

Abstract
Background
Malignant gliomas are complex systems containing a number of factors that drive tumor initiation and progression, including genetic aberrations that lead to extensive cellular heterogeneity within the neoplastic compartment. Mouse models recapitulate these genetic aberrations, but readily observable heterogeneity remains challenging.
Methods
To interrogate cellular heterogeneity in mouse glioma models, we utilized a replication-competent avian sarcoma-leukosis virus long terminal repeat with splice acceptor/tumor virus A (RCAS-tva) system to generate spontaneous mouse gliomas that contained a Sox2-enhanced green fluorescent protein (EGFP) reporter. Glial fibrillary acidic protein-tva mice were crossed with Sox2–EGFP mice, and tumors were initiated that contained a subpopulation of Sox2–EGFP-high cells enriched for tumor-initiating cell properties such as self-renewal, multilineage differentiation potential, and perivascular localization.
Results
Following implantation into recipient mice, Sox2–EGFP-high cells generated tumors containing Sox2–EGFP-high and Sox2–EGFP-low cells. Kinomic analysis of Sox2–EGFP-high cells revealed activation of known glioma signaling pathways that are strongly correlated with patient survival including platelet-derived growth factor receptor beta, phosphoinositide-3 kinase, and vascular endothelial growth factor. Our functional analysis identified active feline sarcoma (Fes) signaling in Sox2–EGFP-high cells. Fes negatively correlated with glioma patient survival and was coexpressed with Sox2-positive cells in glioma xenografts and primary patient-derived tissue.
Conclusions
Our RCAS-tva/Sox2-EGFP model will empower closer examination of cellular heterogeneity and will be useful for identifying novel glioma pathways as well as testing preclinical treatment efficacy.
Keywords: glioma, intratumoral heterogeneity, RCAS/tva, Sox2
Gliomas are the most common malignant and lethal primary adult brain tumors and are characterized by an invasive phenotype and a high degree of cellular heterogeneity. Gliomas encompass a class of tumors ranging in malignancy from benign pilocytic astrocytoma to highly lethal glioblastoma (GBM).1 Despite aggressive treatment, which includes surgical resection followed by radiation and chemotherapy, recurrence is nearly universal.2 The median survival time for patients diagnosed with high-grade astrocytoma (WHO grade III and IV) and grade III oligodendroglioma is 1 year and 3.5 years, respectively.2,3 Due to this poor prognosis, there is an immediate need to develop more effective therapeutics for these tumors.
Difficulties with glioma treatment are in part due to the high degree of cellular heterogeneity found within these tumors. Generation of intratumoral heterogeneity is associated with tumor initiating cells (TICs), a subpopulation capable of reconstituting the parental tumor upon secondary transplantation into recipient mice.4–7 TICs can self-renew or differentiate toward multiple lineages to maintain the heterogeneity of the original tumor and can survive both chemo- and radiotherapies to drive recurrence following treatment.8–10 TICs reside in distinct microenvironments or niches that provide instructive signals controlling the balance between self-renewal and differentiation.11 In gliomas, the perivascular and hypoxic niches contribute to maintenance of a TIC phenotype.12,13 Because current treatment options are mainly effective against non-TICs, the development of multimodal treatments that target TICs in combination with conventional therapies has the potential to greatly impact patient survival.
One important intracellular marker associated with TICs is sex determining region Y box 2 (Sox2), a member of the high mobility group box family of transcription factors. Sox2 is frequently amplified or overexpressed in gliomas14,15 and is a component of a core set of 4 transcription factors capable of reprogramming differentiated GBM cells towards a TIC state.16 Sox2 depletion inhibits proliferation and self-renewal in neurosphere formation assays14,17 and prevents differentiated GBM cells from acquiring TIC phenotypes.18 Additional evidence suggests that tight control of endogenous Sox2 levels may be required to control cell fate and promote tumorigenesis19 and that targeting of Sox2 decreases TIC tumorigenic potential.17 Furthermore, overexpression of Sox2 increased GBM cell migration and invasion, while targeting of Sox2 inhibited GBM motility.14 This essential role for Sox2 in GBM is also present in other brain tumors including medulloblastoma, in which Sox2-positive cells drive tumor recurrence in the sonic hedgehog molecular subtype.20 These data strongly suggest the importance of understanding Sox2 effects on TIC phenotypes and brain tumor biology.
The paradigm for investigating GBM relies on interrogation of tumor cells via culture-based assays or genetically engineered mouse models. Both methods have been useful for examining tumor progression but are deficient at accurately modeling the complexity found in primary tumors. Mouse models, such as a replication-competent avian sarcoma-leukosis virus long terminal repeat with splice acceptor/tumor virus A (RCAS-tva), induce de novo glioma formation in an in vivo environment.21 By expressing tva from a cell-type-specific promoter, oncogenic mutations can be delivered via avian retrovirus with a high degree of cell specificity. For example, overexpression of constitutively active epidermal growth factor receptor (EGFR) in a glial fibrillary acidic protein (GFAP)-tva background leads to the development of gliomas by transforming glial progenitors and mature astrocytes.21 RCAS delivery of platelet-derived growth factor beta (PDGF-β) in GFAP–tva-expressing astrocytes increases proliferation and dedifferentiation and promotes glioma formation in approximately 40% of animals.22 GFAP-tva models have uncovered key aspects of tumor progression, providing benefits for target identification and as models for evaluation of preclinical therapies.23 Although the GFAP-tva models have yet to recapitulate the cellular hierarchy associated with TIC models observed in human patients,24 reporter systems in mouse models have been shown to be effective for evaluating intratumoral heterogeneity including reporters based on stem cell factors.10,20 Together, these data suggest the importance of developing a Sox2 reporter mouse model of glioma to evaluate intratumoral heterogeneity.
To accomplish this goal, we constructed a reporter system for glioma intratumoral heterogeneity by crossing GFAP-tva mice21 with mice expressing enhanced green florescence protein (EGFP) under the control of the endogenous Sox2 promoter.25 Prior characterization of Sox2-EGFP mice determined that EGF-positive neural progenitors express Sox2 protein and suggested the importance of Sox2 as a neural stem cell marker associated with self-renewal and multilineage differentiation.25 Using the GFAP–tva-positive/Sox2–EGFP-positive mice with avian retroviral introduction of PDGFβ, we found that tumors contained a Sox2–EGFP-high subpopulation that localized to a perivascular niche, differentiated to multiple cell lineages, and could self-renew.4,5,12 This reporter system facilitates the interrogation of Sox2 signaling and biology in malignant gliomas as well as the examination of cellular heterogeneity.
Materials and Methods
Generation of GFAP-tva–positive/Sox2–EGFP-positive Mice
GFAP-tva and Sox2-EGFP mice were maintained as previously described21,26 and crossed to generate GFAP-tva/Sox2-EGFP double-positive mice. All animal studies were done in accordance with approved Cleveland Clinic Institutional Animal Use and Care Committee (IACUC) protocols. GFAP–tva-positive/Sox2–EGFP-positive mice were genotyped by PCR amplification of genomic DNA using the following primers:
Sox2-EGFP primers:
IMR0872: 5′-AAGTTCATCTGCACCACCG-3′
IMR1416: 5′-TCCTTGAAGAAGATGGTGCG-3′
IMR7338: 5′-CTAGGCCACAGAATTGAATTGAAAGATCT-3′
IMR7339: 5′-GTAGGTGGAAATTCTAGCATCATCC-3′
IMR1111: 5′-CTGCTGCCCGGTAACGTGACCCG-3′
GFAP-tva primers:
IMR1112: 5′-GCCCTGGGGGAAGGTCCTGCCC-3′
Common: 5′-AGAGCTCCGGGTTCTCTCTC-3′
WT: 5′-GGGAGGAAGGAACTCCACTC-3′
Cycling conditions consisted at 94°C for 3 minutes; 30 cycles at 94°C for 30 seconds, 66°C for 1 minute, and 72°C for 1 minute plus an annealing step of 72°C for 10 minutes.
Generation of Sox2–EGFP-high Glioma Cells and Allografts
DF1 cells (spontaneously transformed chicken fibroblasts, ATCC CRL-12203) were transfected with RCAS-PDGF virus as previously described21 and used to initiate tumors in GFAP–tva-positive/Sox2–EGFP-positive mice. Mice were allowed to survive until the development of neurological signs, at which point tumor-bearing brains were harvested and tumors microdissected based on gross appearance. Tissue was dissociated to single cells using a papain-based dissociation system (Worthington Biochemical), and Sox2–EGFP-high glioma cells were subsequently isolated via flow cytometry. To generate allografts, 5000 Sox2–EGFP-high glioma cells were injected into the right forebrains of 4–6 week-old athymic nude mice in accordance with a Cleveland Clinic IACUC-approved protocol and treated as above.
Cell Culture Conditions
Sox2–EGFP-high glioma cells were maintained as tumorspheres in Neurobasal medium (Life Technologies) supplemented with B27, EGF, and basic fibroglast growth factor (bFGF), 20 ng/µL each). Differentiation of Sox2–EGFP-positive cells was performed in Dulbecco's Modified Eagle's medium supplemented with 10% fetal bovine serum (FBS).
Immunohistochemical Staining
Fluorescence imaging was performed as previously described.26 Human GBM and intracranial xenografts were fixed in 4% paraformaldehyde, incubated overnight in 30% sucrose, and embedded in optimal cutting temperature (OCT) compound (Tissue Tek). Samples were sectioned at a thickness of 10 µm for subsequent staining analysis. Sox2 was detected using endogenous fluorescence, and blood vessels were visualized by anti-rat CD31 antibody (Abcam). Proximity of Sox2-positive cells to blood vessels was evaluated across at least 3 histological sections and calculated based on the distance from the center of the cell to the edge of the CD31-positive blood vessel. Phospho-feline sarcoma (p-Fes) was detected using an anti-rabbit antibody (Y713, Sigma) in parallel with Sox2 using an anti-mouse Sox2 antibody (R&D Systems) and stained with the relevant fluorescent secondary antibodies. For in vitro staining, cells were fixed in 4% paraformaldehyde and stained as described above. Human TICs were enriched on the basis of CD133 expression as previously described.8 Nuclei were counterstained with Hoechst 33342 prior to visualization using an SP-5 confocal microscope with a 63 × objective (Leica Microsystems) for in vivo analysis and an EVOS microscope with a 10 × objective (Life Technologies) for in vitro analysis. Images were assembled using Photoshop software (Adobe).
Tumorsphere Formation Assay
Tumorsphere formation assays were performed as previously described.26 Briefly, Sox2–EGFP-high and -low cells were plated in decreasing numbers to 1 cell/well. The number of wells with tumorspheres was determined after 14 days, and the percentage of TICs was determined using ELDA software (http://bioinf.wehi.edu.au/software/elda/).
In Vivo Tumor Initiation
For in vivo tumor initiation analysis, Sox2–EGFP-high and -low cells were enriched by flow cytometry as described above and sorted into Neurobasal medium (containing no B27 or growth factors) to recover for an hour. Cells were then counted, and 2000 cells from each group were transplanted into the right cortex of athymic nude mice as previously described.8 Mice were monitored for the development of neurological signs, at which time they were euthanized, and the presence of a tumor was confirmed.
Differentiation Analysis
Loss of EGFP signal as a result of serum-induced differentiation over 7 days was achieved by sequential imaging using an EVOS microscope. Phase contrast and EGFP-filtered images were merged using Adobe Photoshop. Following differentiation, EGFP cells were fixed to chamber slides with 4% PFA for 15 minutes. Subsequently, cells were blocked in phosphate-buffered saline (PBS) containing 10% normal goat serum and 0.1% Triton X-100 for 30 minutes at room temperature and then at 4°C overnight. Immunostaining for the differentiation markers β3 tubulin (Abcam) and GFAP (Sigma) was performed with mouse IgG1 antibody and relevant secondary antibodies. Nuclei were counterstained with Hoechst 33342. To quantify changes in EGFP expression during differentiation, Sox2–EGFP-high glioma cells were imaged every 6 hours during serum-induced differentiation in an IncuCyte Zoom kinetic imaging system (Essen Biosciences) that quantified EGFP signal intensity.
In Vitro Lineage Tracing
Six-well plates were coated with 1.6% Geltrex overnight. Following removal of Geltrex and 2 PBS washes, Sox2–GFP-positive glioma cells were plated at 100 000 cells/well. Images were captured in 15 minute intervals over the course of 72 hours.
Kinome Screen
The multiplex in vitro kinase assay was performed using PamChips with PamGene Evolve Software on the PamStation12 at the University of Alabama at Birmingham Kinome Core. For this experiment, subcutaneous tumors were initiated from allografted Sox2–EGFP-positive cells infected with a constitutive EF1a-RFP reporter to label tumor cells. Resulting tumors were dissociated using papain, allowed to recover for ∼2 hours, and then sorted via flow cytometry for Sox2–GFP-high/RFP-positive and Sox2–GFP-low/RFP-positive cells. Immediately after sorting, collected cells were centrifuged, washed in PBS, and lysed without any intervening cell culture. Lysates were generated using M-PER mammalian protein extraction reagent (Pierce) and loaded at 15 µg/well onto PamChips for reaction with 144 known tyrosine peptides. A median signal intensity of each spot was measured and normalized by subtracting background over time, and data were analyzed using BioNavigator Microarray software. To identify differentially phosphorylated peptides, qualitative phosphorylation curve differences that exceeded overall trends were assessed. Kinase pathway analysis was completed with GeneGo MetaCore software.
Bioinformatics Analysis
To determine the level of total Fes expression in human tissue, the human protein atlas (www.proteinatlas.org, accessed April 14, 2014) was interrogated. Association between gene expression and glioma patient survival was examined using the National Cancer Institute Repository for Molecular Brain Neoplasia Data (NCI REMBRANDT, https://caintegrator.nci.nih.gov/rembrandt, accessed April 14, 2014), where survival was evaluated based on high (142 patients) and low (70 patients) expression from an intermediate value. Where high is twice and low is half of the intermediate value.
Flow Cytometric Analysis
To determine the coexpression between p-Fes and Sox2, xenografted cells were fixed with 4% paraformaldehyde and stained as described above. Cells were analyzed using BD Fortessa (BD Biosciences), and a minimum of 10 000 events were collected per group. Isotype control antibody staining was analyzed to establish positive gates.
Statistical Analysis
For analysis, reported values were mean values ± standard deviation from studies performed using least 3 biological replicates. Unless otherwise stated, 1-way ANOVA was used to calculate statistical significance; P values are detailed in the text and figure legends. Patient survival analysis was performed using the NCI REMBRANDT database, where survival was calculated by log-rank analysis.
Results
Mouse models of glioma have been limited in highlighting distinct cell populations within the tumor. Because the GFAP-tva system can reproducibly generate de novo gliomas in mice and Sox2 is characterized as a TIC marker important for glioma growth and tumorigenic potential, we sought to generate a system to model intratumoral heterogeneity using Sox2-EGFP in GFAP-tva mice. We crossed GFAP–tva-positive and Sox2–EGFP-positive mice (Fig. 1A) and confirmed the presence of both tva and EGFP in resulting progeny (Fig. 1B). GFAP-tva and Sox2-EGFP could be distinguished from wild-type GFAP and Sox2 by differences in PCR amplicon size when separated on an agarose gel. Tumors were initiated in GFAP-tva/Sox2-EGFP mice by orthotopic injection with chicken fibroblast (DF1) cells containing RCAS-PDGFβ virus.
Fig. 1.

PDGF overexpression induces tumor formation in GFAP–tva-positive/Sox2–EGFP-positive mice. (A) Schematic of breeding strategy yielding GFAP–tva-positive/Sox2–EGFP-positive mice bearing gliomas initiated via PDGF overexpression. The Holland GFAP–tva-positive and RCAS-PDGF model system was modified to include a transgene in which one copy of the Sox2 allele was replaced with EGFP. (B) GFAP–tva-positive/Sox2–EGFP-positive mice were genotyped to confirm the presence of EGFP and tva.
Sox2–EGFP-high glioma cells isolated from mice bearing neurological symptoms were orthotopically injected into immunocompromised mice to confirm their tumorigenic capacity and generate allografts for further use (Fig. 2A and B). Sox2-EGFP cells were also confirmed to express endogenous Sox2 both in vivo and in vitro (Supplementary material, Fig. 1). Heterogeneity in resulting allografts was confirmed by fluorescence-activated cell sorting analysis and fluorescent microscopy, which showed 22%–35% Sox2–EGFP-positive glioma cells (Fig. 2C–E), which is within the range of Sox2-positive cells in xenografts27 as well as in GFAP-tva mice.28 Additionally, this heterogeneity appeared to be intrinsic to the tumor, as we did not observe Sox2-EGFP cells in the normal brain parenchyma outside of the previously reported neurogenic niche locations.25 To determine if Sox2–EGFP-positive cells may be supported by specific tumor microenvironments, we determined whether the EGFP-positive population was found adjacent to blood vessels in a perivascular niche.12 We stained tumor sections for the endothelial cell marker CD31 and examined Sox2-EGFP localization by fluorescent microscopy. Sox2–EGFP-high glioma cells preferentially associated with the vasculature compared with Sox2–EGFP-low cells, and 20% of Sox2–EGFP-positive cells were located <10 µm from a blood vessel (Fig. 2F and G). These results indicate that PDGF overexpression leads the formation of gliomas in GFAP–tva-positive/Sox2–EGFP-positive mice and that Sox2–EGFP-high cells give rise to Sox2–EGFP-high and Sox2–EGFP-low tumor cells. Additionally, these data indicate that Sox2–EGFP-high cells preferentially localize adjacent to the vasculature.
Fig. 2.

Sox2–EGFP-high glioma cells give rise to heterogeneous tumors and display perivascular localization. (A) Schematic demonstrating propagation of Sox2–EGFP-high glioma cells through intracranial injection into athymic nude mice to generate an allograft. (B) Representative histology of an allografted tumor showed by hematoxylin and eosin staining. (C) Analysis of tumor cells immediately after dissociation demonstrated that Sox2–EGFP-positive glioma cells make up a fraction of the bulk tumor. (D) Representative immunofluorescent images of allografts resulting from Sox2–EGFP-positive glioma cells. Images demonstrate intratumoral heterogeneity with respect to Sox2-EGFP expression. (E) Quantification of the percentage of Sox2–EGFP-high glioma cells in immunofluorescent images of tumor sections. (F) Representative immunofluorescent images demonstrating perivascular localization of Sox2–EGFP-high glioma cells in allografts. (G) Analysis of blood vessel proximity showing that Sox2–EGFP-high glioma cells preferentially associate with the tumor vasculature. Scale bar represents 50 µm for panel D and 10 µm for panel E. Data represent mean ± standard deviation, ***P < .001 as assessed by 1-way ANOVA.
Since Sox2 is a previously established TIC marker, we next determined whether Sox2–EGFP-high glioma cells were enriched for TIC phenotypes, including the ability to self-renew and differentiate. In vitro limiting-dilution analysis, which assesses self-renewal, proliferation, and survival, revealed increased tumorsphere formation in EGFP-high cells compared with EGFP-low cells (Fig. 3A). To further validate this finding, we sorted Sox2-EGFP cells based on expression of integrin α6, a marker known to enrich for TICs in human cells.26 Integrin α6-high cells were also enriched for tumorsphere formation (Fig. 3A), demonstrating that sorting on TIC markers enriches for self-renewal in a subpopulation of GFAP-tva/Sox2-EGFP cells. To evaluate if Sox2–EGFP-high cells were more tumorigenic than Sox2–EGFP-low cells, we transplanted 2000 sorted cells from each group into recipient mice. We found no significant difference in tumor initiation capacity between Sox2–EGFP-high and -low cells, with Sox2–EGFP-low cells giving rise to tumor slightly faster. This is consistent with previous reports using the GFAP-tva model in which the population of cells that has increased self-renewal displayed reduced tumor initiation capacity in vivo.29
Fig. 3.

Sox2–EGFP-positive glioma cells display some functional characteristics of cancer stem cells. (A) In vitro limiting-dilution assay to determine self-renewal in Sox2–EGFP-high cells sorted for EGFP and integrin α6 expression. (B) Kaplan-Meier survival curve demonstrates no difference in tumor initiation between Sox2–EGFP-high (green line) and -low (black line) glioma cells. (C) Representative images of tumorsphere-forming Sox2–EGFP-high glioma cells in the absence or presence of the prodifferentiation agent fetal bovine serum (FBS). Sox2–EGFP-high glioma cells lost EGFP expression in the presence of 10% FBS and gave rise to neural (β3 tubulin-positive) and astrocyte (GFAP-positive) lineages. (D) Quantification of EGFP signal over one week of differentiation in the presence of 10% FBS. Graph is representative of quadruplicate samples.
Multilineage differentiation is a process involving downregulation of stem cell factors such as Sox2 and increased expression of differentiation factors such as the astrocyte marker GFAP or the neuronal marker β3-tubulin.4 To determine whether Sox2–EGFP-positive glioma cells behave similarly, we differentiated Sox2–EGFP-high tumorspheres using FBS. Over the course of one week, we observed gradual loss of EGFP signal and concomitant changes in morphology (Fig. 3C and D). After 7 days of FBS treatment, EGFP glioma cells stained positive for β3-tubulin and GFAP (Fig. 3C). Quantification of EGFP expression revealed a gradual loss of EGFP signal intensity over the duration of the differentiation, with expression dropping by 60% (Fig. 3D). This loss in EGFP was confirmed at the single cell level, and we observed a concomitant decrease in EGFP and endogenous Sox2 over a serum-induced differentiation time course (Supplementary material, Fig. 2). These data demonstrate that Sox2–EGFP-high cells lose signal upon differentiation, indicating that they can report the stem cell state.
Aberrant signaling is a hallmark of cancer cells, and the amplification and overexpression of Sox2 in glioma cells suggests the importance of Sox2-regulated pathways for tumor maintenance and progression. Although changes in gene expression can provide valuable information for downstream target identification, the ability to target protein function, and particularly kinases, through small molecule inhibitors has the potential to provide a more direct route to therapy development. To determine whether kinase activity differed between Sox2–EGFP-high and Sox2–EGFP-low glioma cells, we subjected lysates from cells sorted for EGFP directly from allografts, without intervening cell culture, to kinomic analysis with a focus on tyrosine phosphorylation. We observed several peptides at which phosphorylation occurred at a higher frequency in Sox2–EGFP-high cells such as Fes, a nonreceptor tyrosine kinase,30 PDK1, PDGFRβ, and RET (Table 1, Fig. 4A). To determine if these peptides were informative for clinical outcome in glioma, we used the NCI REMBRANDT to correlate expression with patient survival (Fig. 4B). Expression of Fes, PDK1, and PDGFRβ each strongly correlated with poor survival. Because PDK1 and PDGFRβ have previously been described in glioma,31,32 we focused on Fes for further analysis.
Table 1.
Protein tyrosine kinase peptides altered with increased Sox2
| Protein | Phosphorylated Tyr | Percent Increase | Correlation with Glioma Survival |
|---|---|---|---|
| Feline sarcoma (Fes) | 713 | 65.3% | Negative correlation |
| Phosphatidylinositol 3-kinase (PI3K) regulatory subunit alpha | 607 | 69.7% | No correlation |
| 3-phosphoinositide-dependent protein kinase 1 (PDK1) | 9 | 78.7% | Negative correlation |
| Beta-type platelet-derived growth factor receptor precursor (PDGRFβ) | 579, 581 | 66.2% | Negative correlation |
| Proto-oncogene tyrosine-protein kinase receptor ret precursor (RET) | 1029 | 66.7% | No correlation |
Fig. 4.

Sox2–EGFP-high cells are preferentially enriched for known glioma signaling pathways. (A) Kinetics of PDGFRβ and Fes phosphorylation in cells sorted for EGFP expression. (B) Kaplan-Meier plot showing patient survival is strongly correlated with Fes expression. Immunofluorescent staining (C and D) of p-Fes and Sox2 expression in primary patient-derived tissue (08-0457, C) and human-derived xenograft (T4121, D) samples show an overlap between p-Fes and Sox2. Flow cytometric analysis (E) confirms elevated Sox2 expression in p-Fes positive cells (as indicated by red cells). Scale bar represents 10 µm.
Fes has been recently identified as both an oncogene in leukemia, breast, and prostate cancers and a tumor suppressor in colorectal cancer.33–36 To examine Fes signaling in human tumors, we stained for an autophosphorylation site of Fes (Fes-Y713) and Sox2 in a primary patient specimen and xenografted tumor. We detected phospho-Fes coexpressed with Sox2 in both glioma xenografts and glioma patient tissue (Fig. 4C and D). We confirmed this coexpression using flow cytometry and found that the majority of p-Fes positive cells expressed Sox2 ranging from 62%–96% across 3 GBM xenografts (Fig. 4D, Supplementary material, Fig. 3). We evaluated if p-Fes was enriched in TIC by comparing p-Fes and Sox2 expression in CD133-positive and –negative cells and found an enrichment of p-Fes in CD133-positive cells (Supplementary material, Fig. 4). Finally, we evaluated Fes expression in The Human Genome Atlas and found that total Fes was preferentially expressed in glioma compared with normal brain (Supplementary material, Fig. 5). This analysis indicates that Sox2–EGFP-high cells contain signaling aberrations that are informative for human glioma prognosis.
To further analyze signaling pathways preferentially activated in Sox2–EGFP-positive cells, phosphopeptides from the kinomic analysis were mapped to the Shortest Paths network (Fig. 5A). Predicted involvement of several key elements in glioma progression was indicated, such as phosphatase and tensin homolog (PTEN), RAC-alpha serine/threonine-protein kinas (Akt), and c-Src.37 Additionally, Sox2–EGFP-positive phosphopeptides were used to predict upstream kinases in their respective signaling pathways (Fig. 5B). PTEN and c-Src also appear in this map, along with other key drivers of glioma formation including p53 and members of the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway.37,38 These data indicate that Sox2–EGFP-high cells can be used to identify novel elements of gliomagenesis as possible therapeutic targets.
Fig. 5.
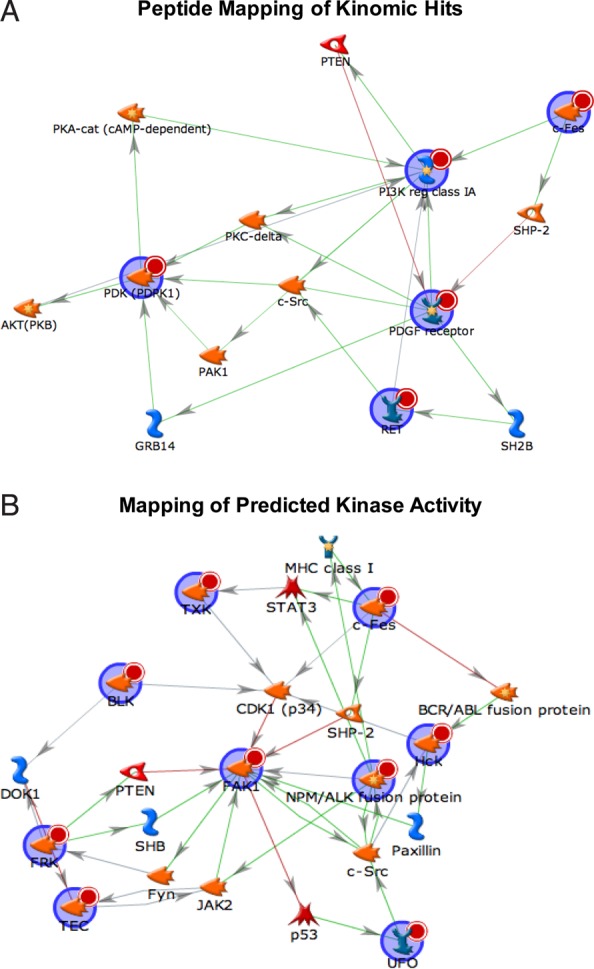
Kinomic analysis establishes active signaling networks in Sox2–EGFP-high cells. (A) Signaling map based on top hits from kinomic analysis. Proteins from the kinomic screen are denoted by a red circle, agonistic relationships are denoted by green arrows, and antagonistic relationships are denoted by a red arrow. (B) Map of kinase activity predicted by kinomic analysis.
Discussion
Examination of the pathology of cancer requires experimental paradigms that accurately represent the original disease state. Culture-based assays yield useful information in the initial stages of discovery, but they are insufficient for recapitulating key aspects of tumorigenesis including spatial orientation, scaffolding, vascularization, and cellular heterogeneity. Xenograft models have proven to be useful for delineating the efficacy of preclinical therapies, but they too fail to fully recapitulate the initial stages of tumor development and stromal interactions between tumor cells and their native microenvironment. In contrast, spontaneous tumorigenesis models using transgenic mice allow orthotopic de novo tumor formation that most closely resembles human tumorigenesis. Moreover, using cell type-specific systems, transformation events can be restricted to a desired cell population. We have modeled intratumoral heterogeneity in malignant gliomas with respect to TICs by introducing a Sox2-EGFP reporter into a GFAP-tva background.
Modifications of the RCAS/tva model have allowed examination of the role of tumor suppressor deletions such as p53, Nf1,23 PTEN,39 and Ink4a/Arf concomitant with hyperactive growth factor signaling.40 These models have proven useful for uncovering aberrant signaling molecules associated with glioma formation23,39,40 and have provided evidence that the GBM cell of origin may arise from neural stem cells in the subventricular zone of the adult brain.41 A recent study sought to examine glioma cellular heterogeneity by expressing VenusYFP from the ID1 promoter in an RCAS–tva/Arf−/− background.29 Surprisingly, the results identified a population of Id1-low cells that do not undergo self-renewal but are rather enriched for tumor formation in an orthotopic transplantation model. Interestingly, our results indicated no difference in tumor formation between cells sorted for EGFP expression (Fig. 3B). While we cannot eliminate the possibility that loss of a Sox2 allele to allow EGFP expression differentially impacts glioma formation and tumorigenic potential, we believe that our data, in combination with the literature, indicate a mouse model-specific phenotype. There is an extensively well-characterized link between self-renewal and tumor initiation potential in human patient-derived xenografts.5,8
Although not informative for tumor initiation, Sox2-EGFP cells maintained many of the several TIC phenotypes. Importantly, a pure population of Sox2–EGFP-high cells gave rise to a heterogeneous tumor characterized by both Sox2–EGFP-high and Sox2–EGFP-low cells. The ability to visualize Sox2–EGFP-high cells in a heterogeneous tumor is a powerful tool that opens the possibility for spatiotemporal examination of these cells in vivo.27 Our lineage tracing analysis indicated that we could successfully examine TIC cell division at an individual cell level, a methodology that could mimic conditions observed in an in vivo setting.
We have provided evidence that Sox2–EGFP-high cells reside in the perivascular niche, indicating that these cells may also be useful for studying other TIC niches such as the hypoxic tumor core and the invasive edge of the tumor. Furthermore, we have demonstrated that Sox2–EGFP cells can act as a reporter for cell differentiation. This property makes them well suited for assessing preclinical therapies that disrupt the TIC state. Similarly, our lineage tracing analysis indicated that we can successfully examine TIC cell division at an individual cell level. This analysis could be expanded to include various culture conditions that mimic those observed in the native tumor microenvironment.
Results from the kinomic analysis of Sox2–EGFP-positive cells identified pathways that are preferentially upregulated in TICs and correlate strongly with patient survival. Our identification of increased PDGFRβ, PI3K, and VEGF signaling in Sox2–EGFP-high cells confirmed previous reports of their roles in TIC maintenance.42–44 These data indicate that Sox2-EGFP cells are useful for examining TICs at the subcellular level. Additionally, the effect of preclinical treatments that target a specific signaling node could immediately be tied to the TIC state by simple visualization of EGFP signal intensity.
In addition to previously identified glioma markers, our kinomic screen revealed increased Fes activity in Sox2–EGFP-high cells. Fes and its homolog Fer are the only 2 members of a class of nonreceptor tyrosine kinases distinguished by an N-terminal F-BAR domain. Fes expression is normally limited to myeloid, vascular endothelial, and neuronal cells, where it induces differentiation, proliferation, angiogenesis, and inflammation.38,45 Little is known about the role of Fes in malignant glioma, but it has been identified as an oncogene in leukemia and in solid tumors of the breast and prostate.33,34,36,46 Conversely, Fes has also been linked to tumor suppression in colorectal cancer.35 We validated the kinomic and bioinformatics analyses by detecting Fes in a patient-derived xenograft cell line and multiple primary patient samples.
The identification of Fes in our kinomic screen reveals a novel role for this class of nonreceptor tyrosine kinases in glioma. The role of Fes in cancer progression is yet to be defined, but our data suggest that Fes may contribute to gliomagenesis based on its low expression in the non-neoplastic brain and elevation in GBM. Based on its expression in Sox2-positive cells, Fes may contribute to the maintenance of these cells. Fes is expressed ubiquitously and has been shown to play a role in multiple cancer types including prostate, breast, and colon, where it has been tied to IL-6/STAT3 signaling, integrin-dependent metastasis, and cell-cycle progression, respectively.47–49 Fer was also shown to be important in PDGFR-induced STAT3 activation in mouse fibroblast cells and tumor growth in PDGFβ-transformed cells, indicating that Fer may be active in cancers with increased PDGFR signaling.50 The results of our kinomic analysis indicate that our model can be used to identify novel TIC signaling pathways.
TICs present a unique challenge in treating malignant gliomas. Surgical resection in the brain is insufficient to remove all cancer cells, and the TICs left behind are thought to drive tumor recurrence. This problem is compounded by TIC resistance to postsurgical chemo- or radiotherapies.8–10 Recent findings have indicated that chemotherapy has the potential to induce self-renewal in glioma cells,51 further underscoring the importance of determining glioma recurrence mechanisms. By faithfully modeling the TIC state in vivo, therapies can be developed that effectively target these cells. Using a well-established mouse model, we created a system that identifies TICs in malignant glioma by expression of a Sox2-EGFP reporter. We have demonstrated that Sox2–EGFP-high cells are phenotypically similar to patient-derived TICs with respect to niche location, differentiation, modes of cell division, and self-renewal. Taken together, our novel model system utilizing Sox2–EGFP-high cells in a GFAP-tva background can be used as a tool for validating several aspects of glioma initiation and maintenance, identifying novel glioma signaling abnormalities, and evaluating preclinical treatments.
Supplementary Material
Funding
This manuscript has been supported by National Institutes of Health grants K99/R00 CA157948 (.J.D.L.), R01 NS083629 (J.D.L.), and R01 CA1515122 (A.B.H.).
Supplementary Material
Acknowledgments
We thank the members of the Lathia laboratory for constructive comments on the experimental design and manuscript. We thank Cathy Shemo, Moneen Morgan, Patrick Barrett, and Sage O′Bryant for flow cytometry assistance. We thank Drs. Mahendra S. Rao (New York Stem Cell Foundation) and Mark P. Mattson (NIH) for kindly providing the Sox2-EGFP mice and Drs. Dolores Hambardzumyan (Department of Neurosciences, Lerner Research Institute) and Eric Holland (Fred Hutchinson Cancer Research Center) for kindly providing the GFAP-tva mice and advice on use of the model. We thank Dr. Roger McLendon (Duke University) for providing glioma sections for immunostaining analysis (generated from funds from the Pediatric Brain Tumor Foundation and 1P01-CA154291). These studies were supported by National Institutes of Health grants K99/R00 CA157948 (J.D.L.), R01 NS083629 (J.D.L.), R01 CA1515122 (A.B.H.), and R21 CA185712 (University of Alabama Birmingham Kinomics Core). Work in the Lathia lab is also supported by the Lerner Research Institute, Sontag Foundation Voices Against Brain Cancer, Ohio Cancer Research Associates, a V Scholar Award from the V Foundation for Cancer Research, Grant IRG-91-022-18 to the Case Comprehensive Cancer Center from the American Cancer Society, and the Cleveland Clinic Product Development Fund.
Conflict of interest statement. None declared.
References
- 1.Huse JT, Holland EC. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat Rev Cancer. 2010;10:319–331. doi: 10.1038/nrc2818. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 3.Charles N, Ozawa T, Squatrito M, et al. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell. 2010;6:141–152. doi: 10.1016/j.stem.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 5.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 6.Ignatova TN, Kukekov VG, Laywell ED, et al. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia. 2002;39:193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- 7.Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 8.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 9.Liu G, Yuan X, Zeng Z, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J, Li Y, Yu TS, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lathia JD, Heddleston JM, Venere M, et al. Deadly teamwork: neural cancer stem cells and the tumor microenvironment. Cell Stem Cell. 2011;8:482–485. doi: 10.1016/j.stem.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 13.Li Z, Bao S, Wu Q, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alonso MM, Diez-Valle R, Manterola L, et al. Genetic and epigenetic modifications of Sox2 contribute to the invasive phenotype of malignant gliomas. PloS One. 2011;6:e26740. doi: 10.1371/journal.pone.0026740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Annovazzi L, Mellai M, Caldera V, et al. SOX2 expression and amplification in gliomas and glioma cell lines. Cancer Genomics Proteomics. 2011;8:139–147. [PubMed] [Google Scholar]
- 16.Suva ML, Rheinbay E, Gillespie SM, et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell. 2014;157:580–594. doi: 10.1016/j.cell.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gangemi RM, Griffero F, Marubbi D, et al. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells. 2009;27:40–48. doi: 10.1634/stemcells.2008-0493. [DOI] [PubMed] [Google Scholar]
- 18.Berezovsky AD, Poisson LM, Cherba D, et al. Sox2 promotes malignancy in glioblastoma by regulating plasticity and astrocytic differentiation. Neoplasia. 2014;16:193–206. doi: 10.1016/j.neo.2014.03.006. 206 e119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cox JL, Wilder PJ, Desler M, et al. Elevating SOX2 levels deleteriously affects the growth of medulloblastoma and glioblastoma cells. PloS One. 2012;7:e44087. doi: 10.1371/journal.pone.0044087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vanner RJ, Remke M, Gallo M, et al. Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell. 2014;26:33–47. doi: 10.1016/j.ccr.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holland EC, Hively WP, DePinho RA, et al. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998;12:3675–3685. doi: 10.1101/gad.12.23.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai C, Celestino JC, Okada Y, et al. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001;15:1913–1925. doi: 10.1101/gad.903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holland EC. A mouse model for glioma: biology, pathology, and therapeutic opportunities. Toxicol Pathol. 2000;28:171–177. doi: 10.1177/019262330002800122. [DOI] [PubMed] [Google Scholar]
- 24.Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 25.Ellis P, Fagan BM, Magness ST, et al. SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev Neurosci. 2004;26:148–165. doi: 10.1159/000082134. [DOI] [PubMed] [Google Scholar]
- 26.Lathia JD, Gallagher J, Heddleston JM, et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell. 2010;6:421–432. doi: 10.1016/j.stem.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lathia JD, Gallagher J, Myers JT, et al. Direct in vivo evidence for tumor propagation by glioblastoma cancer stem cells. PloS One. 2011;6:e24807. doi: 10.1371/journal.pone.0024807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polajeva J, Swartling FJ, Jiang Y, et al. miRNA-21 is developmentally regulated in mouse brain and is co-expressed with SOX2 in glioma. BMC Cancer. 2012;12:378. doi: 10.1186/1471-2407-12-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barrett LE, Granot Z, Coker C, et al. Self-renewal does not predict tumor growth potential in mouse models of high-grade glioma. Cancer Cell. 2012;21:11–24. doi: 10.1016/j.ccr.2011.11.025. [DOI] [PubMed] [Google Scholar]
- 30.Muller AJ, Scherle PA. Targeting the mechanisms of tumoral immune tolerance with small-molecule inhibitors. Nat Rev Cancer. 2006;6:613–625. doi: 10.1038/nrc1929. [DOI] [PubMed] [Google Scholar]
- 31.Goidts V, Bageritz J, Puccio L, et al. RNAi screening in glioma stem-like cells identifies PFKFB4 as a key molecule important for cancer cell survival. Oncogene. 2012;31:3235–3243. doi: 10.1038/onc.2011.490. [DOI] [PubMed] [Google Scholar]
- 32.Kim Y, Kim E, Wu Q, et al. Platelet-derived growth factor receptors differentially inform intertumoral and intratumoral heterogeneity. Genes Dev. 2012;26:1247–1262. doi: 10.1101/gad.193565.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Youn JI, Nagaraj S, Collazo M, et al. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sottoriva A, Spiteri I, Piccirillo SG, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA. 2013;110:4009–4014. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Triozzi PL, Aldrich W, Achberger S, et al. Differential effects of low-dose decitabine on immune effector and suppressor responses in melanoma-bearing mice. Cancer Immunol Immunother. 2012;61:1441–1450. doi: 10.1007/s00262-012-1204-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kohanbash G, McKaveney K, Sakaki M, et al. GM-CSF promotes the immunosuppressive activity of glioma-infiltrating myeloid cells through interleukin-4 receptor-alpha. Cancer Res. 2013;73:6413–6423. doi: 10.1158/0008-5472.CAN-12-4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Z, Wang H, Eyler CE, et al. Turning cancer stem cells inside out: an exploration of glioma stem cell signaling pathways. J Biol Chem. 2009;284:16705–16709. doi: 10.1074/jbc.R900013200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greer P. Closing in on the biological functions of Fps/Fes and Fer. Nat Rev Mol Cell Biol. 2002;3:278–289. doi: 10.1038/nrm783. [DOI] [PubMed] [Google Scholar]
- 39.Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schulte A, Gunther HS, Martens T, et al. Glioblastoma stem-like cell lines with either maintenance or loss of high-level EGFR amplification, generated via modulation of ligand concentration. Clin Cancer Res. 2012;18:1901–1913. doi: 10.1158/1078-0432.CCR-11-3084. [DOI] [PubMed] [Google Scholar]
- 41.Kinsella P, Howley R, Doolan P, et al. Characterization and response of newly developed high-grade glioma cultures to the tyrosine kinase inhibitors, erlotinib, gefitinib and imatinib. Exp Cell Res. 2012;318:641–652. doi: 10.1016/j.yexcr.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 42.Ping YF, Yao XH, Jiang JY, et al. The chemokine CXCL12 and its receptor CXCR4 promote glioma stem cell-mediated VEGF production and tumour angiogenesis via PI3K/AKT signalling. J Pathol. 2011;224:344–354. doi: 10.1002/path.2908. [DOI] [PubMed] [Google Scholar]
- 43.Bleau AM, Hambardzumyan D, Ozawa T, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4:226–235. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oka N, Soeda A, Inagaki A, et al. VEGF promotes tumorigenesis and angiogenesis of human glioblastoma stem cells. Biochem Biophys Res Commun. 2007;360:553–559. doi: 10.1016/j.bbrc.2007.06.094. [DOI] [PubMed] [Google Scholar]
- 45.Kim J, Feldman RA. Activated Fes protein tyrosine kinase induces terminal macrophage differentiation of myeloid progenitors (U937 cells) and activation of the transcription factor PU.1. Mol Cell Biol. 2002;22:1903–1918. doi: 10.1128/MCB.22.6.1903-1918.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feldman RA, Gabrilove JL, Tam JP, et al. Specific expression of the human cellular fps/fes-encoded protein NCP92 in normal and leukemic myeloid cells. Proc Natl Acad Sci USA. 1985;82:2379–2383. doi: 10.1073/pnas.82.8.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zoubeidi A, Rocha J, Zouanat FZ, et al. The Fer tyrosine kinase cooperates with interleukin-6 to activate signal transducer and activator of transcription 3 and promote human prostate cancer cell growth. Mol Cancer Res. 2009;7:142–155. doi: 10.1158/1541-7786.MCR-08-0117. [DOI] [PubMed] [Google Scholar]
- 48.Ivanova IA, Vermeulen JF, Ercan C, et al. FER kinase promotes breast cancer metastasis by regulating alpha6- and beta1-integrin-dependent cell adhesion and anoikis resistance. Oncogene. 2013;32:5582–5592. doi: 10.1038/onc.2013.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Makovski A, Yaffe E, Shpungin S, et al. Down-regulation of Fer induces ROS levels accompanied by ATM and p53 activation in colon carcinoma cells. Cell Signal. 2012;24:1369–1374. doi: 10.1016/j.cellsig.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 50.Lennartsson J, Ma H, Wardega P, et al. The Fer tyrosine kinase is important for platelet-derived growth factor-BB-induced signal transducer and activator of transcription 3 (STAT3) protein phosphorylation, colony formation in soft agar, and tumor growth in vivo. J Biol Chem. 2013;288:15736–15744. doi: 10.1074/jbc.M113.476424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Auffinger B, Tobias AL, Han Y, et al. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014;21:1119–1131. doi: 10.1038/cdd.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.