of non-rapid eye movement (NREM) and REM EEG in normal young adult and elderly subjects. Clin Neurophysiol. 2001;112:1540–1552. doi: 10.1016/s1388-2457(01)00570-3. [DOI] [PubMed] [Google Scholar]
- 56.Zheng H, et al. Sources of variability in epidemiological studies of sleep using repeated nights of in-home polysomnography: SWAN Sleep Study. J Clin Sleep Med. 2012;8:87–96. doi: 10.5664/jcsm.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cohen DA, Robertson EM. Preventing interference between different memory tasks. Nat Neurosci. 2011;14:953–955. doi: 10.1038/nn.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jack CR, Jr, et al. Neurology. 2013;80:890–896. doi: 10.1212/WNL.0b013e3182840bbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Villemagne VL, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12:357–367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- 60.Elman JA, et al. Neural compensation in older people with brain amyloid-beta deposition. Nat Neurosci. 2014;17:1316–1318. doi: 10.1038/nn.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oh H, Madison C, Haight TJ, Markley C, Jagust WJ. Effects of age and beta-amyloid on cognitive changes in normal elderly people. Neurobiol Aging. 2012;33:2746–2755. doi: 10.1016/j.neurobiolaging.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Desikan RS, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–980. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 63.Bradshaw EM, et al. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013;16:848–850. doi: 10.1038/nn.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hedden T, et al. Cognitive profile of amyloid burden and white matter hyperintensities in cognitively normal older adults. J Neurosci. 2012;32:16233–16242. doi: 10.1523/JNEUROSCI.2462-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hedden T, et al. Failure to modulate attentional control in advanced aging linked to white matter pathology. Cereb Cortex. 2012;22:1038–1051. doi: 10.1093/cercor/bhr172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lieberman MD, Cunningham WA. Type I and Type II error concerns in fMRI research: re-balancing the scale. Soc Cogn Affect Neurosci. 2009;4:423–428. doi: 10.1093/scan/nsp052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lancaster JL, et al. Bias between MNI and talairach coordinates analyzed using the ICBM-152 brain template. Human Brain Mapping. 2007;28:1194–1205. doi: 10.1002/hbm.20345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rechtschaffen A, Kales A. A manual of standardized terminology, techniques an scoring system of sleep stages in human subjects. UCLA Brain Information Services; Los Angeles: 1968. [Google Scholar]
- 69.Mander BA, Santhanam S, Saletin JM, Walker MP. Wake deterioration and sleep restoration of human learning. Curr Biol. 2011;21:R183–184. doi: 10.1016/j.cub.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saletin JM, van der Helm E, Walker MP. Structural brain correlates of human sleep oscillations. Neuroimage. 2013;83:658–668. doi: 10.1016/j.neuroimage.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mazziotta J, et al. A probabilistic atlas and reference system for the human brain: International Consortium for Brain Mapping (ICBM) Philos Trans R Soc Lond B Biol Sci. 2001;356:1293–1322. doi: 10.1098/rstb.2001.0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.