

Abstract
Background
Glioblastoma (GBM) is poorly responsive to current chemotherapy. The nuclear transporter exportin 1 (XPO1, CRM1) is often highly expressed in GBM, which may portend a poor prognosis. Here, we determine the efficacy of novel selective inhibitors of nuclear export (SINE) specific to XPO1 in preclinical models of GBM.
Methods
Seven patient-derived GBM lines were treated with 3 SINE compounds (KPT-251, KPT-276, and Selinexor) in neurosphere culture conditions. KPT-276 and Selinexor were also evaluated in a murine orthotopic patient-derived xenograft (PDX) model of GBM. Cell cycle effects were assayed by flow cytometry in vitro and immunohistochemistry in vivo. Apoptosis was determined by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) and caspase 3/7 activity assays.
Results
Treatment of GBM neurosphere cultures with KPT-276, Selinexor, and KPT-251 revealed dose-responsive growth inhibition in all 7 GBM lines [range of half-maximal inhibitory concentration (IC50), 6–354 nM]. In an orthotopic PDX model, treatment with KPT-276 and Selinexor demonstrated pharmacodynamic efficacy, significantly suppressed tumor growth, and prolonged animal survival. Cellular proliferation was not altered with SINE treatment. Instead, induction of apoptosis was apparent both in vitro and in vivo with SINE treatment, without overt evidence of neurotoxicity.
Conclusions
SINE compounds show preclinical efficacy utilizing in vitro and in vivo models of GBM, with induction of apoptosis as the mechanism of action. Selinexor is now in early clinical trials in solid and hematological malignancies. Based on these preclinical data and excellent brain penetration, we have initiated clinical trials of Selinexor in patients with relapsed GBM.
Keywords: apoptosis, exportin 1, glioblastoma, patient-derived xenograft, SINE
Glioblastoma (GBM) is the predominant subtype of World Health Organization grade IV glial tumors.1 GBM is the most common malignant primary brain tumor in adults, with over 10 000 new cases diagnosed each year in the United States.2 Despite optimal treatment with surgery, radiation, and temozolomide, the median survival is only 15 months.3 Following disease progression, most chemotherapeutic agents have minimal activity.2 GBM also occurs in children. Five-year overall survival of supratentorial GBM, which is more common in older children, is 10%–20%.4 A subset of diffuse intrinsic pontine glioma (DIPG), which is more common in younger children, is also classified as GBM, and long-term survival of this disease is <5%.4 The dearth of effective chemotherapy for GBM clearly represents an unmet need.
Exportin 1 (XPO1, CRM1), one of 7 mammalian nuclear export proteins, mediates the export of ∼220 proteins,5 as well as mRNAs.6 Amongst its myriad effects, XPO1 is the sole nuclear exporter of many tumor suppressor proteins, including tumor protein 53 (TP53; p53), retinoblastoma 1 (Rb1; pRb), and cyclin-dependent kinase inhibitor 1B (CDKN1B; p27).7 XPO1 is overexpressed in many cancers,8–10 leading to mislocalization and consequently functional inactivation of tumor suppressor proteins by their translocation outside the nucleus in these diseases. Inhibition of XPO1 as a therapeutic approach has been hampered by inhibitors, such as leptomycin, that were found to be highly toxic to both cancerous and normal cells.11 Presently, though, a novel class of small-molecule, druglike, orally available XPO1 inhibitors, the selective inhibitors of nuclear export (SINE), has shown a high degree of preclinical efficacy and specificity in several hematological and solid malignancies, including acute myeloid leukemia,12,13 T-cell acute lymphoblastic leukemia,14 multiple myeloma,15 melanoma,16 and renal cancer.17 One SINE compound, Selinexor (KPT-330), has advanced to early phase clinical trials.
GBM is one of the tumors in which XPO1 is known to be overexpressed, and the degree of expression correlates directly with glioma tumor grade and inversely with length of survival.18 Given the clear and immediate need for new GBM therapies, and the relationship of these tumors with XPO1 and its clients, we examined the potential efficacy of SINE compounds in preclinical models of GBM.
Methods
Selinexor Pharmacokinetics
A 1 mg/mL IV solution of Selinexor was prepared by vortexing and sonicating the compound serially in a solution containing 10% N-methyl-2-pyrrolidone (Sigma), 10% Solutol HS 15 (BASF), and 80% 10% hydroxypropyl-beta-cyclodextrin (Seebio) in saline. A 1 mg/mL suspension for rat enteral dosing was prepared by vortexing the compound serially in a solution containing 1% sterile water and 99% 1% Pluronic F-68 (BASF) and 1% Povidone K30 (BASF) in water. A 5 mg/mL suspension for monkey enteral dosing was prepared in 0.5% Pluronic F68 solution. Selected for the experiment were 9 Sprague-Dawley rats (SLAC Laboratory) 210–230 g in weight, and 2 male cynomolgus monkeys (Hainan Jinggang Biotech) 2–4 kg in weight. Three rats were administered 5 mg/kg Selinexor IV, and 6 received 10 mg/kg Selinexor by nasogastric tube (3 for time point analysis and 3 for terminal sampling of plasma and brain). Both monkeys received 10 mg/kg Selinexor by nasogastric tube. Blood samples were collected from the rat tail vein and monkey cephalic or saphenous veins at designated time points and stored on wet ice. Animals were euthanized, and brain tissue was harvested and snap-frozen in dry ice. Brain tissue was homogenized in phosphate buffered saline. Aliquots of plasma and homogenized brain were added to Diclofenac 200 mg/mL in acetonitrile, and Selinexor concentration was then measured by ultra-performance liquid chromatography–tandem mass spectrometry. All studies were performed under protocols approved by the Institutional Animal Care and Use Committee.
GBM Tumor Microarray Scoring
GBM tumor microarray #324.1.GBM, containing 4 sections each of 65 GBM tumors and 3 normal brain controls, was stained with anti-XPO1 antibody (Santa Cruz SC-5595) as described below. The tumor microarray was then examined by 2 independent scorers, and each sample was graded based on the percentage of tumor cells (for tumors) or neurons (for controls) staining positive, per the following system: 0 (<5%), 1+ (5%–25%), 2+ (26%–75%), and 3+ (>75%).
Tumor Lines and Culture Conditions
Four primary human adult GBM lines (BT 145, BT 159, BT 172, all derived from tumors resected at initial diagnosis at Brigham and Women's Hospital, and AGBM1, provided by Dr Michelle Monje, Stanford University) and 3 primary human pediatric GBM lines (BT 245, derived from a supratentorial GBM resected at initial diagnosis at Boston Children's Hospital, and DIPG 4 and DIPG 6, derived from previously irradiated DIPGs at autopsy, provided by Dr Monje) were grown in ultra-low attachment flasks (Corning) in neurosphere (suspension) culture conditions.19 All lines were tested by molecular profiling. Molecular characteristics of these lines were determined by array comparative genomic hybridization, using the Agilent 1 M feature array, and are shown in Supplementary Table S1. NeuroCult NS-A media (Stemcell Technologies) supplemented with penicillin-streptomycin (1:100), heparin (2 μg/mL), human epidermal growth factor (EGF; 20 ng/mL), and human basic fibroblast growth factor (FGFb; 10 ng/mL) was used to maintain all lines except for AGBM1, DIPG 4, and DIPG 6; these lines were maintained in Neurobasal-A medium mixed 1:1 with Dulbecco's modified Eagle's medium/F-12 supplemented with 1:100 HEPES [4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid] 1 M, Minimum Essential Medium Eagle (MEM) sodium pyruvate 100 mM, MEM with Non-Essential Amino Acids 10 mM, Glutamax-I, and antibiotic-antimycotic (all Invitrogen), with B27-A supplement 50x (1:50, Invitrogen), heparin (2 μg/mL), and human EGF, FGFb, and platelet derived growth factor–AB (Shenandoah Biotech) (all 20 ng/mL). For all experiments, cells were plated at a concentration of 2 × 105 cells/mL of media. Prior to all endpoint measurements, neurospheres were dispersed by trituration using a micropipette.
Dose-Response Curves
Three structurally similar SINE compounds, KPT-276, Selinexor, and KPT-251, were provided by Karyopharm Therapeutics, and 10 mM solutions of each were made in dimethyl sulfoxide (DMSO). Serial log 10 dilutions were used to make a compound plate with concentrations ranging from 10 mM to 100 nM. Cells were plated in 96-well format with 2 × 104 cells/well, and a pin transfer robot (Janus) was used to transfer 100 nL of compound solution into each well, with 3 replicates per condition. Cell viability was measured after 5 days of continuous drug exposure by CellTiter Blue fluorescence assay (Promega). Relative viability was determined using viability results from cells treated with DMSO alone as a control. Values of half-maximal inhibitory concentration (IC50) and 90% maximal inhibitory concentration (IC90) were calculated using the curve fitting function in Prism (GraphPad).
Radiation and Selinexor Combination
BT 145, BT 159, BT 172, BT 245, and DIPG 4 cells were plated in 96-well format. Compound plates were made with 5 dose levels of Selinexor for each line, including the IC50; 2 dose levels above and 2 dose levels below the IC50; and a DMSO-only control. The compound was delivered by robot pinning of 100 nL per well as described above. On days 2, 3, and 4, cells were irradiated with an open field at 220 kVp and 13 mA using a 35-cm source-to-surface distance on a Small Animal Radiation Research Platform.20 Radiation conditions included ⅓ Gy daily, ⅔ Gy daily, and 1 Gy daily, as well as no radiation. There were 6 replicate wells per condition. Viability was measured on day 7 by CellTiter Blue as above.
Orthotopic Patient-Derived Xenografts
BT 145 cells were infected with the pMMP–LucNeo retrovirus, then selected for infection with G418 500 μg/mL. Prior to injection, cells were resuspended in phosphate buffered saline at a concentration of 1.8 × 105 cells/μL. Forty nonobese diabetic severe combined immunodeficient–interleukin 2 receptor gamma null mice (NOD-SCID gamma [NSG] mice; Jackson Labs) were anesthetized with isoflurane and immobilized using ear bars and a mouthpiece connected to a stereotactic frame. A hole in the skull was made with a 24-gauge needle 2 mm to the right of the bregma. A 33-gauge needle attached to the stereotactic apparatus was then passed through the skull and re-zeroed. The needle was inserted to a depth of 2.5 mm; it was then retracted to 2.0 mm for injection of 1 μL of cell mixture, targeting the right striatum. Mice were subjected to bioluminescence imaging (BLI) 1 month after injections and then every other week for the remainder of the study. Nine and a half weeks after implantation, mice were divided into treatment groups (n = 10 per group) as follows: KPT-276 at 50 mg/kg, Selinexor at 20 mg/kg, and vehicle at 10 mL/kg. Compounds were administered via oral gavage 3 times a week (Monday, Wednesday, Friday). On the basis of prior studies,14,15 doses were escalated after 1 week of treatment as follows: KPT-276 to 75 mg/kg and Selinexor to 25 mg/kg. Compounds continued to be administered 3 times a week for the duration of the study. At treatment day 56, animals from each group with the highest and lowest BLI values were sacrificed, and brains were submitted for neuropathologic examination. On day 61 of treatment, 4–5 mice per group whose BLI levels were closest to the median for their group underwent brain MRI. MRI was performed using a Biospec 7T scanner (Bruker BioSpin), with tumor volume determined from 1-mm-thick T2 images. Mice were sacrificed once they displayed neurological symptoms or became moribund. All studies were performed under protocols approved by the Institutional Animal Care and Use Committee.
Staining, Immunohistochemistry, and Immunofluorescence
The brains from the highest and lowest bioluminescent animals in each treatment group at treatment day 56 were sectioned with razor blades coronally into 2-mm-thick blocks. Staining, immunohistochemistry (IHC), and immunofluorescence (IF) were performed on 4-micron-thick paraffin sections. Hematoxylin and eosin (H&E) staining was performed by the Harvard Medical School Rodent Histopathology Core. Luxol fast blue–cresyl violet staining was performed by the Brigham and Women's Neuropathology Core. For IHC and IF, deparaffinized sections were subjected to antigen retrieval with 1 mM Na citrate. Sections were blocked with Dako peroxidase for 10 min. Diluted per institutional protocols (generally 1:200 or per manufacturer recommendations if different) and incubated overnight at 4°C were primary antibodies to human-specific nuclear mitotic apparatus protein 1 (NUMA1; Epitomics S2825), marker of proliferation Ki-67 (MKI67; Vector VP-RM04), glial fibrillary acidic protein (GFAP; Abcam ab7260), tubulin beta 3 (TUBB3; Covance MMS-435P), Rb1 (BD 554136), TP53 (Immunotech 1767), CDKN1B (CST 2552P), CDKN2A (Ventana 9517), myeloid cell leukemia 1 (MCL1; CST 4572), XPO1 (Santa Cruz 5595), and cluster of differentiation 31 (CD31; Abcam 28364). After washing in Tris-buffered saline and 0.05% Tween 20, anti-rabbit or anti-mouse secondary (Dako) was appropriately added for 1 h at room temperature. For IHC, slides were then counterstained with Mayer's hematoxylin and fixed with Permount. For IF, secondary antibodies included Alexa Rb 488 for NUMA1 and Ms 555 for MKI67 (both Invitrogen); slides were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) and fixed with Vectashield. Staining for terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick end labeling (TUNEL) was done with the TUNEL DeadEnd Fluorometric System (Promega), according to manufacturer instructions for formalin-fixed paraffin embedded tissue. For cell counts, multiple 60× fields from tumor-involved areas in each brain were imaged, and cells were counted manually.
Western Blot
Cells in neurosphere culture were treated for 48 h at the IC50 and twice the IC90 levels of KPT-276 and Selinexor compared with DMSO control (MCL1 expression) or treated 7 days at the IC50 concentrations of Selinexor compared with DMSO control (XPO1 expression with SINE treatment). Protein lysates were then made by adding 20 μL of radioimmunoprecipitation assay buffer with 1:100 Halt protease/phosphatase (Pierce). Protein concentration was measured by Bradford assay, and 20 μg of protein per sample was run on a NuPAGE Mini 10% 1 mm thickness Bis-Tris gel. Transfer was then performed to Immobilon-P polyvinylidene difluoride membrane (Millipore). The membrane was then incubated in 5% milk with 1:1000 MCL1 antibody (CST 4572) or 1:200 XPO1 antibody (SC 5595) overnight at 4°C and developed with SuperSignal West Femto chemiluminescent (Thermo).
Propidium Iodide Flow Cytometry
Neurospheres were treated in 6-well format (4 × 105 cells/condition) for 5 days at the IC50 and twice the IC90 concentrations compared with DMSO controls. They were then washed in Dulbecco's phosphate buffered saline and fixed in 70% ethanol for 30 min at room temperature. Cells were then stained with 0.01 mg/mL propidium iodide solution containing 0.1 mg/mL RNase, and incubated at 37°C for 30 min. Propidium iodide staining was then measured by flow cytometry on a BD LSRFortessa machine and analyzed with ModFit.
Measurement of Apoptosis In vitro
BT 145 and BT 159 cells in neurosphere culture were treated in 6-well format for 5 days with the IC90 and twice the IC90 concentrations of KPT-276 and Selinexor compared with DMSO control. Cells were then transferred to chamber slides (BD) and allowed to settle. TUNEL staining was then performed using the TUNEL DeadEnd Fluorometric System (Promega) according to manufacturer instructions. Since most cells remained in suspension, a minimum volume of 150 μL was maintained in each chamber during staining, removal of chambers, and coverslipping. BT 145, BT 159, BT 172, BT 245, and DIPG 4 cells were plated in 96-well format. A compound plate was created with Selinexor in 4 dose levels separated by half-log10 increments, ranging from 10 μM to 316 nM, along with DMSO-only control. The compound was delivered by robot pinning of 100-nL aliquots as above. There were 6 replicate wells per condition. Apoptosis was measured using the Caspase Glo 3/7 Assay (Promega) at 48 h of treatment after 2 h of incubation with the Caspase Glo reagent at room temperature.
Data Analysis
Statistical analysis was done by 2-way ANOVA for BLI and by a log-rank test for survival studies. For MRI tumor volume, MKI67+ percentage, TUNEL+ (in vitro and in vivo) percentage, and MCL1+ percentage, the 1-tailed, unpaired t-test was used to compare means of endpoint measurements for treated versus control cells. *P = .005–.05; **P = .0005–.005; ***P< .0005.
Results
Assessment of Pharmacokinetics of Selinexor
KPT-251, KPT-276, and Selinexor (KPT-330) share the same chemical warhead but differ in their pharmacokinetic properties. The properties of KPT-251 and KPT-276 have been previously described.15 The physicochemical properties of Selinexor (Fig. 1A) are consistent with features associated with existing central nervous system drugs,21 and the compound would be predicted to penetrate the blood–brain barrier. The pharmacokinetics of Selinexor in rats revealed peak serum levels from 0.5 to 2 h after oral administration (Fig. 1B). Assessment of brain penetration 2 h after single oral dosing of Selinexor revealed brain:plasma ratios averaging 0.72 in rats and 0.61 in cynomolgus monkeys (Fig. 1C). Together, these results support the use of Selinexor for a neuro-oncology indication.
Fig. 1.
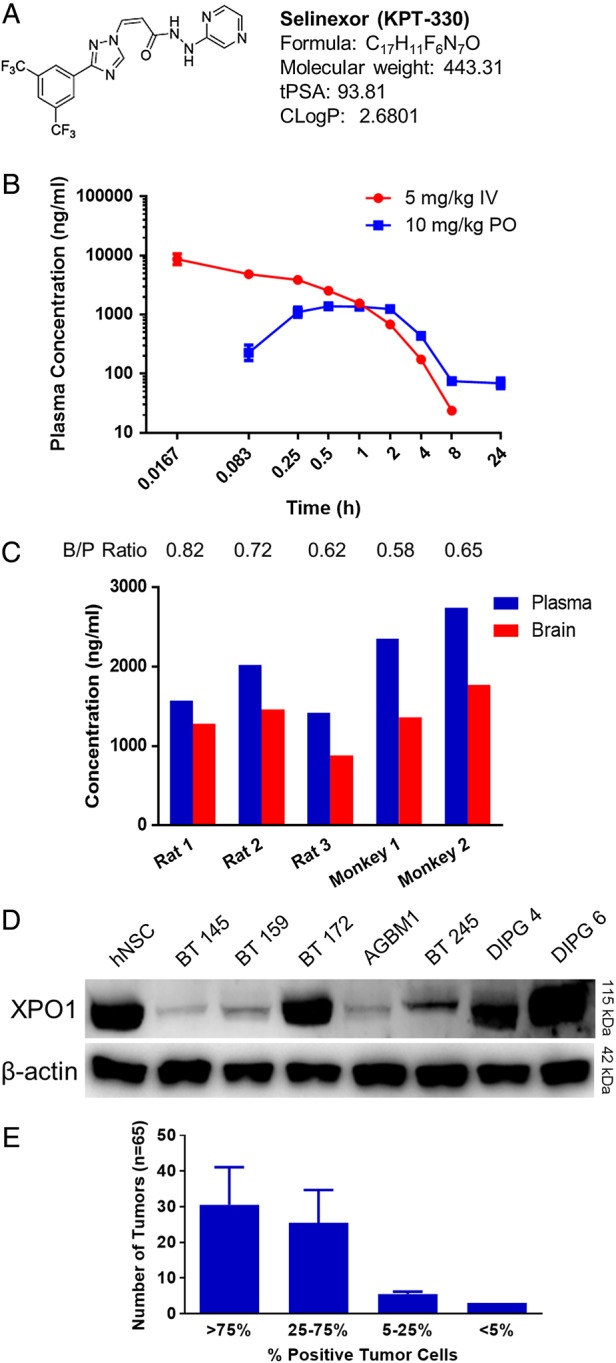
Selinexor (KPT-330) pharmacokinetics and XPO1 expression. (A) Chemical structure and key properties of Selinexor related to brain penetration. (B) Plasma concentration of Selinexor over time after single IV and oral doses in rats. (C) Brain vs plasma (B/P) concentrations of Selinexor in rat and monkey test subjects 2 h after a single oral dose. (D) Western blot for XPO1 in all tumor lines without treatment, with hNSC control. (E) Results of scoring of a GBM tumor microarray for percentage of tumor cells staining positive for XPO1, expressed as mean number of samples per category ± SD.
Exportin 1 Expression in Cell Lines and Other GBM Samples
We assessed baseline expression of XPO1 in a panel of 4 adult (BT 145, BT 159, BT 172, and AGBM1) and 3 pediatric (BT 245, DIPG 4, and DIPG 6) patient-derived, low-passage GBM lines propagated in neurosphere culture conditions, using western blot compared with normal human neuronal stem cells (hNSC). XPO1 expression was detectable in all lines, but levels were variable (Fig. 1D). BT 172 and both DIPG lines showed expression at or above the level in hNSC, while the remainder of the lines showed expression less than hNSC. XPO1 expression was also assessed by IHC of a GBM tumor microarray. XPO1 expression was present in nearly all tumor samples, and most tumors showed XPO1 expression in 25%–100% of tumor cells (Fig. 1E); of the 3 normal brain controls on the tumor microarray, 1 showed 25%–75% positivity, and the other 2 showed >75% positivity of neurons. These results demonstrate consistently positive but variable levels of expression of XPO1 in hNSC and GBM.
SINE Compounds Inhibit Ex vivo GBM Cultures at Sub-Micromolar Concentrations
To assess efficacy of SINE compounds on GBM cells in vitro, we utilized the 7 primary human GBM lines previously mentioned, treated in neurosphere culture conditions (Fig. 2A) with 3 SINE compounds (KPT-276, Selinexor, and KPT-251) spanning a concentration range from 0.1 nM to 100 μM. Viability was measured after 5 days of treatment and normalized to DMSO controls (Fig. 2B). IC50 values were generally consistent across the compounds and lines, and all were within a 2-log range (6–354 nM; Supplementary Table S2). These IC50 values are similar to those found for KPT-185, KPT-251, and Selinexor in preclinical models of other cancer types, including acute myeloid leukemia,12,13 T-cell acute lymphoblastic leukemia,14 multiple myeloma,15 and melanoma,16 and are lower than those for KPT-185 in renal cell carcinoma.17 Since the 3 tested compounds performed similarly in these studies, 2 were taken forward for subsequent studies: KPT-276, due to its higher lipophilicity (relevant to neuro-oncology application), and Selinexor, the compound currently in clinical trials.
Fig. 2.
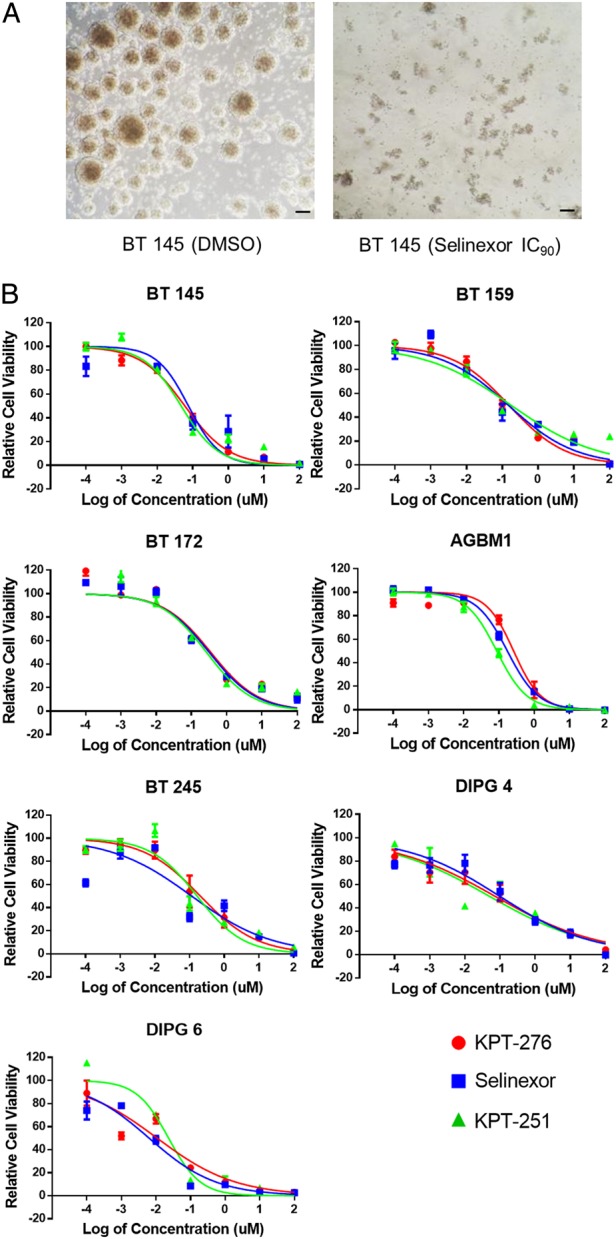
In vitro effects of SINE compounds. (A) Representative light microscopy images of BT 145 DMSO control neurospheres vs those treated with the IC90 concentration of Selinexor (scale bars, 100 μm). (B) Dose-response curves of 7 primary human GBM lines to 3 SINE compounds, shown as mean ± SEM fluorescence intensity level relative to DMSO control.
Effects of SINE Treatment in a Patient-Derived GBM Xenograft Model
To determine whether SINE compounds are able to reach therapeutically relevant levels in the brain, we utilized a patient-derived xenograft (PDX) model by stereotactic injection of 1.8 × 105 luciferase-expressing BT 145 cells into the right striatum of NSG mice. Tumors were followed by serial bioluminescence imaging (BLI). At 9.5 weeks post-implantation, when tumors were steadily growing, mice were divided into 3 treatment groups of 10 mice each and treated with 3 times weekly oral dosing of Selinexor, KPT-276, or vehicle. Weight loss is a known clinical effect of SINE therapy, and thus mice demonstrating >10% weight loss had treatment held until they regained lost weight. Only 2 mice total, both in the Selinexor treatment group, required treatment to be held, missing 9 and 3 doses, respectively. Treatments were continued until mice developed neurological symptoms, at which point they were sacrificed.
Intracranial tumor growth, measured by BLI, was significantly decreased in the groups treated with KPT-276 and Selinexor compared with the vehicle group (P < .001 for both groups; Fig. 3A). At treatment day 56, when the first mice from the vehicle group demonstrated neurological symptoms, the mice from each group with the highest and lowest BLI values were sacrificed so that existence and mechanism of differences in tumor growth at this time point could be explored histologically. On treatment day 61, a subset of mice from each group with BLI levels closest to the respective group medians underwent brain MRI as an independent measure of tumor burden. Tumor volumes reconstructed from MRI confirmed significant reduction in tumor burden in SINE-treated animals compared with vehicle-treated controls (KPT-276 P = .03, Selinexor P = .0004; Fig. 3B). Treatment with KPT-276 and Selinexor significantly prolonged survival compared with vehicle treatment in mice (KPT-276 P < .001, Selinexor P = .001; Fig. 3C).
Fig. 3.
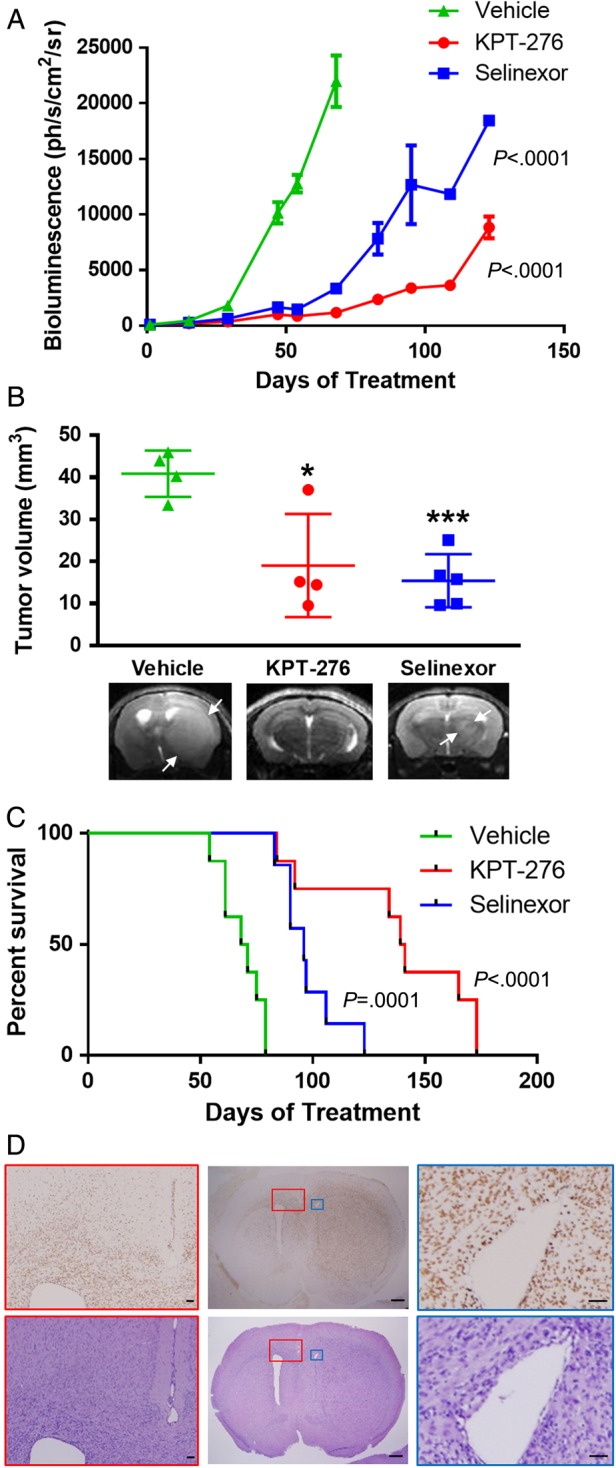
In vivo efficacy of SINE compounds. (A) Tumor growth, measured by mean BLI ± SEM, over time from start of treatment in groups of mice with intracranial GBM xenografts treated with Selinexor, KPT-276, or vehicle. (B) Comparison of tumor volumes (individual values plotted, with bars showing mean ± SD), measured on MRI, for subsets of mice from each group done on treatment day 61, with representative T2 MRIs. (C) Kaplan–Meier curve of mouse survival over time from start of treatment for each group. (D) Representative H&E and human-specific NUMA1 IHC images of a vehicle-treated mouse brain from the PDX study, with a horizontal section in the center showing the main tumor mass with diffuse tumor infiltration and midline shift (scale bars, 1 mm), and inset views of the corpus callosum and lateral ventricle contralateral to the tumor (red boxes; scale bars, 100 μm) and lateral ventricle ipsilateral to the tumor, showing compression (blue boxes; scale bars, 100 μm).
Histopathologic examination of tumors showed invasion of glioma cells into the normal brain parenchyma, consistent with known behavior of GBM, as seen by H&E staining, and IHC with a human-specific NUMA1 antibody to highlight tumor cells (Fig. 3D and Supplementary Fig. S1A). The ratio of tumor cell density between the main tumor mass and invasive areas of the tumor was similar when vehicle-treated mice were compared with mice treated with KPT-276 (P = .13) or Selinexor (P = .20) (Supplementary Fig. S1B). There were no overt neurological symptoms attributable to drug treatment, and Luxol fast blue–cresyl violet staining of the brain hemisphere contralateral to the tumor showed a normal appearance of myelin in all treatment and control groups (Supplementary Fig. S1C). CD31 IHC for angiogenesis showed no difference between treatment and control groups (Supplementary Fig. S1C).
Pharmacodynamic Efficacy of SINE Treatment
To determine pharmacodynamic efficacy of SINE treatment, we first examined the effect of treatment on XPO1 protein expression. As described in other disease models, including multiple myeloma15 and Philadelphia chromosome-positive leukemias,22 XPO1 protein levels decrease secondary to treatment by SINE compounds. Using XPO1 IHC on tumor samples from the PDX study, we found consistent expression in neurons across the vehicle- and SINE-treated animals, but a diminution of XPO1 expression in tumor cells with SINE treatment compared with vehicle treatment (Fig. 4A). We then validated this finding in vitro through western blot. Cells were treated in neurosphere culture for 7 days. BT 145, BT 159, and BT 245 cells treated at their IC50 doses of Selinexor showed a mild decrease in XPO1 expression compared with cells treated with DMSO alone (Fig. 4B).
Fig. 4.

Pharmacodynamic efficacy and lack of proliferation effect of SINE compounds. (A) Representative images of XPO1 IHC for each of the murine intracranial xenograft groups (scale bars, 20 μm). (B) Western blot for XPO1 in 3 tumor lines treated with the IC50 dose of Selinexor compared with DMSO control; GAPDH, glyceraldehyde 3-phosphate dehydrogenase. (C) Representative images of NUMA1 IHC for each of the murine intracranial xenograft groups (scale bars, 10 μm). (D) Representative images of MCL1 IHC staining for each of the murine intracranial xenograft groups (scale bars, 20 μm). (E) Mean ± SD percentages of MCL1+ cells for each of the murine intracranial xenograft groups. (F) Western blot for MCL1 in AGBM1 cells treated with IC50 and double the IC90 levels of KPT-276 and Selinexor vs control. (G) Percentage of cells in each cell cycle phase, as determined by propidium iodide flow cytometry, after treatment of 2 tumor lines in neurosphere culture with KPT-276 or Selinexor at the line-specific IC90 or double the IC90 concentration vs control. (H) Representative merged images of NUMA1 and MKI67 IF for each of the murine intracranial xenograft groups (scale bars, 20 μm). (I) Mean ± SD percentage of NUMA1+ cells that were also MKI67+ for each of the murine intracranial xenograft groups.
Next, we performed IHC for NUMA1 on tumor samples from the PDX study. XPO1 is known to affect the cellular localization of NUMA1,23 an abundant component of the nuclear matrix in its normal state. NUMA1 localization in tumor cells clearly transitions from diffuse in vehicle-treated animals to intensely nuclear in animals treated with KPT-276 and Selinexor (Fig. 4C). Together, these results demonstrate that Selinexor treatment resulted in XPO1 inhibition in vivo in orthotopic GBM models.
We also used IHC to examine the localization of a number of XPO1 client proteins, including regulators of cell cycle (Rb1, CDKN2A, CDKN1B) and apoptosis (TP53). IHC revealed similar staining patterns for Rb1, CDKN2A, CDKN1B, and TP53 in vehicle compared with SINE-treated tumor sections (Supplementary Fig. S2A). We also examined expression of MCL1, an anti-apoptotic member of the BCL2 family that is a known regulator of apoptosis in GBM.24 It is not known to be an XPO1 client but may be highly dependent on mRNA shuttling due to a short half-life.25 The percentage of MCL1-positive cells was significantly decreased in animals treated with KPT-276 and Selinexor compared with vehicle-treated controls (KPT-276 P = 2 × 10−8, Selinexor P = .02; Fig. 4D and E). To validate these results, we performed protein quantification of MCL1 by western blot in 3 tumor lines in neurosphere culture, BT 145, BT 159, and AGBM1, treated with the IC50 and twice the IC90 doses of KPT-276 and Selinexor compared with DMSO, in order to evaluate a partial and near-total response versus control. AGBM1 cells exhibited a dose-dependent diminution of MCL1 levels with increasing concentrations of both SINE compounds (Fig. 4F); however, diminution of protein abundance with treatment was not observed in the other lines and therefore is not a generalizable explanation for increased apoptosis in GBM cells.
SINE Compounds Do Not Cause Cell Cycle Arrest or Differentiation in GBM
To determine whether SINE compounds impact proliferation in GBM, we first assessed the cell cycle distribution in SINE-treated cells compared with DMSO control in neurosphere culture. BT 145 and BT 159 tumor cells were treated with the IC90 and twice the IC90 concentrations of KPT-276 and Selinexor. Flow cytometric analysis of propidium iodide–stained cells revealed no significant effect of treatment on the percentage of cells in S-phase in either line (Fig. 4G). Next, we assessed the effects of SINE compounds on proliferation in sections of tumors harvested from the PDX treatment study. We co-stained tumor sections with antibodies for human NUMA1 and MKI67 (Fig. 4H). The percentage of NUMA1-positive cells that were also MKI67 positive was not significantly different in either the KPT-276 or Selinexor groups compared with vehicle (P = .48 for both groups; Fig. 4I). Finally, we performed IHC for GFAP, a glial marker, and TUBB3 (Tuj1), a neuronal marker, in brains of SINE-treated compared with vehicle-treated mice from the PDX experiment (Supplementary Fig. S2B). Staining appeared similar among the 3 groups, suggesting that terminal differentiation was not a significant cause of decreased growth from treatment.
Treatment With SINE Compounds Induces Apoptosis in GBM In vitro and In vivo
To assess apoptosis in SINE-treated cells, we first determined the percentage of apoptotic cells in BT 145 and BT 159 neurosphere culture samples treated with Selinexor IC90 concentration compared with DMSO controls. Cells were treated in neurosphere culture format, then transferred to slides, stained with TUNEL, and counted under IF microscopy (Fig. 5A). The percentage of apoptotic cells was significantly greater in treated compared with control samples of BT 145 (11.5% vs 4.3%, P = .009) and showed a trend toward significance in BT 159 (42.5% vs 23.3%, P = .06) (Fig. 5B). In addition, we assessed SINE-induced apoptosis in vitro in BT 145, BT 159, BT 172, BT 245, and DIPG 4 via caspase 3/7 activity measurement after 48 h of treatment with a range of Selinexor concentrations from 10−0.5 to 101 μM compared with DMSO control. All lines showed a significant increase in apoptosis at the highest Selinexor concentration compared with DMSO control, and most also showed a significant dose-responsive effect (Fig. 5C).
Fig. 5.

SINE compounds induce apoptosis in GBM. (A) Representative images of DAPI (nuclear) and TUNEL IF (scale bars, 100 μm), and (B) mean ± SD percentage of TUNEL+ cells in BT 145 and BT 159 cells in neurosphere culture treated with Selinexor compared with DMSO control. (C) Levels of apoptosis, as measured by caspase 3/7 activity after 48 h of treatment, in adult and pediatric tumor lines treated in vitro with a standard range of Selinexor doses compared with control, expressed as mean ± SEM luminescence levels. (D) Representative images of fields of NUMA1 IHC staining and TUNEL IF (scale bars, 100 μm), and (E) mean ± SD percentage of TUNEL+ cells for each of the murine intracranial xenograft groups.
We next assessed apoptosis induction in vivo by determining the percentage of TUNEL-positive tumor cells in the BT 145 PDX sections (Fig. 5D). The percentage of apoptotic cells was significantly higher in the KPT-276 group than in the vehicle group (3.2% vs 1.2%, P = .007; Fig. 5E). The fraction of apoptotic cells in Selinexor-treated animals was elevated (mean 3.4%), but results were not statistically significant. Together, these data demonstrate that the antiglioblastoma effects of SINE treatment are due to induction of apoptosis, and not antiproliferation, both in vitro and in vivo.
Selinexor and Radiation Therapy Combination
In anticipation of translation to human clinical trials, we next explored the potential for Selinexor to be combined with an other therapeutic modality used to treat GBM. Since radiation therapy (RT) is standard of care at diagnosis in both adult and pediatric GBM,26 we examined the interaction between RT and Selinexor in vitro. BT 145, BT 159, BT 172, BT 245, and DIPG 4 cells were treated with 5 dose levels of Selinexor, each line's IC50 and 2 levels bracketing the IC50 above and below, as well as DMSO only as a control. Cell plates were subjected to various RT doses on days 2–4 of drug treatment, including ⅓ Gy daily, ⅔ Gy daily, and 1 Gy daily, as well as no RT. Cell viability was then measured on day 7. For cell lines that showed minimal response to these RT doses (BT 145 and DIPG 4), the viability at the high and low RT doses and no RT is shown (Fig. 6A). For cell lines that demonstrated monotherapeutic response to radiation (BT 159, BT 172, and BT 245), viability at the RT dose most closely approximating the IC50 is displayed (Fig. 6B). In addition, an “additive effect line” is displayed, with points calculated by multiplying the relative viability of cells with that RT dose by the relative viability of cells at that drug level in the no-RT condition. This line thus shows the expected viability of cells assuming Selinexor and RT have nonoverlapping mechanisms of action. In aggregate, combinatorial treatment with Selinexor and RT appeared to show an additive effect, with little evidence of either antagonism or synergy.
Fig. 6.
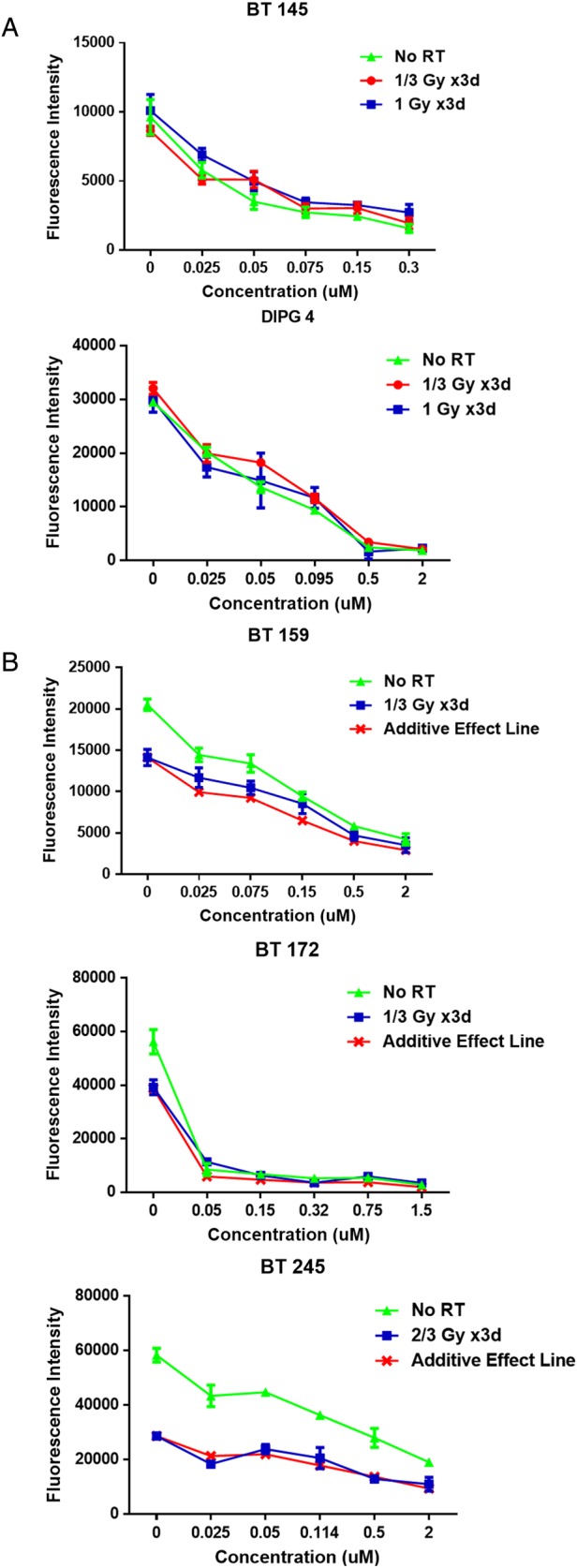
Radiation in combination with Selinexor. (A) Dose-response curves for 2 cell lines resistant to RT, expressed as mean ± SEM fluorescence intensity for various RT conditions across a range of Selinexor concentrations. (B) Dose-response curves for 3 cell lines sensitive to RT, expressed as mean ± SEM fluorescence intensity for no RT and the RT dose closest to the IC50 for each line, with an additive effect line showing a calculated curve assuming nonoverlapping mechanisms of action of Selinexor and RT.
Discussion
In this study, we demonstrate the efficacy of SINE compounds in preclinical models of GBM. We find sub-micromolar IC50 values for 3 SINE compounds across a panel of primary human GBM lines tested in neurosphere culture conditions. Using a PDX model of GBM, we demonstrate pharmacodynamic efficacy of orally administered Selinexor and KPT-276, and antitumor efficacy. The mechanism of action of SINE compounds in GBM appears to be through induction of apoptosis, and not through cell cycle arrest or terminal differentiation. Induction of apoptosis is likely due to pleiotropic effects of XPO1 inhibition on both protein and mRNA transport.
The primary human tumor lines used in this study are biologically heterogeneous (Supplementary Table S1), consistent with GBM as a whole. The uniform efficacy of SINE compounds across these lines, representing several predominant genotypes, as well as adult and pediatric disease, encouragingly suggests that this treatment strategy could be generalizable to a diverse set of GBM subclasses. While expression of the SINE target XPO1 was variable across the cell lines studied, the responsiveness of the lines to SINE treatment in vitro did not appear to correlate with the level of XPO1 expression. Of note, plasma levels of Selinexor achieved in clinical trials are ∼10-fold higher than the IC50 concentrations found in our studies, which is promising in terms of achievement of therapeutic effect.
One line, BT 145, was then used to assess target engagement in vivo due to its known ability to establish orthotopic tumors within several months of injection.27 Results from this model demonstrate that SINE compounds can achieve pharmacodynamic and therapeutic efficacy in the protected environment of the brain. The decrease in XPO1 expression with SINE treatment, a finding replicated across cell lines in vitro, is likely due to protease degradation of XPO1 following SINE treatment and has been seen in other disease models.15,22 The increased effect of KPT-276 over Selinexor found in the PDX model may be due to the fact that KPT-276 is more lipophilic, giving it an advantage in penetration in the lipid-rich environment of the brain.15 It is also encouraging that symptomatic and histopathologic effects of treatment were minimal, suggesting no overt neurotoxicity with SINE treatment. A structurally related SINE compound, KPT-185, has been shown to form a slowly reversible covalent bond (t1/2 ∼24 h) with cysteine 528 of XPO1, in contrast to the completely irreversible nature of the bond formed with the natural XPO1 inhibitor leptomycin B, which may account for the improved therapeutic window of SINE compounds compared with this predecessor.28 Anorexia and weight loss, which were observed in mice treated with Selinexor, are known side effects from preclinical and phase I trials of Selinexor and have been palliated with appetite-stimulating agents and supportive care.
Preclinical studies of SINE compounds in other tumors have found diverse mechanisms of action, including induction of cell cycle arrest, apoptosis, and differentiation. The majority of the evidence we present here suggests apoptosis induction as the primary mechanism of SINE compounds in GBM, an effect that was observed across the diverse cell lines used in this study. One potential mechanism contributing to induction of apoptosis is decreased expression of the anti-apoptotic BCL2 family member MCL1, which could be due to nuclear sequestration of its mRNA by XPO1 inhibition. However, these findings were seen in only 1 of 3 tumor lines tested, and therefore we do not believe MCL1 diminution to be the sole mechanism of action of XPO1 inhibition in GBM. Rather, these variable results across tumor lines give further evidence that the molecular basis for induction of apoptosis is pleiotropic and likely to include both protein and mRNA effects of XPO1 inhibition. Identifying the mechanisms responsible for SINE induction of apoptosis in GBM is a key goal of future studies.
The preclinical study presented here, along with the favorable CNS penetration properties of Selinexor, establish a compelling rationale for proceeding with clinical testing of Selinexor in patients with GBM. Although there are clear biological differences between adult and pediatric GBM and between pediatric supratentorial tumors and DIPG, the pleiotropic effects of XPO1 inhibition may be advantageous in this setting, since our data suggest generalizability of efficacy across patient ages and genetic subtypes of GBM. Accordingly, we have initiated a phase II trial of Selinexor in adult patients with recurrent GBM and are developing a pediatric phase I trial that will include children with recurrent GBM/DIPG. Furthermore, our data suggest an additive relationship between Selinexor and RT, the latter of which is a standard treatment for adult and pediatric patients with primary GBM and is increasingly being used at recurrence as well.29,30 This indicates that future clinical trials of combination therapy may also be possible.
Supplementary Material
Funding
This work was supported by St. Baldrick's Foundation (A.L.K., A.L.G.), Pedals for Pediatrics (A.L.G.), TeamConnor (A.L.G.), and the Stahl Family Foundation (A.L.G.).
Supplementary Material
Acknowledgments
The authors wish to thank Dr Michelle Monje for providing 2 GBM lines and molecular characterization data for these lines; Dr Yanping Sun for MRI support; Dr Allison O′Neill for technical advice; Dr Yosef Landesman for advice regarding pharmacodynamics; Silvia Escudero, Catherine Gallagher, and Dr Loren Walensky for advice and support regarding MCL1 inhibition; Dr Brian Crompton, Michael McKeown, Roodolph St Pierre, and Justin Roberts for assistance with dose-response testing and pin transfer; Ludmila Flores for assistance with cell irradiation; the Rodent Histopathology Core at Harvard Medical School; the Flow Cytometry Core and the Radiation Oncology SARRP Pre-Clinical Irradiation Facility at Dana-Farber Cancer Institute; Christine Unitt and the Specialized Histopathology Core at Brigham and Women's Hospital; and Drs Mark Kieran, Jay Bradner, and Stuart Orkin for their advice and support of this work.
Conflict of interest statement. D.M. and S.S. are employees of, and hold equity in, Karyopharm Therapeutics.
References
- 1.Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492–507. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. [DOI] [PubMed] [Google Scholar]
- 4.Fangusaro J. Pediatric high-grade gliomas and diffuse intrinsic pontine gliomas. J Child Neurol. 2009;24(11):1409–1417. [DOI] [PubMed] [Google Scholar]
- 5.Xu D, Grishin NV, Chook YM. NESdb: a database of NES-containing CRM1 cargoes. Mol Biol Cell. 2012;23(18):3673–3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siddiqui N, Borden KL. mRNA export and cancer. Wiley Interdiscip Rev RNA. 2012;3(1):13–25. [DOI] [PubMed] [Google Scholar]
- 7.Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol. 2012;83(8):1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang WY, Yue L, Qiu WS, et al. Prognostic value of CRM1 in pancreas cancer. Clin Invest Med. 2009;32(6):E315. [PubMed] [Google Scholar]
- 9.Noske A, Weichert W, Niesporek S, et al. Expression of the nuclear export protein chromosomal region maintenance/exportin 1/Xpo1 is a prognostic factor in human ovarian cancer. Cancer. 2008;112(8):1733–1743. [DOI] [PubMed] [Google Scholar]
- 10.Yao Y, Dong Y, Lin F, et al. The expression of CRM1 is associated with prognosis in human osteosarcoma. Oncol Rep. 2009;21(1):229–235. [PubMed] [Google Scholar]
- 11.Mutka SC, Yang WQ, Dong SD, et al. Identification of nuclear export inhibitors with potent anticancer activity in vivo. Cancer Res. 2009;69(2):510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Etchin J, Sun Q, Kentsis A, et al. Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia. 2013;27(1):66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ranganathan P, Yu X, Na C, et al. Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood. 2012;120(9):1765–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Etchin J, Sanda T, Mansour MR, et al. KPT-330 inhibitor of CRM1 (XPO1)-mediated nuclear export has selective anti-leukaemic activity in preclinical models of T-cell acute lymphoblastic leukaemia and acute myeloid leukaemia. Brit J Haematol. 2013;161(1):117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tai YT, Landesman Y, Acharya C, et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: molecular mechanisms and therapeutic implications. Leukemia. 2014;28(1):155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salas Fragomeni RA, Chung HW, Landesman Y, et al. CRM1 and BRAF inhibition synergize and induce tumor regression in BRAF-mutant melanoma. Mol Cancer Ther. 2013;12(7):1171–1179. [DOI] [PubMed] [Google Scholar]
- 17.Inoue H, Kauffman M, Shacham S, et al. CRM1 blockade by selective inhibitors of nuclear export attenuates kidney cancer growth. J Urol. 2013;189(6):2317–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen A, Wang Y, Zhao Y, et al. Expression of CRM1 in human gliomas and its significance in p27 expression and clinical prognosis. Neurosurgery. 2009;65(1):153–159; discussion 159–160. [DOI] [PubMed] [Google Scholar]
- 19.Mehta S, Huillard E, Kesari S, et al. The central nervous system-restricted transcription factor olig2 opposes p53 responses to genotoxic damage in neural progenitors and malignant glioma. Cancer Cell. 2011;19(3):359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong J, Armour E, Kazanzides P, et al. High-resolution, small animal radiation research platform with x-ray tomographic guidance capabilities. Int J Radiat Oncol Biol Phys. 2008;71(5):1591–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pajouhesh H, Lenz GR. Medicinal chemical properties of successful central nervous system drugs. NeuroRx. 2005;2(4):541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker CJ, Oaks JJ, Santhanam R, et al. Preclinical and clinical efficacy of XPO1/CRM1 inhibition by the karyopherin inhibitor KPT-330 in Ph+ leukemias. Blood. 2013;122(17):3034–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rousselet A. Inhibiting Crm1 causes the formation of excess acentriolar spindle poles containing NuMA and B23, but does not affect centrosome numbers. Biol Cell. 2009;101(12):679–693. [DOI] [PubMed] [Google Scholar]
- 24.Sheng Z, Li L, Zhu LJ, et al. A genome-wide RNA interference screen reveals an essential CREB3L2-ATF5-MCL1 survival pathway in malignant glioma with therapeutic implications. Nat Med. 2010;16(6):671–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maurer U, Charvet C, Wagman AS, et al. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006;21(6):749–760. [DOI] [PubMed] [Google Scholar]
- 26.Sturm D, Bender S, Jones DT, et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer. 2014;14(2):92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gruber Filbin M, Dabral SK, Pazyra-Murphy MF, et al. Coordinate activation of Shh and PI3K signaling in PTEN-deficient glioblastoma: new therapeutic opportunities. Nat Med. 2013;19(11):1518–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun Q, Carrasco YP, Hu Y, et al. Nuclear export inhibition through covalent conjugation and hydrolysis of Leptomycin B by CRM1. Proc Natl Acad Sci U S A. 2013;110(4):1303–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Balducci M, Diletto B, Chiesa S, et al. Low-dose fractionated radiotherapy and concomitant chemotherapy for recurrent or progressive glioblastoma: final report of a pilot study. Strahlenther Onkol. 2014;190(4):370–376. [DOI] [PubMed] [Google Scholar]
- 30.Fontanilla HP, Pinnix CC, Ketonen LM, et al. Palliative reirradiation for progressive diffuse intrinsic pontine glioma. Am J Clin Oncol. 2012;35(1):51–57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.