

Abstract
Background
Leptomeningeal metastases occur in 2%–5% of patients with breast cancer and have an exceptionally poor prognosis. The blood–brain and blood–meningeal barriers severely inhibit successful chemotherapy. We have developed a straightforward method to induce antitumor memory T-cells using a Her2/neu targeted vesicular stomatitis virus. We sought to determine whether viral infection of meningeal tumor could attract antitumor memory T-cells to eradicate the tumors.
Methods
Meningeal implants in mice were studied using treatment trials and analyses of immune cells in the tumors.
Results
This paper demonstrates that there is a blood–meningeal barrier to bringing therapeutic memory T-cells to meningeal tumors. The barrier can be overcome by viral infection of the tumor. Viral infection of the meningeal tumors followed by memory T-cell transfer resulted in 89% cure of meningeal tumor in 2 different mouse strains. Viral infection produced increased infiltration and proliferation of transferred memory T-cells in the meningeal tumors. Following viral infection, the leukocyte infiltration in meninges and tumor shifted from predominantly macrophages to predominantly T-cells. Finally, this paper shows that successful viral therapy of peritoneal tumors generates memory CD8 T-cells that prevent establishment of tumor in the meninges of these same animals.
Conclusions
These results support the hypothesis that a virally based immunization strategy can be used to both prevent and treat meningeal metastases. The meningeal barriers to cancer therapy may be much more permeable to treatment based on cells than treatment based on drugs or molecules.
Keywords: blood-meningeal barrier, memory T-cells, meningeal tumor, viral therapy, VSV
Metastasis to the CNS is common, with an annual incidence in the United States of ∼170 000 cases, and is associated with a poor median survival of ∼7 months.1 Brain metastases from breast cancer occur in 10%–16% of cases and leptomeningeal (LM) metastases in 2%–5%. Prognosis of LM metastases is exceptionally poor, with a median survival of 3–4 months.1–4 The incidence of brain and LM metastases in patients with breast cancer overexpressing human epidermal growth factor receptor 2 has been increasing because the monoclonal antibody, Herceptin, is often effective in controlling systemic disease but does not cross the blood–brain or blood–meningeal barrier. The brain and meninges are sanctuary sites.5–8 The blood–brain and blood–meningeal barriers to molecules consist of tight endothelial cell junctions and tight epithelial cell junctions in arachnoid, ependymal, and choroid plexuses as well as limited pinocytosis in endothelial cells and efficient efflux transporters that move drugs from the brain to the blood side of the endothelial cell.9,10 The brain and meningeal barriers to cells are more complicated and much less studied. Important variables include immune cell type, activation state of the cells, cellular expression of selectins and integrins, secretion of cytokines and chemokines, and anatomic compartment of the CNS.11–13 The cellular and molecular mechanisms underlying immune surveillance of the CNS are just beginning to be elucidated, and the application of this understanding to therapy of CNS metastases has barely begun.14
The aim of this study was to determine whether there exists a barrier to immune cell therapy of LM metastases and, if so, whether viral infection of the metastases can overcome the barrier. Our long-term therapeutic goal is to generate antitumor immunity prior to surgical removal of the primary breast tumor by infecting the tumor mass with a replicating recombinant vesicular stomatitis virus (rrVSV) that preferentially infects Her2/neu-expressing cells. We have previously shown that such therapy cures established peritoneal tumor implants and generated antitumor T-cells that are curative when transferred to host animals with established peritoneal tumors.15,16 In this paper we show that the same antitumor memory T-cells that can cure peritoneal tumors are much less effective against LM implants, demonstrating experimentally that there is a relative barrier to therapeutic antitumor T-cells in the meninges. However, this barrier can be overcome by viral infection of the meningeal implants, which induces proliferation of antitumor memory T-cells and increases their accumulation in meningeal tumors. At the same time, there is a marked reduction in tumor-associated macrophages and an increase in the ratio of meningeal T-cells to macrophages. The end result is elimination of the tumors and cure of the animals. Importantly, this work was done in mice with a normal diversity of T-cells and should therefore be directly relevant to the clinical situation. In addition, we find that mice cured of peritoneal tumors are resistant to rechallenge in the meninges, providing hope not only that therapeutic vaccination at the time of primary tumor excision can produce memory T-cells that can be induced to travel to the meninges and cure growing metastatic lesions after they are discovered but that these T-cells might prevent establishment of LM metastases in the first place.
Materials and Methods
Cells, Antibodies, Chemicals, and Animals
D2F2/E2 cells, a mouse mammary tumor line that has been stably transfected with a vector expressing the human Her2/neu gene and its parent cell line, D2F2, were a generous gift from Dr Wei-Zen Wei, Karmanos Cancer Institute, Wayne State University. MC38/E2 cells, a mouse colon carcinoma tumor line that has been stably transfected with a vector expressing the human Her2/neu gene and its parent cell line, MC38, were a generous gift from Dr Manuel Penichet, UCLA. Anti–cytotoxic T lymphocyte antigen 4 (CTLA4; 9H10) was obtained commercially (#BE0131, BioXcell Fermentation/Purification Services). Anti-CD8 (2.43) and anti-CD4 (GK1.5) ascites were prepared from hybridomas obtained from the American Type Culture Collection. Animal studies with implanted D2F2/E2 cells were conducted using female Bagg albino (Balb)/c mice, and studies with implanted MC38/E2 cells used female C57/Bl6 mice (Taconic). Mice were 8–20 weeks of age and weighed 20–25 g. Thy-1.2 Balb/c mice were obtained from Taconic. A mating pair of Thy-1.1 Balb/c mice were purchased from the Jackson Laboratory (strain name: CBy.PL(B6)-Thy1a/ScrJ; stock number: 005443) and bred on site. Animal studies were approved by the institutional animal research and care committee.
Replicating Recombinant Vesicular Stomatitis Virus
The rrVSV targeted to cells expressing Her2/neu was created from vector components as previously described and with generous contributions from Dr John K. Rose, Dr Irvin S. Y. Chen, and Genentech.15
Cell Collection
Cells were harvested from spleens, lymph nodes, and lungs by standard techniques. The entire brain was harvested, including cerebellum, brainstem, and attached meninges, minced with scissors, ground through a 70-μM nylon cell strainer, and washed with phosphate buffered saline (PBS). The cells were suspended in 20 mL of 30% Percoll (#17-0891, GE Healthcare) and placed over 10 mL of 70% Percoll in a 50-mL conical centrifuge tube. The tube was centrifuged at 390 g for 20 min at 4°C, and 5 mL was harvested from the Percoll interface and then washed twice with PBS.
Depletion in vivo of T-cells was as previously described.15 Flow cytometry was as previously described.16
For histopatholgy, we used standard techniques of formalin fixation/paraffin embedding and hematoxylin and eosin staining.
Immunohistochemistry
Immunohistochemistry (IHC) was performed on whole brains that were harvested, embedded, sectioned, and stained using standard techniques. At least 10 images of randomly chosen tumor tissue and surrounding normal brain tissue were acquired from each animal. The density (expressed as cells per square millimeter) of positively staining cells in normal and malignant tissue was determined by image analysis (MetaMorph 7.2, Molecular Devices).
Cured Animals and Production of Antitumor and Antivirus Memory T-Cells
Transfer experiments required spleen cells from cured mice. These mice were produced by implanting female Balb/c Thy-1.2 mice intraperitoneally (i.p.) with 2 × 106 D2F2/E2 cells in 300 µL PBS. On day 3 they were treated with rrVSV, 1 × 108 i.p.; on day 4 with 200 µg anti-CTLA4 monoclonal antibody; and on day 5 with cyclophosphamide (CPM), ∼100 mg/kg. The animals were considered cured if they survived for 100 days after tumor.
Meningeal Implants
Animals received isoflurane anesthesia. The hair was shaved from the posterior neck and the skin prepped with iodine and alcohol. The head was flexed and 20 µL of cells or treatment were inserted into the CSF of the cisterna magna (CM) slightly lateral to the midline just inferior to the occipital bone of the skull using an insulin syringe and needle (NDC #08287-28).
Treatment Trials
Peritoneal or meningeal tumors were established as noted in the sections on cured animals and meningeal implants. Adoptive transfer of splenocytes from naïve and cured animals were i.v. administered. Animals were sacrificed if they developed any signs of weakness or disability. The animals were considered cured if they survived for 100 days after i.p. implants and 70 days after CM implants.
Statistics
The log-rank statistic was used to compare survival among the treatment groups. A one-tailed t-test was used to compare area under the curve for cellular accumulation over time in various tissues for experimental and control groups. An unpaired one-tailed t-test was used to compare percent accumulation of T-cells and macrophages in meninges, with and without virus administration. GraphPad Prism software was used to analyze the data.
Results
We previously showed that rrVSV therapy of implanted peritoneal tumors generates therapeutic antitumor memory T-cells.16 We now determine how to use these cells to eradicate meningeal tumors.
Transferred Antitumor Memory T-Cells Cure Peritoneal Tumors More Readily Than Meningeal Tumors
Peritoneal tumors were established as previously described.15 Meningeal tumors were established by implanting 2 × 105 cells percutaneously into the cisterna magna. Tumors grew largest in the olfactory region but also grew in the meninges throughout the cerebrum and cerebellum (Supplementary Fig. S1). Tumors grew rapidly, and untreated average duration of survival was only 15 days.
Three days following tumor implantation, mice received spleen cells i.v. from cured donor animals (henceforth called cured donors). One donor was used per recipient, but all donor cells were pooled so that recipient animals with peritoneal or meningeal tumors received donor cells from the same pooled collection. Each host received 4–6 × 107 donor cells. As previously described, host animals, experimental and control, in all studies were pretreated 1 day before transfer with a single dose of CPM at 100–125 mg/kg to facilitate cell transfer.16 Memory T-cells were much more effective in eliminating peritoneal tumor than meningeal tumor (Fig. 1A; P = .0003). Transferred antitumor memory T-cells increased survival by at least 25 days and cured 60% of mice with peritoneal tumors, but cured only 20% of mice with meningeal tumors and only increased survival by a few days. Transferred spleen cells from naïve animals (henceforth called naïve donors) were completely ineffective against peritoneal or meningeal tumors, as expected. Untreated animals implanted CM at the same time with 2–6 × 104 cells showed a short survival time and a very narrow survival range (Fig. 1A).
Fig. 1.
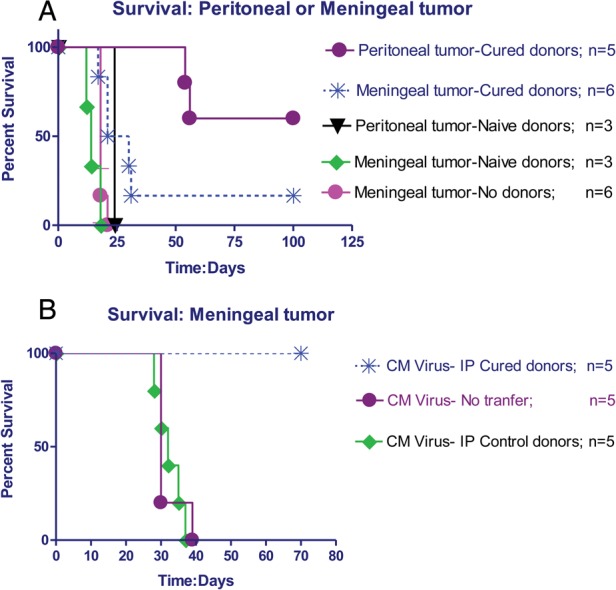
(A) Survival following treatment of peritoneal or meningeal tumors with cured donors. Mice were implanted with D2F2/E2 tumor cells in the peritoneum or the meninges and treated 3 days later with spleen cells from either cured or naïve donors. Cured donors significantly increased survival in peritoneal tumors compared with meningeal tumors (P = .0003, log-rank statistic). Naïve donors were not effective in either model. Untreated animals showed a short survival time and a very narrow survival range. (B) Survival following treatment of meningeal tumors with cured donors combined with direct viral infection of the meningeal tumors. Experimental mice received meningeal virus 1 day and cured donors 3 days after tumor implantation. Control mice received no donor cells or donor cells from mice treated with virus but not implanted with peritoneal tumor. All animals received i.p. CPM to facilitate cell transfer. N = 5 for all groups. Cured donors with viral infection of the meninges significantly increased survival compared with virus infection alone (P = 0.0016) or virus infection with control donors (P = .0018). Cured donors with viral infection of the meningeal tumors significantly increased survival of meningeal tumors compared with cured donors without virus infection (curve in 1A compared with curve in 1B; P = .0086).
These results support the idea that there is a relative blood–meningeal barrier to cellular immune therapy. We next attempted to overcome this barrier by direct viral infection of the meningeal tumors.
Transferred Antitumor Memory T-Cells Cure Leptomeningeal Tumors After Viral Infection of the Tumors
Meningeal tumors were established in Balb/c mice and treated as above with the addition that 1 day after tumor implant the animals received rrVSV CM (2 × 106 infectious dose (ID)) and i.v. (2 × 107 ID). This therapy was remarkably effective, resulting in cure of all 5 animals (Fig. 1B), and significantly improved survival compared with treatment with virus alone (P = .0016). All control animals who received virus alone developed neurological deficits and were sacrificed at a mean of 31.8 days. In order to prove that antitumor and not just antivirus memory T-cells were critical to the therapeutic response, we treated one set of 5 mice with donor cells from animals that had been infected with virus but never implanted with tumor. All animals died at a mean of 32.4 days, significantly worse than the experimental group treated with cured donors (P = .0018), indicating that specific antitumor memory T-cells were necessary for successful therapy. Further evidence came from one donor whose i.p. tumor was cured by treatment with anti-CTLA4 and CPM alone but no virus. Transferred spleen cells from this animal combined with viral infection of the meningeal tumor cured meningeal tumor in 1 host animal, indicating that antitumor memory T-cells without antivirus T-cells were curative. These results generalized to a different mouse strain. Cures were achieved in C57/Bl6 mice who were implanted CM with MC38/E2 and treated as above with virus and spleen cells from cured donors; 2 from donors cured with virus plus anti-CTLA4 and CPM and 2 from donors cured with anti-CTLA4 and CPM alone and no virus. Three of the 4 treated animals were cured. One treated animal died after 49 days. One control that received no treatment died at 28 days. In both strains of mice, cured animals behaved normally and showed no adverse effects of CM administration of rrVSV. We felt it most likely that viral infection of tumor was attracting circulating T-cells to the CSF and leading to the elimination of tumor by antitumor memory T-cells.
Virus Infection of Meningeal Tumor Attracts Antitumor Memory T-Cells to the Meninges and Tumor
Experiments utilizing both flow cytometry and IHC confirmed that viral infection of tumor recruited transferred memory T-cells to the meninges and tumor. Meningeal tumors were established in Thy-1.1 Balb/c mice, and spleen cells were transferred from Thy-1.2 Balb/c cured donors. The experimental group of mice received CM virus on days 7–9 after tumor implant and transferred spleen cells from cured donors 2 days later. Control animals did not receive CM virus. Some animals also received i.v. virus, which had no influence on transferred T-cells in the brain and was ignored in the analysis. Brains were harvested 3 days after cell transfer. In one set of experiments, flow cytometry was performed on mononuclear inflammatory cells isolated from whole brains including meninges by Percoll gradient separation.17 The virus treated group (n = 4) had 17.9% transferred T-cells in brain inflammatory cells compared with 6.8% in the control group (n = 6, P = .0005; Fig. 2, top left). The transferred CD4 T-cells were 11.4% in the treated group compared with 4.1% in the control group (P = .0005; Fig. 2, middle left). The transferred CD8 T-cells were 4.0% in the treated group compared with 2.0% in the control group (P = .013; Fig. 2, bottom left).
Fig. 2.

Accumulation of donor T-cells in meninges following treatment with CM virus. Balb/c Thy-1.1 mice were implanted with CM tumor and 9–11 days later received spleen cells from cured Thy-1.2 donors. CM virus was administered 2 days before cells in the experimental (n = 4) but not the control group (n = 6). Mononuclear inflammatory cells were harvested from brain and meninges 3 days after cell transfer and flow cytometry used to determine the percentage of donor total T-cells, CD4, and CD8 T-cells (mean with SEM). Data on left show significantly increased accumulation of total, CD4, and CD8 donor T-cells in CM virus treated animals compared with controls. Graphs on the right are representative from one experimental and one control animal.
In a separate experiment, transferred cells were counted in meninges and brain using IHC. The cell density for transferred T-cells averaged 290.3/mm2 in the CM virus treated group (n = 3), which was significantly higher than the 44.0/mm2 in the control group (n = 5, P = .002). This histological experiment demonstrated that most of the transferred T-cells were in the meninges within or near the tumor rather than spread randomly in meninges. Few if any transferred T-cells were in the tumor-free brain (Fig. 3). Inflammatory cells isolated from whole brains will henceforth be considered meningeal.
Fig. 3.

Highly selective localization of adoptively transferred T-cells into virally infected meningeal metastases. Spleen cells from cured animals were adoptively transferred into animals with meningeal tumors. CM virus was administered 2 days before cells in the experimental (n = 3) but not the control group (n = 5). Animals were sacrificed 3 days after cell transfer. A significantly higher density of donor T-cells was found in the meningeal tumors in animals treated with CM virus compared with control. (A–C) Sections of brain from virus treated animals. Numerous phycoerythrin-stained (red) donor T-cells were found in most tumors growing in the meninges on the brain surface. Hardly any donor T-cells were seen in the adjacent normal brain tissue. (D–F) Sections of brain from control animals. Only a few donor T-cells (red) were found in brain tumors from control animals. Bars in A and D = 200 microns. Bars in B, C, E and F = 50 microns.
The next set of experiments determined that following viral infection of CM tumors, transferred cells from cured donors were more likely to enter meninges than transferred cells from naïve donors and that maximal entry to meninges occurred 4–7 days following transfer. Similar preferential accumulation of transferred cells from cured mice was seen in the lungs and spleen but not the mesenteric lymph nodes. Meningeal tumors were established in Thy-1.1 Balb/c mice and treated as above with i.v. and CM virus and transferred spleen cells from either cured or naïve donor Thy-1.2 Balb/c mice. At various times after cell transfer, host animals were sacrificed and mononuclear inflammatory cells harvested from the entire brain, spleen, and right lung. Total cell count was determined and flow cytometry was used to quantify the percent transferred CD4 and CD8 CD4 T-cells. Figure 4 shows that CD4 T-cells from cured donors accumulated in meninges, lung, and spleen significantly more than CD4 T-cells from naïve donors; CD8 T-cells from cured donors accumulated in meninges significantly more than CD8 T-cells from naïve donors (n = 3 for most time points) (P = .04 for meninges CD4 T-cells; P = .03 for meninges CD8 T-cells; P = .0066 for lung CD4 T-cells; P = .08 for lung CD8 T-cells; P = .02 for spleen CD4 T-cells; P = .05 for spleen CD8 T-cells). As expected, no relative accumulation was noted in mesenteric lymph nodes (n = 2 for almost all time points). Accumulation was apparent at 4 days after cell transfer, peaked at 7 days, and was still present at 11 days.
Fig. 4.

Time course of accumulation of cured and naïve donor T-cells in brain, lung, spleen, and mesenteric lymph nodes (LN). Following CM implants of D2F2/E2 tumor, mice were treated with CM virus and then received either cured donor or cured naïve spleen cells. Animals were sacrificed at indicated times following cell transfer and inflammatory cells harvested from organs and counted. Flow cytometry was used to determine percentage of donor CD4 and CD8 T-cells (n = 3 for almost all time points except mesenteric lymph nodes. This organ had n = 2 for all time points except 18 h). A one-tailed t-test was used to compare area under the curve for cellular accumulation over time in various tissues for experimental and control groups. (A) Mean data from brains of all animals showing significantly increased accumulation of CD4 and CD8 T-cells from cured compared with naïve donors. Representative graphs from 1 naïve and 1 cured donor are shown on the right. (B) Mean data from lung, spleen, and mesenteric lymph nodes showing significantly increased accumulation of CD4 T-cells from cured compared with naïve donors in lung and spleen..
Virus Infection of Meningeal Tumor Results in Proliferation of Antitumor Memory T-Cells in Meninges, Lung, and Spleen
T-cell replication was assessed by labeling donor Thy-1.2 cells with carboxyfluorescein succinimidyl ester, harvesting tissues at various times after cell transfer, and using flow cytometry to identify replicated T-cells by diluted fluorescence and positive staining for Thy-1.2. T-cell proliferation was greater in donor cells from cured animals than from naïve animals, indicating a rapid response from memory antivirus and antitumor T-cells (n = 2–6 for each time point; Supplementary Fig. S2). Proliferation was maximal 3–5 days following cell transfer and followed a similar temporal pattern in meninges, lung, and spleen, suggesting memory T-cell division independently in each of these organs. The meninges were the site of the tumor implant and viral infection. The spleen and lung are lymphoid organs that filter antigens released from the brain into the blood.
An interesting corollary of the increased T-cells in the meninges and tumors was a corresponding decrease in the percentage of macrophages. Tumors were implanted CM, and 7 days later experimental animals were treated with CM virus. Control animals received no treatment. Flow cytometry of brain and meningeal mononuclear inflammatory cells 5–6 days after viral administration showed that the percentage of macrophages were high in the brains and meninges of control animals but decreased markedly in experimental animals treated with virus (P = .0049; Fig. 5). Absolute numbers of macrophages were also 2.7-fold higher in control animals than in virus treated animals. At the same time, the percentage of T-cells in the brain and meninges increased in virus treated animals (P = .0001; Fig. 5).
Fig. 5.

Effect of CM viral infection on tumor associated meningeal macrophages. Seven days following CM tumor implants with D2F2/E2, experimental animals received CM virus and control animals did not. Brain and meninges were harvested 5–6 days later and analyzed for the presence of macrophages (F4/80 positive) and T-cells (Thy-1.2 or CD3e positive) byflow cytometry. Data on left show significantly increased T-cells in the animals receiving virus, and graphs on right are representative from 2 animals. An unpaired one-tailed t-test was used to compare percent accumulation of T-cells and macrophages in meninges, with and without virus administration.
Successful Viral Therapy of Peritoneal Tumors Generates Memory CD8 T-Cells That Prevent Establishment of Tumor in the Meninges
The work reported above was done with transferred memory cells in an established tumor model. We now show that successful viral therapy of peritoneal tumors generated memory T-cells that could prevent establishment of meningeal tumors without the requirement for viral infection. Mice cured of peritoneal D2F2/E2 tumor with viral therapy as reported above were challenged with D2F2/E2 in the cisterna magna. Figure 6 shows that 5 of 7 animals survived and 2 died at 34 and 39 days after challenge. All control animals died 18 days after challenge. Challenge was then performed in cured animals that were depleted of CD4 T-cells, CD8 T-cells, or both. Animals depleted of both CD4 and CD8 T-cells died promptly after challenge. Most animals with CD8 depletion also died promptly. Interestingly, animals with CD4 depletion usually survived, indicating that the major memory cell type preventing meningeal neoplastic implantation in this model system was CD8 T-cells. In the absence of CD8 T-cells, CD4 cells could not prevent or treat neoplastic implantation.
Fig. 6.

Survival following CM challenge. Animals cured of i.p. tumors (survivors) using standard treatment with rrVSV, anti-CTLA4, and CPM were challenged with CM tumor, as were controls. Antitumor memory CD8 T-cells prevented establishment of meningeal tumors in survivors.
Discussion
The results in this paper support the use of targeted viral infection of tumor to generate antitumor memory T-cells to prevent or treat leptomeningeal metastases. Current therapy is ineffective because surgery can rarely remove all metastases safely, radiation therapy has a poor therapeutic index in the brain, and drug treatments are inhibited by a robust multilayered and multifunctional blood–brain and blood–meningeal barrier to molecules.18 In contrast, memory T-cells and other immune cells have no difficulty penetrating meninges and brain when attracted by the appropriate inflammatory signals.12,19 This paper establishes several crucial proofs of principle as follows:
Targeted virus infection of tumor can generate therapeutic memory T-cells. In clinical practice this means that a single off-the-shelf reagent, the virus, can be used to treat any patient. Expensive individually tailored therapy is not required. Following virus infection of tumor, each patient will generate the most potent immune response from his or her T-cell repertoire.
Memory T-cells can prevent establishment of meningeal tumors. The ideal clinical case would be to infect breast tumors before surgical removal and generate antitumor memory T-cells, which prevent metastases from initially implanting in meninges. The metastatic cells to be blocked could potentially come from subclinical sites in lymph nodes, lymph organs, or lung.
Memory T-cells can treat established meningeal tumors following viral infection of the tumors. Once again, antitumor memory T-cells would be generated by viral infection of the initial breast tumor but in this case subclinical meningeal metastases would already be established. Clinically, surveillance imaging would detect growing lesions, and viral infection of the tumor in the CNS would attract a memory response to the tumor.
The results in this paper show that the memory T-cells are capable of finding multiple geographically separated tumor collections of various sizes and completely eradicating them. This result was achieved in 2 mouse strains, Balb/c and C57/Bl6, with very different genetic immunologic biases.20
There remain several preclinical steps to be accomplished before this therapy can be considered clinically. This model system is not fully syngeneic because the implanted mouse cells have been engineered to express the human Her2/neu receptor. The model is instructive because the animals do not generate an effective immune response, and untreated tumor growth is progressive and lethal, but translation to the clinic could founder on a weaker immune response. We are currently developing a fully syngeneic model system to more conclusively establish the concepts demonstrated in this paper. Although repeat viral infection in the CNS is possible because neutralizing antibodies formed in response to the initial infection cannot penetrate the blood–brain or blood–meningeal barrier, we recognize that stimulating an immune response by infecting the brain with a replicating virus, albeit targeted and much attenuated, is not an ideal solution for the clinic. Further work is needed to develop an effective nonreplicating pseudovirus or other immune stimulant to attract memory T-cells to the CNS. For maximum clinical utility, the results in this paper must be generalized to parenchymal brain tumors as well as meningeal tumors. We used a meningeal model in these studies to avoid the confounding factor of trauma at the tumor site. In this model, the tumors implant at multiple locations in the meninges far from the injection site. We are developing a model of parenchymal disease by carotid artery injection that will also avoid the problem of local trauma at the tumor site. Finally, toxicity studies will be required for all components of the proposed therapy.
Our results support previous work showing that there is a relative brain barrier to a cellular immune response.13,21–23 Viral infection of the tumor was able to overcome this barrier probably by provoking an inflammatory response in the meninges that released chemokines attracting CD4 and CD8 T-cells and by lysis of tumor cells releasing tumor and viral antigens that could activate memory T-cells in the draining lymphoid organs. Time course studies showed that virus infection produced a 2.6-fold increase in total transferred T-cells in the meninges. The increase in memory T-cells must be much greater because of the diluting effect of the nonspecific T-cells. This effect was seen on day 3 following cell transfer at the same time that cell proliferation of transferred memory cells was just beginning, indicating that the increase at that time point was due to enhanced trafficking and accumulation and not enhanced local proliferation. Proliferation of memory T-cells was robust between days 3 and 5 after cell transfer and occurred simultaneously in meninges, lung, and spleen. This suggests that antigen presentation was occurring simultaneously in these 3 organs. Antigen release from the meninges occurs via CSF drainage into nasal lymphatics and into the dural venous sinuses. Lymphatic drainage into cervical lymph nodes was not assessed here, but the results in this paper reinforce the view that the spleen and lung harvest meningeal antigens from the blood and present them effectively to circulating memory T-cells.19,24 This model system did not find evidence that T-cells must first become licensed in the lung before entering the brain.24 The results also support the view that local antigen presenting cells in the meninges stimulate memory T-cells that survey the CSF.11
Concomitant with the increase of T-cells in tumor and meninges, we noted a decrease in the percentage of macrophages. Tumors in many tissues and especially in the brain have a large proportion of tumor-associated macrophages.25,26 These macrophages have an M2 phenotype that supports tumor growth and inhibits immunologic reaction against the tumor by a wide variety of mechanisms.27–30 Further work is required to assess whether the therapeutic effect of viral infection in the meninges is in part due to reduction of the effects of suppressor macrophages. The work is complicated by the fact that macrophages in meninges and brain are a combination of yolk sac–derived microglia and bone marrow–derived monocytes,31 and current surface markers do not clearly differentiate these populations.
In most transfer experiments, cured donor cells contained a mixture of antitumor and antivirus memory T-cells. We were able to prove that antitumor memory T-cells were necessary and sufficient for the therapeutic response in some situations by 2 experiments. In one, donor cells from animals that were infected with virus but never implanted with tumor were not able to effect cure of CM tumor. In the other, donor cells from animals who were cured of i.p. tumors by CTLA4 and CPM but no virus cured CM tumor in 2 animals and prolonged life in a third, even though the donor animals had never received virus. These results do not exclude an important therapeutic role for the antivirus memory T-cells in stimulating a strong pro-inflammatory response in the CSF in the D2F2/E2 model, and further work is required to differentiate the effects of the antitumor and the antivirus T-cells. We are confident that T-cells are the therapeutic cells in the donor spleen cells because we have shown that memory cells are required for cure and that antibody from B-cells will not adequately cross the blood–brain barrier to cure meningeal tumors; in previous work we showed that cure of i.p. tumors was achieved by transferring T-cells,16 and in the current work we show that prevention of CM implantation is abrogated by depletion of T-cells.
Supplementary Material
Funding
This work was supported by US Army Medical Research Breast Cancer IDEA awards to I.B. (no. BC101672) and P.B. (no. BC101672P1), and by a grant from the Emmerling Fund of the Pittsburgh Foundation to I.B.
Supplementary Material
Acknowledgments
We thank Drs Wei-Zen Wei, John K. Rose, Irvin S. Y. Chen, Manual Penichet, and Genentech Inc., who very generously supplied materials as noted in the text. This project used the UPCI Cell and Tissue Imaging Facility, which is supported in part by the US National Institutes of Health (P30CA047904).
Conflict of interest statement. I.B. and P.W-D.: US Patent Application 7,429,481 entitled “Targeting viruses using a modified Sindbis0 glycoprotein” awarded September 30, 2008. The targeted virus is being tested in this animal work. I.B. and P.W-D. have no affiliation with any company.Y.G., M.A.B., and P.H.B. have no conflicts of interest to report.
References
- 1.Dawood S, Gonzalez-Angulo AM. Progress in the biological understanding and management of breast cancer–associated central nervous system metastases. Oncologist. 2013;18(6):675–684. doi: 10.1634/theoncologist.2012-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Azevedo CRAS, Cruz MRS, Chinen LTD, et al. Meningeal carcinomatosis in breast cancer: prognostic factors and outcome. J Neurooncol. 2011;104(2):565–572. doi: 10.1007/s11060-010-0524-y. [DOI] [PubMed] [Google Scholar]
- 3.Le Rhun E, Taillibert S, Zairi F, et al. A retrospective case series of 103 consecutive patients with leptomeningeal metastasis and breast cancer. J Neurooncol. 2013;113(1):83–92. doi: 10.1007/s11060-013-1092-8. [DOI] [PubMed] [Google Scholar]
- 4.Altundag K, Bondy ML, Mirza NQ, et al. Clinicopathologic characteristics and prognostic factors in 420 metastatic breast cancer patients with central nervous system metastasis. Cancer. 2007;110(12):2640–2647. doi: 10.1002/cncr.23088. [DOI] [PubMed] [Google Scholar]
- 5.Le Rhun E, Taillibert S, Zairi F, et al. Clinicopathological features of breast cancers predict the development of leptomeningeal metastases: a case-control study. J Neurooncol. 2011;105(2):309–315. doi: 10.1007/s11060-011-0592-7. [DOI] [PubMed] [Google Scholar]
- 6.Gutierrez M, Lyazidi S, Brasseur L, et al. Leptomeningeal meningitis related to breast cancer overexpressing HER2: is there a place for a more specific treatment? Bull Cancer. 2011;98(4):417–424. doi: 10.1684/bdc.2011.1341. [DOI] [PubMed] [Google Scholar]
- 7.Park YH, Park MJ, Ji SH, et al. Trastuzumab treatment improves brain metastasis outcomes through control and durable prolongation of systemic extracranial disease in HER2-overexpressing breast cancer patients. Br J Cancer. 2009;100(6):894–900. doi: 10.1038/sj.bjc.6604941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weil RJ, Palmieri DC, Bronder JL, et al. Breast cancer metastasis to the central nervous system. Am J Pathol. 2005;167(4):913–920. doi: 10.1016/S0002-9440(10)61180-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barshes N, Demopoulos A, Engelhard HH. Anatomy and physiology of the leptomeninges and CSF space. Cancer Treat Res. 2005;125:1–16. doi: 10.1007/0-387-24199-x_1. [DOI] [PubMed] [Google Scholar]
- 10.Daneman R. The blood–brain barrier in health and disease. Ann Neurol. 2012;72(5):648–672. doi: 10.1002/ana.23648. [DOI] [PubMed] [Google Scholar]
- 11.Bartholomaus I, Kawakami N, Odoardi F, et al. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature. 2009;462(7269):94–98. doi: 10.1038/nature08478. [DOI] [PubMed] [Google Scholar]
- 12.Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol. 2012;33(12):579–589. doi: 10.1016/j.it.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 13.Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Invest. 2010;120(5):1368–1379. doi: 10.1172/JCI41911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liau LM, Prins RM, Kiertscher SM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11(15):5515–5525. doi: 10.1158/1078-0432.CCR-05-0464. [DOI] [PubMed] [Google Scholar]
- 15.Gao Y, Whitaker-Dowling P, Griffin JA, et al. Recombinant vesicular stomatitis virus targeted to Her2/neu combined with anti-CTLA4 antibody eliminates implanted mammary tumors. Cancer Gene Ther. 2009;16(1):44–52. doi: 10.1038/cgt.2008.55. [DOI] [PubMed] [Google Scholar]
- 16.Gao Y, Whitaker-Dowling P, Griffin JA, et al. Treatment with targeted vesicular stomatitis virus generates therapeutic multifunctional anti-tumor memory CD4T cells. Cancer Gene Ther. 2012;19(4):282–291. doi: 10.1038/cgt.2011.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reichmann G, Villegas EN, Craig L, et al. The CD28/B7 interaction is not required for resistance to Toxoplasma gondii in the brain but contributes to the development of immunopathology. J Immunol. 1999;163(6):3354–3362. [PubMed] [Google Scholar]
- 18.Maher EA, Mietz J, Arteaga CL, et al. Brain metastasis: opportunities in basic and translational research. Cancer Res. 2009;69(15):6015–6020. doi: 10.1158/0008-5472.CAN-08-4347. [DOI] [PubMed] [Google Scholar]
- 19.Prins RM, Shu CJ, Radu CG, et al. Anti-tumor activity and trafficking of self, tumor-specific T cells against tumors located in the brain. Cancer Immunol Immunother. 2008;57(9):1279–1289. doi: 10.1007/s00262-008-0461-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schulte S, Sukhova GK, Libby P. Genetically programmed biases in Th1 and Th2 immune responses modulate atherogenesis. Am J Pathol. 2008;172(6):1500–1508. doi: 10.2353/ajpath.2008.070776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol. 2007;28(1):12–18. doi: 10.1016/j.it.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 22.Wekerle H. Immune protection of the brain—efficient and delicate. J Infect Dis. 2002;186(Suppl 2):S140–S144. doi: 10.1086/344937. [DOI] [PubMed] [Google Scholar]
- 23.Hickey WF. Basic principles of immunological surveillance of the normal central nervous system. Glia. 2001;36(2):118–124. doi: 10.1002/glia.1101. [DOI] [PubMed] [Google Scholar]
- 24.Odoardi F, Sie C, Streyl K, et al. T cells become licensed in the lung to enter the central nervous system. Nature. 2012;488(7413):675–679. doi: 10.1038/nature11337. [DOI] [PubMed] [Google Scholar]
- 25.Charles NA, Holland EC, Gilbertson R, et al. The brain tumor microenvironment. Glia. 2011;59(8):1169–1180. doi: 10.1002/glia.21136. [DOI] [PubMed] [Google Scholar]
- 26.Nagaraj S, Youn JI, Gabrilovich DI. Reciprocal Relationship between Myeloid-Derived Suppressor Cells and T Cells. J Immunol. 2013;191(1):17–23. doi: 10.4049/jimmunol.1300654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmieder A, Michel J, Schonhaar K, et al. Differentiation and gene expression profile of tumor-associated macrophages. Semin Cancer Biol. 2012;22(4):289–297. doi: 10.1016/j.semcancer.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 28.Laoui D, Movahedi K, Van Overmeire E, et al. Tumor-associated macrophages in breast cancer: distinct subsets, distinct functions. Int J Dev Biol. 2011;55(7–9):861–867. doi: 10.1387/ijdb.113371dl. [DOI] [PubMed] [Google Scholar]
- 29.Ostrand-Rosenberg S, Sinha P, Beury DW, et al. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol. 2012;22(4):275–281. doi: 10.1016/j.semcancer.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arina A, Schreiber K, Binder DC, et al. Adoptively transferred immune T cells eradicate established tumors despite cancer-induced immune suppression. J Immunol. 2014;192(3):1286–1293. doi: 10.4049/jimmunol.1202498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomez PE, Schulz C, Geissmann F. Development and homeostasis of “resident” myeloid cells: the case of the microglia. Glia. 2013;61(1):112–120. doi: 10.1002/glia.22393. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.