

Abstract
Type 2 diabetes mellitus is a leading health issue worldwide. Among cases of diabetes mellitus nephropathy (DN), the major complication of type 2 diabetes mellitus, the nephrotic phenotype is often intractable to clinical intervention and demonstrates the rapid decline of renal function to end-stage renal disease. We recently identified the gene for glypican-5 (GPC5), a cell-surface heparan sulfate proteoglycan, as conferring susceptibility for acquired nephrotic syndrome and additionally identified an association through a genome-wide association study between a variant in GPC5 and DN of type 2 diabetes mellitus. In vivo and in vitro data showed a progressive increase of GPC5 in type 2 DN along with severity; the excess was derived from glomerular mesangial cells. In this study, diabetic kidney showed that accumulation of fibroblast growth factor (Fgf)2 strikingly induced progressive proteinuria that was avoided in Gpc5 knockdown mice. The efficacy of Gpc5 inhibition was exerted through expression of the Fgf receptors 3 and 4 provoked in the diabetic kidney attributively. Extraglomerular Fgf2 was pathogenic in DN, and the deterrence of Gpc5 effectively inhibited the glomerular accumulation of Fgf2, the subsequent increase of mesangial extracellular matrix, and the podocytes' small GTPase activity. These findings elucidate the pivotal role of GPC5, identified as a susceptible gene in the genome-wide association study, in hyperglycemia-induced glomerulopathy.
Among diabetes mellitus nephropathy (DN) cases, intractable dysfunction rapidly propagating to end-stage renal disease often accompanies the nephrotic proteinuria phenotype. Our recent study implicated glypican-5 (GPC5) as a susceptible gene for acquired nephrotic syndrome and demonstrated its functional significance by using podocyte-specific knockout mice; a genome-wide association study of DN satisfying the criterion of nephrotic syndrome also clarified GPC5 as the candidate gene [odds ratio 1.45 (95% CI, 1.18–1.79)].1 Glypicans share 14 conserved and unique cysteine residues and covalently link to the cell membrane via a glycosylphosphatidylinositol anchor.2 Glypican is a heparan sulfate proteoglycan, which facilitates selective protein–protein interactions establishing transient cell-signal platforms in concert with lipid rafts; glypican plays a critical role in regulating the signaling of Wnt; hedgehogs; bone morphogenetic protein; and, especially, of fibroblast growth factor (FGF)2.3
FGF2 is a highly conserved 18-kDa cationic protein released from the cytosolic storage site through plasma membrane disruptions.4,5 In the kidney, the release of FGF2 is probable from mesangial cells6 and podocytes.7 It stimulates in vitro proliferation of mesangial cells,8 endothelial cells,9 and podocytes.7 An excess of FGF2 clearly injures podocytes in vivo and induces proteinuria.10,11 Based on the information from the genome-wide association study that GPC5 is a susceptible gene, this study was undertaken to clarify the functional role of GPC5 in DN and related pathways.
Materials and Methods
All the chemical compounds are purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan) and culture medium and related supplements were purchased from Life Technologies (Carlsbad, CA) unless otherwise specified.
Human Specimens
For human glomerulus staining, human tissues were obtained from Asterand (Detroit, MI) and used with written informed consent and institutional review board approval. Human kidney tissues were obtained from diabetic patients and nondiabetic controls during surgery (Table 1).
Table 1.
Basic Characteristics of Human Subjects
| Characteristic | Control group | DN group |
|---|---|---|
| n | 2 | 5 |
| Sex (F/M)∗ | 1/1 | 1/4 |
| Age, years† | 28.5 ± 3.5 | 62.0 ± 17.1 |
| Proteinuria, grams per 24 hours† | 0.1 ± 0.1 | 5.1 ± 3.5 |
| Serum creatinine, mg/dL† | 0.9 ± 0.5 | 1.5 ± 0.2 |
F, female; M, male; DN, diabetes mellitus nephropathy.
Data are expressed as numbers of subjects.
Data are expressed as means ± SEM.
Animals
We obtained 8- to 12-week-old C57BL/6 and C57BL/6Cr Ins2+/C96Y (Akita)12 mice (Japan SLC, Tokyo). Podocyte-specific Gpc5 knockdown mice and Gpc5 systemic knockdown mice were produced as reported previously.1 All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals13 and were approved by The University of Tokyo Institutional Review Board.
Cell Culture
MES13 mouse mesangial cell line (ATCC, Manassas, VA) was cultured in a 3:1 mixture of Dulbecco's modified Eagle's medium and Ham's F12 medium. This mixture was supplemented with 5% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin.
Conditionally immortalized mouse podocytes were described previously14 and were characterized by positive staining for Wilms tumor 1, synaptopodin, podocin, CD-associated protein, nephrin, ezrin, and podocalyxin. These cells were grown in RPMI 1640 medium containing 10% fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, 1 mmol/L sodium pyruvate, 10 mmol/L HEPES buffer, and 0.075% sodium bicarbonate. For passaging the cells, podocytes were grown under growth-permissive conditions, which involves growing cells at 33°C in the presence of 50 U/mL interferon-γ. For podocytes to acquire a differentiated and quiescent phenotype, cells were grown under restrictive conditions at 37°C without interferon-γ for 10 days.
A rat mesangial cell line had been established by our group and was reported previously.12,15 Characterization of rat cultured mesangial cells was performed using antibodies of Thy-1.1. Cells were cultured in RPMI 1640 medium supplemented with 20% fetal bovine serum, 0.66 U/mL insulin, 100 U/mL penicillin, and 100 μg/mL streptomycin.
Immunofluorescence Staining
Frozen sections cut in 8-μm pieces were dried and fixed with ice-cold acetone at −20°C for 20 minutes. Primary antibodies were FGF receptor (FGFR)1, FGFR2, FGFR3, FGFR4, FGF2 (Abcam, Cambridge, UK), GPC5 (R&D Systems, Minneapolis, MN), and nephrin (Acris Antibodies, Herford, Germany). After washing them with phosphate-buffered saline (PBS), we applied fluorescent-conjugated (Alexa488, Alexa555, or Alexa633) secondary antibody (Thermo Fisher Scientific, South San Francisco, CA) for 60 minutes. The sections were then examined using confocal microscopy (LSM 510 META NLO imaging system; Carl Zeiss, Oberkochen, Germany).1,16
Immunoelectron Microscopy Analysis
Immunotransmission electron microscopic analysis was performed using the pre-embedding method. Briefly, 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) was used for the fixation of sliced C57BL/6 kidney, and the overnight and dehydration procedure used was the sucrose gradient method from 10% to 30%. The kidney was embedded in Tissue-Tek (Sakura Finetek, Tokyo, Japan) and sliced to 8 μm. After blocking processes, the first antibody [GPC5 antibody (R&D Systems) described in Immunofluorescnece Staining] was applied for 2 days. After a rigorous wash with PBS, the second biotinylated antibody was applied for 1 hour. Then avidin-conjugated horseradish peroxidase was applied after a rigorous wash with PBS. After the colorimetric reaction, the sample was fixed again in osmium tetroxide and transferred to Epon (Miller-Stephenson, Fairfield, CT). Ultrathin sections were examined by means of transmission electron microscopy (JEOL, Tokyo, Japan).
In Vitro Analysis of FGFRs, FGF2, and GPC5 Expression
Cells were incubated in each glucose concentration for 72 hours, and each osmolality was adjusted using d-mannitol (Sigma-Aldrich, St. Louis, MO). Because glucose-free F12 medium is not available, Dulbecco's modified Eagle's medium supplemented with 5% fetal bovine serum was alternatively used for the MES13. Total RNA was isolated using TRIzol (Life Technologies). After performing deoxyribonuclease I treatment, we performed reverse transcription (ImProm-II; Promega, Madison, WI) and quantitative real-time PCR by using the SYBR Green system (Life Technologies) or the Universal ProbeLibrary system (Roche, Basel, Switzerland) (mouse Gpc5 and mouse Fgfr3 only) and normalized for the relative expression of the genes of interest by using β-actin. The quantitative real-time PCR primer sequences are presented in Table 2. All samples were analyzed in triplicate.
Table 2.
Quantitative Real-Time PCR Primer Sequences
| Target gene | Primer sequence |
|---|---|
| Mouse Gpc5 | F: 5′-CTAGTGGAGACTGTGATGACGAA-3′ |
| R: 5′-TTCATGCTGTCTGGCATCC-3′ | |
| Probe#76 (Roche) | |
| Mouse Fgfr3 | F: 5′-GGAGGACGTGGCTGAAGA-3′ |
| R: 5′-CGGAAGCGGACAGTGTTT-3′ | |
| Probe#76 (Roche) | |
| Mouse Fgfr4 | F: 5′-ATGACCGTCGTACACAATCTTAC-3′ |
| R: 5′-TGTCCAGTAGGGTGCTTGC-3′ | |
| Mouse Fgf2 | F: 5′-CGGCTCTACTGCAAGAACG-3′ |
| R: 5′-TGCTTGGAGTTGTAGTTTGACG-3′ | |
| Rat Fn1 | F: 5′-CCGGTGGCTGTCAGTCAGA-3′ |
| R: 5′-CCGTTCCCACTGCTGATTTATC-3′ | |
| Rat Col4a2 | F: 5′-TGATGGAGAAAAAGGAGTCTCAG-3′ |
| R: 5′-GCCTTGGAAACCATCCAG-3′ | |
| Mouse Actb | F: 5′-CGCACCACTGGCATTGTCAT-3′ |
| R: 5′-TTCTCCTTGATGTCACGCAC-3′ | |
| Rat Gapdh | F: 5′-CAAGGTCATCCATGACAACTTTG-3′ |
| R: 5′-GGCCATCCACAGTCTTCTGG-3′ |
F, forward primer; R, reverse primer.
In Situ Hybridization
The mouse kidney was dissected after perfusion, fixed with tissue fixative (Genostaff, Tokyo, Japan), embedded in paraffin, and sectioned at 6 μm. The standard in situ hybridization protocol was used.17 Tissue sections were dewaxed and rehydrated. The sections were fixed in 4% paraformaldehyde for 15 minutes, treated with 8 μg/mL proteinase K (Merck, Whitehouse Station, NJ) for 30 minutes at 37°C, refixed with 4% paraformaldehyde (Sigma-Aldrich), and placed in 0.2 N HCl for 10 minutes. The sections were acetylated by means of incubation in 0.1 mol/L triethanolamine HCl (Sigma-Aldrich), pH 8.0, and 0.25% acetic anhydride for 10 minutes. The sections were then dehydrated again. Hybridization was performed with probes at concentrations of 300 ng/mL in Probe Diluent-1 (Genostaff) at 60°C for 16 hours (GenBank; http://www.ncbi.nlm.nih.gov/nuccore; Accession number NM_008006.2, position 444 to 691, size 248). After hybridization, the sections were washed in 5× HybriWash (Genostaff), equal to 5× standard saline citrate, at 60°C for 20 minutes and then in 50% formamide, 2× HybriWash at 60°C for 20 minutes followed by ribonuclease treatment in 50 μg/mL ribonuclease A (Qiagen, Hilden, Germany) in 10 mmol/L Tris-HCl, pH 8.0, 1 mol/L NaCl, and 1 mmol/L EDTA for 30 minutes at 37°C. Then the sections were washed twice with 2× HybriWash (Genostaff) at 60°C for 20 minutes, twice with 0.2× HybriWash at 60°C for 20 minutes, and once with Tris-buffered saline with 0.1% Tween 20. After treatment with 0.5% blocking reagent (Roche) in Tris-buffered saline with 0.1% Tween 20 for 30 minutes, the sections were incubated with antidigoxigenin alkaline phosphatase conjugate (Roche) diluted 1:1000 with Tris-buffered saline with 0.1% Tween 20 for 2 hours at room temperature. The sections were washed twice with Tris-buffered saline with 0.1% Tween 20 and then incubated in 100 mmol/L NaCl, 50 mmol/L MgCl2, 0.1% Tween 20, and 100 mmol/L Tris-HCl, pH 9.5. Coloring reactions were performed using nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl-phosphate solution (Sigma-Aldrich) overnight and then washed with PBS. The sections were counterstained with Kernechtrot stain solution (Muto Pure Chemicals, Tokyo, Japan) and mounted with CC/Mount (Diagnostic BioSystems, Pleasanton, CA).
Measurement for FGF2 in Vivo
Endogenous FGF2 protein was detected using a mouse basic FGF enzyme-linked immunosorbent assay kit (Ray Biotech, Norcross, GA). To elucidate the accumulation of systemic circulating FGF2 in vivo, we biotinylated recombinant human FGF2 (Kaken Pharmaceutical, Tokyo, Japan) by using a Pierce EZ-Link MicroSulfo-NHS-LC biotinylation kit (Thermo Fisher Scientific, Waltham, MA) according to the protocol and purified it using a G-25 column (GE Healthcare, Little Chalfont, UK). Biotinylated FGF2 (10 μg per mouse) was injected intravenously, and the kidneys were harvested 1 hour later. Frozen sections (8 μm pieces) were dried and fixed with ice-cold acetone at −20°C for 20 minutes. After conjugation with Alexa-555 conjugated streptavidin (Life Technologies), sections were examined using confocal microscopy. We quantified at least five glomeruli in the outer cortex selected from each animal. The signal intensity was measured and standardized with the interstitial background.
Animal Experiment Protocol for Extrarenal FGF2 Infusion
We assigned the animals to one of three groups: C57BL/6, Akita, and systemic Gpc5 knockdown mice. FGF2 mice were injected intravenously on days 0, 1, 2, and 3 (5 μg per animal). Systemic Gpc5 knockdown was undertaken 2 days before the FGF2 injection as reported previously.1,18 Briefly, the mouse Gpc5 siRNA (5′-GCAGGCGCUUAAUCUGGGCAUUGAA-3′, 5′-UUCAAUGCCCAGAUUAAGCGCCUGC-3′) and scrambled siRNA (5′-CCUACAUGACGACGAUGUACCGUGA-3′, 5′-UCACGGUACAUCGUCGUCAUGUAGG-3′) were obtained from Life Technologies as annealed in vivo–grade siRNA. Synthetic siRNA 400 μg dissolved in 0.8 mL PBS was rapidly injected intravenously following the hydrodynamic method.
Analyses of Urinary Albumin and Urinary Creatinine
We determined the urinary albumin levels with an enzyme-linked immunosorbent assay by using a murine microalbuminuria enzyme-linked immunosorbent assay kit (Albuwell M; Exocell, Philadelphia, PA) and measured the urine creatinine concentration with Nescoat VLII CRE (Alfresa Pharma, Osaka, Japan). Albuminuria was determined at the ratio of urinary albumin to urinary creatinine (mg/g Cre). All procedures were performed in accordance with the manufacturer's protocols.
Extracellular Matrix Expression by FGF2 Signal Transduction in the Mesangial Cell Line
Rat mesangial cells were transfected either with Stealth RNAi Negative Control Med GC (Life Technologies) or with predesigned Gpc5 siRNA targeted against Gpc5 (NM_001107285.1; 5′-GCAGGCGCUUAAUCUGGGCAUUGAA-3′, 5′-UUCAAUGCCCAGAUUAAGCGCCUGC-3′; Life Technologies) by using Lipofectamine 2000 (Life Technologies). After transfection, cells were subjected to different concentrations of glucose (300 or 600 mg/dL) for 48 hours with subsequent incubation by using different concentrations of FGF2 for 24 hours. Then mRNA was harvested to examine fibronectin 1(Fn1) and collagen type IV (Col4a2) expression.
Rac1 Activation Assay in the Mouse Podocyte Cell Line
Mouse podocyte cells were subjected to different concentrations of glucose (300 or 600 mg/dL) for 96 hours with subsequent incubation with 25 nmol/L FGFR antagonist PD173074 (Sigma-Aldrich) or dimethyl sulfoxide for 3 hours. Then the protein lysate was harvested to examine active Rac1 levels (Rac1 G-LISA Activation Assay Biochem Kit; Cytoskeleton, Denver, CO).
Statistical Analysis
Data are expressed as means ± SEM. Unless otherwise specified, differences in each group were tested using an unpaired t-test for the two-group comparison. Statistical significance was inferred for P < 0.05. One-way analysis of variance followed by Bonferroni post hoc comparison tests were performed in statistical analyses for comparison of the three groups (Dr. SPSS II version 11.01; IBM, Armonk, NY).
Results
GPC5 Expression Increases in Diabetes
Immunofluorescence analysis of diabetic glomeruli showed that higher expressions of GPC5 were proportional to diabetes severity (Figure 1A). The expression pattern of GPC5 in DN was not limited to the glomerular capillary region but was also seen in the mesangial area along the progression to nodular glomerular sclerosis. The signal intensity was increased in mild DN cases (204% ± 37%) and severe DN cases (271% ± 43%) compared with the control (100% ± 32%).
Figure 1.

Expression pattern of GPC5 in developing diabetic albuminuria. A: Immunofluorescence staining of human kidney shows diabetes mellitus nephropathy (DN). Representative images in periodic acid-Schiff (PAS) stain, low-power field (LPF) and high-power field (HPF), are shown. GPC5 (green) was stained with Alexa 488 fluorescence. Nephrin (red) was costained with Alexa 633; images from the same sections were merged to form an overlapping image (yellow). Arrows depict GPC5 expression in nodular glomerular sclerosis. B: Immunofluorescence staining of kidney from C57BL/6 mice, podocyte-specific Gpc5 knockdown (Δ) C57BL/6 mice, Akita mice, and podocyte-specific Gpc5 knockdown (Δ) mice that were inbred with Akita mice. Gpc5 (green) was stained with Alexa 488. Nephrin (red) was counterstained with Alexa 633. An arrow depicts total absence of intraglomerular Gpc5 staining on podocyte in Podocyte-Gpc5Δ C57BL/6 whereas intraglomerular Gpc5 staining is seen in Podocyte-Gpc5Δ Akita, and this arrow indicates mesangial region. Images from the same sections were merged to form an overlapping image. C: Localization of Gpc5 in kidney sections by means of immunoelectron microscopy from 10-week-old C57BL/6 mice, Akita mice, and normal IgG. Electron micrographs show localization of Gpc5 by using a pre-embedding method. D: Gpc5 mRNA expression levels in cultured mouse podocyte and mesangial cell lines after three different administrations of the 72-hour glucose challenge [90 (white bars), 270 (gray bars), or 450 mg/dL (black bars)]. Osmolality was controlled with d-mannitol. Data are expressed as means ± SEM. n = 5 (D). ∗∗Post hoc P < 0.01 versus 90 mg/dL.
GPC5 Expression in Podocytes and Mesangial Cells
The glomerular localization of Gpc5 was examined in C57BL/6 and in C57BL/6Cr Ins2+/C96Y (Akita) mice19; localization of Gpc5 appeared in podocytes and in the paramesangial matrix (Figure 1, B and C). Consistent with previous findings,1 podocyte-specific Gpc5 knockdown mice (Figure 1B) revealed that most expression in the glomerulus was podocyte derived. Gpc5 expression of nonpodocyte origin was dramatically increased (182% ± 32%) in podocyte-GPC5Δ Akita mice compared with that in podocyte-GPC5Δ control mice (Figure 1B). In Akita mice, Gpc5 localization was more accentuated at the paramesangial area (Figure 1C). In vitro analysis further revealed that a high glucose level affected the expression level of Gpc5 in the MES13 mouse mesangial cell line and mouse podocyte cell line.14 In response to a 72-hour exposure of the cells to elevated glucose concentrations, there was a dose-dependent increase in Gpc5 mRNA expression in the mesangial cell line but not in the podocyte cell line (Figure 1D). This increase was significant at the concentration of 450 mg/dL glucose compared with 90 mg/dL glucose. These results suggest that GPC5 is mainly produced in the podocyte, and it is located on the cell surface of podocytes and secreted into the paramesangial matrix in nondiabetic states. In a high-glucose state, not only podocytes but also mesangial cells conspicuously express GPC5.
FGF2 and Proteinuria in Diabetic Mice and Gpc5 Knockdown Mice
Next we examined whether a challenge with exogenous FGF2 would induce progressive proteinuria in diabetic mice, and conversely, whether inbreeding podocyte-specific Gpc5 knockdown mice1 with the Akita strain could mitigate the proteinuria. Mice without diabetes (C57BL/6) showed no proteinuria after an injection of recombinant human FGF2, as reported previously.1 However, the same protocol of FGF2 injection induced proteinuria in Akita diabetic mice (Figure 2). Systemic Gpc5 siRNA knockdown mice had virtually no induction of proteinuria.
Figure 2.
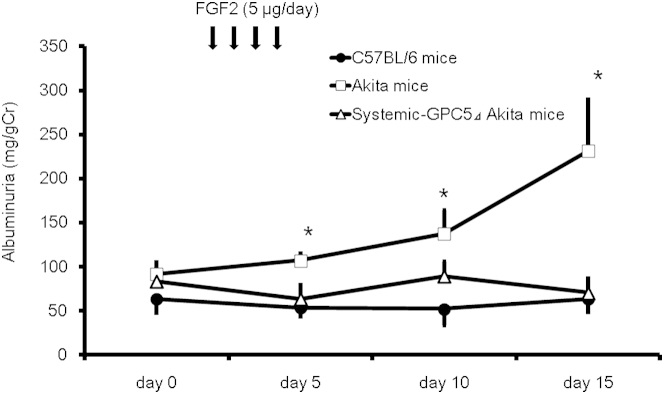
Pathogenetic role of Gpc5 and fibroblast growth factor 2 in developing diabetic albuminuria in mice. Albuminuria (milligrams of urinary albumin to grams of creatinine) was increased significantly in Akita mice after FGF2 injection; days 5, 10, and 15, respectively, compared with that of day 0. This increase was repressed significantly to the level of nondiabetic C57BL/6 by the knockdown of Gpc5. Data are expressed as means ± SEM. n = 6. ∗P < 0.05 versus systemic Gpc5 knockdown.
Expression of Fgfr3 and Fgfr4 Increases in Diabetes
The expression levels of FGFRs in nondiabetic mice and diabetic mice were examined using Akita mice and C57BL/6 mice as controls. Immunofluorescence staining revealed the increase of Fgfr3 and Fgfr4 in Akita mice, which was not seen in C57BL/6 nondiabetic mice (Figure 3). The expression levels of FGFRs under various concentrations of glucose (90, 270, and 450 mg/dL) were examined using the mouse podocyte cell line and mouse mesangial cell line. Fgfr3 and Fgfr4 expression increased significantly under high-glucose conditions in both cell lines (in the podocyte cell line at 270 mg/dL and in the mesangial cell line at 450 mg/dL) (Figure 4A). Moreover, human diabetic glomeruli showed higher expressions of FGFR3 and FGFR4 in proportion to diabetes severity (Figure 4B).
Figure 3.

Expressions of fibroblast growth factor receptor (FGFR)3 and FGFR4 increase in diabetic mice. Immunofluorescence staining of kidney from C57BL/6 mice and Akita mice is shown. Fgfr1, Fgfr2, Fgfr3, and Fgfr4 (green) were stained with Alexa 488 fluorescence. Nephrin (red) was counterstained with Alexa 633. Images from the same sections were merged to form an overlapping image (yellow; right column). Arrows depict increased FGFR3 and FGFR4 expression in the glomerulus.
Figure 4.

Expression of Fgfr3 and Fgfr4 increases in a high-glucose medium in vitro and in a human specimen. A: Fgfr3 and Fgfr4 mRNA expression levels in a cultured mouse podocyte cell line and mesangial cell line after three different administrations of a 72-hour glucose challenge [90 (white bars), 270 (gray bars), or 450 mg/dL (black bars)] are shown. Osmolality was controlled using d-mannitol. B: Immunofluorescence staining of the human kidney is shown. Lanes from the left: Nondiabetic control, patient with mild diabetes mellitus nephropathy (DN), and patient with severe DN. Top panels are representative images in periodic acid-Schiff (PAS) stain. FGFR3 and FGFR4 (green) were stained with Alexa 488 fluorescence. Arrows depict increased expression of FGFR in the glomerulus. Data are expressed as means ± SEM (A). n = 6 (A). Post hoc ∗P < 0.05, ∗∗P < 0.01 versus 90 mg/dL.
Accumulation of Systemic Circulating FGF2 in Diabetic Mice
Immunofluorescence staining revealed that the protein level of FGF2 in the Akita glomerulus was more prominent than that in the C57BL/6 nondiabetic mice (Figure 5A). However, in situ hybridization showed no evident induction of the Fgf2 gene transcript in the diabetic kidney (Figure 5B). In vitro assays in which the mouse podocyte cell line and mesangial cell line were used showed no increase in mRNA (Figure 5C). These results meant that the Fgf2 protein found in the glomeruli would derive from an extracellular origin other than podocytes and mesangial cells in these settings. For clarification, we biotinylated human recombinant FGF2 (labeled FGF2) and injected it systemically into both Akita and C57BL/6 mice. A greater accumulation of signal intensity at the glomerulus was remarkably detectable in Akita mice (Figure 5D). When labeled FGF2 was administered to systemic Gpc5 siRNA knockdown Akita mice, a minimal accumulation of labeled FGF2 was found in the glomeruli (Figure 5D). This observation supports our inference that extraglomerular FGF2 accumulates GPC5 dependently in the glomerulus during the progression of DN.
Figure 5.

Accumulation of extraglomerular fibroblast growth factor (FGF)2 in diabetic glomerulus and efficacy of Gpc5 deletion. A: FGF2 protein levels in whole lysates of the kidney of C57BL/6 and Akita mice are shown (left). Immunofluorescence staining of kidney from C57BL/6 and Akita mice (right). FGF2 (green) was stained with Alexa 488 fluorescence, nephrin (red) was costained with Alexa 633, and images from the same sections were merged to form an overlapping image (yellow). Arrow depicts increased FGF2 expression in the glomerulus. B:In situ hybridization for FGF2 mRNA in kidney from C57BL/6 and Akita mice. Placenta (E17.5) was used for positive control (arrows); images with sense probe were used for negative control. C:FGF2 mRNA expression levels in a cultured mouse podocyte cell line and mesangial cell line after three different administrations of a 72-hour glucose challenge (90, 270, or 450 mg/dL). Osmolality was controlled using d-mannitol. D: Human recombinant biotinylated FGF2 was injected intravenously in C57BL/6, Akita, and systemic Gpc5 knockdown (Δ) Akita mice. The prebiotinylated FGF2 was detected with the biotin-avidin reaction. Although a strong background signal of biotin was detected in the tubular area (upper panels), the signal from injected FGF2 was detected clearly in the glomerulus (arrows; lower panel), especially in diabetic Akita mice. Average fluorescent (Alexa 555) signals in glomeruli were calculated. E and F: Fibronectin (E) and collagen type IV (F) mRNA expression levels in a cultured rat mesangial cell line subjected to different concentrations of glucose for 48 hours with subsequent incubation with different concentrations of FGF2 for 24 hours. Osmolality was controlled with d-mannitol. G: Mouse podocyte cells were subjected to different concentrations of glucose for 96 hours with subsequent incubation using FGF receptor (FGFR) antagonist or dimethyl sulfoxide for 3 hours. Then protein lysate was harvested to examine active Rac1. Osmolality was controlled with d-mannitol. Data are expressed as means ± SEM (A, C–F). n = 5 (A, D–F); n = 6 (C). ∗P < 0.05, ∗∗P < 0.01.
Pathological Effect of FGF2 on the Glomerular Cells
In DN, the mesangial cell proliferation and mesangial matrix expansion was reportedly observed with the increase of fibronectin and collagen type IV in mesangial cells.20–22 Similarly in the rat mesangial cell line,12 high glucose concentrations (600 mg/dL) markedly induced fibronectin (Figure 5E) and collagen type IV (Figure 5F) expression co-incubated with FGF2. This induction was blocked remarkably by Gpc5 knockdown mice. In podocytes, diabetes induces hypertrophy, apoptosis, structural changes, and extracellular matrix synthesis.23 Rac1 is reportedly necessary for the modulation of cell adhesion and motility by fibronectin. We examined Rac1 activation in mouse podocytes. High glucose concentrations (600 mg/dL) markedly activated Rac1 (Figure 5G), and this induction was blocked by an FGFR antagonist.24
Discussion
Nephrotic proteinuria in DN is an ominous sign of progression to end-stage renal disease accompanied with intractable dropsy. Here, the glomerulus in cases of DN shows increased Gpc5 expression. The glomerular distribution of Gpc5 in DN was not limited to the capillary podocyte area but became more evident in the paramesangial area as DN progressed. This finding provided a unique clue about Gpc5 distribution in DN; therefore, further analysis was conducted. Gpc5 was found mostly in the glomerular capillary, more properly the glomerular endothelial cells and the basal side of the foot process (Figure 1C), in C57BL/6, as reported in previous research.1 This finding was again clarified by using Gpc5 knockdown mice. In contrast, the glomerular localization of Gpc5 in Akita mice was not abrogated completely in Gpc5 knockdown Akita mice but was rather clearly seen in the paramesangial area. This observation was consistent with findings in human DN and the in vitro analysis, leading us to the interpretation that the increase of glomerular GPC5 is mostly derived from the mesangial region.
In physiological conditions, heparan sulfate proteoglycans, including GPC5, contribute negative charge in the slit membrane. Once glucose concentration is increased, mesangial cells also start to express GPC5 and contribute to keep a reservoir for detrimental growth factors such as FGF2. Several different factors are well known to induce mesangial cell proliferation and matrix expansion, but the potential involvement of FGF2 was examined in the diabetic kidney because FGF2 is the best studied pathway in relation to GPC5.
A previous study in rodents demonstrated clearly the characteristics of FGF2 to unveil the potential malicious pathopoiesis in experimental membranous nephropathy.10 More specifically, FGF2 might enhance the disease process of nephropathies. Therefore, we next administered FGF2 to Akita diabetic mice and found the emergence and acceleration of proteinuria.25 This treatment did not develop proteinuria in nondiabetic control mice, C57BL/6, in our previous report,1 and the current findings again confirmed the impact of FGF2 on diabetes. Systemic Gpc5 knockdown by siRNA injection prevented the emergence of proteinuria in diabetic mice. Therefore, some type of Fgf receptor is participating in this pathogenicity. Results of in vitro and in vivo analyses suggest that Fgfr3 and Fgfr4 are candidate receptors, and similar results have been confirmed in human DN glomeruli as well. We discovered for the first time that human glomeruli in DN were receptive to FGF2 by using FGFR3 and FGFR4. We found that the kidneys obtained from diabetic Akita mice showed concentrations of Fgf2 that were twice as high as those from the kidneys of nondiabetic C57BL/6 mice. This sign of an underlying disorder accounts for the topping effect of Fgf2 to develop proteinuria in diabetic mice, as shown in Figure 2.
FGF2 has been reported in vitro in glomerular apparatus such as endothelial and mesangial cells.6,26–29 In previous reports, intraglomerular FGF2 proteins were increased in DN kidney specimens.27,30 Therefore, we sought to detect intraglomerular gene expression of FGF2. However, our glomerular analysis using in situ hybridization of FGF2 and immortalized cell lines showed virtually undetectable increased expression in the diabetic kidney. In addition, labeled FGF2 showed remarkable accumulation in the glomerular region in a human diabetes model. Only a limited number of studies previously reported FGF2 mRNA transcript in end-stage renal disease.29 According to a report of normal rat kidney, a weak mRNA signal was detected only in parietal glomerular epithelial cells, although FGF2 protein was found in the glomerular capillary.28 Therefore, we concluded the extraglomerular FGF2 was recruited as the causal etiology toward escalation of glomerulopathy in early DN without obvious light microscopic changes.
The FGF2 level in the systemic blood circulation is negligible under normal conditions because of a lack of an amino terminal signal sequence necessary for efficient release of FGF2 from cells. However, an increase in plasma FGF2 levels is expected in the pathophysiological condition of endothelial injury. Regarding diabetes, Stephan et al31 showed that FGF2 is closely involved in glucose-induced vascular dysfunction. Hamed et al32 also reported a marked increase in serum levels of FGF2 in T2DM compared with those of healthy controls. Zimering et al33 measured plasma FGF2 in type 2 diabetes mellitus and reported a significant increase in a subset of diabetic patients with persistent microalbuminuria or overt proteinuria. The pathogenicity that increases FGF2 levels in type 2 diabetes mellitus remains unknown, but macrovascular and microvascular endothelial injuries in type 2 diabetes mellitus are a potential source. In addition, patients with DN not only displayed an elevated serum FGF2 level but also showed increased expression of FGF2 in the tubulointerstitium, as well as a good correlation with the degree of tubulointerstitial injury.30 Both systemic circulating FGF2 and renal tubulointerstitial FGF2 levels are thought to be the source of FGF2 accumulation in glomeruli.
There is a limitation that the FGF2 injection per se resulted in higher concentrations of FGF2 in plasma than that seen in patients, but our results suggest both cell types, podocytes and mesangial cells, are affected by increased levels of FGF2. A higher FGF2 level can induce podocytopathy through the induction of caspase-31 and can simultaneously stimulate cell proliferation and matrix increase in mesangial cells.6,26 In general, Rho regulates the formation of stress fibers, whereas Cdc42 and Rac1 regulate that of filopodia and lamellipodia, respectively. In previous reports, increased active Rac1 signaling in podocytes produced albuminuria.34,35 Induction of Rac1 is reportedly crucial for cell adhesion and motility on the extracellular matrix.24 Intraglomerular imbalance can manifest as a proliferation of mesangial cells and a change of adhesion and motility in podocytes. This pathway will further accelerate the vicious cycle of DN in a nephrotic phenotype.
In diabetic glomerular injury, our findings explain how GPC5 and FGF signals influence podocytes and mesangial cells. Given that glomerular GPC5 is the key molecule for developing proteinuria encompassing both acquired nephrotic syndrome and DN, what will engender a difference in pathological findings apart from the structural change induced by comorbidities? It is noteworthy that mesangial GPC5 expression is virtually absent in the basal condition, but it is increased considerably under hyperglycemic conditions. This response shows a stark contrast to that of podocytes.
However, our study has a few limitations. GPC5 in DN must be evaluated with a large clinical cohort, and this was a small experimental study. The efficacy of the FGFR antagonist in the mouse DN model was not examined in this study, although a previous report indicated that the complete deletion of FGF2 showed an impairment of blood pressure regulation.36 GPC5 is an escort protein, apart from its effects on FGF2, which potentially interacts with other pathways such as those of Wnts and bone morphogenetic proteins. The role of GPC5 in other pathways awaits further investigation in human DN.
Conclusions
In summary, GPC5, extraglomerular circulating FGF2, and intraglomerular FGF receptors are involved in progressive DN. The induction of FGFRs under hyperglycemic conditions is crucial for the progression of DN, but this process can be controlled by the deletion of GPC5. Our results suggest that the urinary GPC5 level might be a useful biomarker for forecasting the acceleration of nephrotic deterioration of DN during clinical follow-up.
Footnotes
Supported by Grants-in-Aid for Scientific Research on the Priority Areas of Comprehensive Genomics 221S0002 (K.Tok.) and Tailor-made Medical Treatment Program (M.N. and E.N.) from the Ministry of Education, Culture, Sports, Science, and Technology; Adaptable and Seamless Technology Transfer Program through Target-Driven R&D from Japan Science and Technology Agency grant AS2617903Q (E.N.); and Grant-in-Aid for Scientific Research (B) 15K09245, Japan Society for the Promotion of Science (E.N.). K.O. received a 2012 Postdoctoral Fellowship for Research Abroad funded by the Japan Society for the Promotion of Science and is supported intramural research programs of the National Institute for Diabetes, Digestive, and Kidney Diseases (ZO-1 DK043308). E.N. is a leading researcher of the Science and Technology Research Partnership for Sustainable Development, Japan Science and Technology Agency/Japan International Cooperation Agency (Tokyo, Japan) grant 10000284.
Disclosures: None declared.
References
- 1.Okamoto K., Tokunaga K., Doi K., Fujita T., Suzuki H., Katoh T., Watanabe T., Nishida N., Mabuchi A., Takahashi A., Kubo M., Maeda S., Nakamura Y., Noiri E. Common variation in GPC5 is associated with acquired nephrotic syndrome. Nat Genet. 2011;43:459–463. doi: 10.1038/ng.792. [DOI] [PubMed] [Google Scholar]
- 2.David G. Integral membrane heparan sulfate proteoglycans. FASEB J. 1993;7:1023–1030. doi: 10.1096/fasebj.7.11.8370471. [DOI] [PubMed] [Google Scholar]
- 3.Mertens G., Van der Schueren B., van den Berghe H., David G. Heparan sulfate expression in polarized epithelial cells: the apical sorting of glypican (GPI-anchored proteoglycan) is inversely related to its heparan sulfate content. J Cell Biol. 1996;132:487–497. doi: 10.1083/jcb.132.3.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.D'Amore P.A. Modes of FGF release in vivo and in vitro. Cancer Metastasis Rev. 1990;9:227–238. doi: 10.1007/BF00046362. [DOI] [PubMed] [Google Scholar]
- 5.Muthukrishnan L., Warder E., McNeil P.L. Basic fibroblast growth factor is efficiently released from a cytolsolic storage site through plasma membrane disruptions of endothelial cells. J Cell Physiol. 1991;148:1–16. doi: 10.1002/jcp.1041480102. [DOI] [PubMed] [Google Scholar]
- 6.Floege J., Eng E., Lindner V., Alpers C.E., Young B.A., Reidy M.A., Johnson R.J. Rat glomerular mesangial cells synthesize basic fibroblast growth factor. Release, upregulated synthesis, and mitogenicity in mesangial proliferative glomerulonephritis. J Clin Invest. 1992;90:2362–2369. doi: 10.1172/JCI116126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeuchi A., Yoshizawa N., Yamamoto M., Sawasaki Y., Oda T., Senoo A., Niwa H., Fuse Y. Basic fibroblast growth factor promotes proliferation of rat glomerular visceral epithelial cells in vitro. Am J Pathol. 1992;141:107–116. [PMC free article] [PubMed] [Google Scholar]
- 8.Issandou M., Darbon J.M. Basic fibroblast growth factor stimulates glomerular mesangial cell proliferation through a protein kinase C-independent pathway. Growth Factors. 1991;5:255–264. doi: 10.3109/08977199109000289. [DOI] [PubMed] [Google Scholar]
- 9.Ballermann B.J. Regulation of bovine glomerular endothelial cell growth in vitro. Am J Physiol. 1989;256:C182–C189. doi: 10.1152/ajpcell.1989.256.1.C182. [DOI] [PubMed] [Google Scholar]
- 10.Floege J., Kriz W., Schulze M., Susani M., Kerjaschki D., Mooney A., Couser W.G., Koch K.M. Basic fibroblast growth factor augments podocyte injury and induces glomerulosclerosis in rats with experimental membranous nephropathy. J Clin Invest. 1995;96:2809–2819. doi: 10.1172/JCI118351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kriz W., Hähnel B., Rösener S., Elger M. Long-term treatment of rats with FGF-2 results in focal segmental glomerulosclerosis. Kidney Int. 1995;48:1435–1450. doi: 10.1038/ki.1995.433. [DOI] [PubMed] [Google Scholar]
- 12.Nosaka K., Nishi T., Imaki H., Suzuki K., Kuwata S., Noiri E., Aizawa C., Kurokawa K. Permeable type I collagen membrane promotes glomerular epithelial cell growth in culture. Kidney Int. 1993;43:470–478. doi: 10.1038/ki.1993.69. [DOI] [PubMed] [Google Scholar]
- 13.Committee for the Update of the Guide for the Care and Use of Laboratory Animals . National Academies Press; Washington, DC: 2011. National Research Council: Guide for the Care and Use of Laboratory Animals: Eighth Edition. [Google Scholar]
- 14.Mundel P., Reiser J., Kriz W. Induction of differentiation in cultured rat and human podocytes. J Am Soc Nephrol. 1997;8:697–705. doi: 10.1681/ASN.V85697. [DOI] [PubMed] [Google Scholar]
- 15.Nosaka K., Takahashi T., Nishi T., Imaki H., Suzuki T., Suzuki K., Kurokawa K., Endou H. An adenosine deaminase inhibitor prevents puromycin aminonucleoside nephrotoxicity. Free Radic Biol Med. 1997;22:597–605. doi: 10.1016/s0891-5849(96)00349-8. [DOI] [PubMed] [Google Scholar]
- 16.Okamoto K., Iwasaki N., Nishimura C., Doi K., Noiri E., Nakamura S., Takizawa M., Ogata M., Fujimaki R., Grarup N., Pisinger C., Borch-Johnsen K., Lauritzen T., Sandbaek A., Hansen T., Yasuda K., Osawa H., Nanjo K., Kadowaki T., Kasuga M., Pedersen O., Fujita T., Kamatani N., Iwamoto Y., Tokunaga K. Identification of KCNJ15 as a susceptibility gene in Asian patients with type 2 diabetes mellitus. Am J Hum Genet. 2010;86:54–64. doi: 10.1016/j.ajhg.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noiri E., Kuwata S., Nosaka K., Tokunaga K., Juji T., Shibata Y., Kurokawa K. Tumor necrosis factor-alpha mRNA expression in lipopolysaccharide-stimulated rat kidney. Chronological analysis of localization. Am J Pathol. 1994;144:1159–1166. [PMC free article] [PubMed] [Google Scholar]
- 18.Okamoto K., Iwasaki N., Doi K., Noiri E., Iwamoto Y., Uchigata Y., Fujita T., Tokunaga K. Inhibition of glucose-stimulated insulin secretion by KCNJ15, a newly identified susceptibility gene for type 2 diabetes. Diabetes. 2012;61:1734–1741. doi: 10.2337/db11-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshioka M., Kayo T., Ikeda T., Koizumi A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes. 1997;46:887–894. doi: 10.2337/diab.46.5.887. [DOI] [PubMed] [Google Scholar]
- 20.Bangstad H.J., Rudberg S., Østerby R. Renal structural changes in patients with type 1 diabetes and microalbuminuria. In: Mogensen C.E., editor. The Kidney and Hypertension in Diabetes. Kluwer Academic Publishers; New York, NY: 2000. pp. 211–224. [Google Scholar]
- 21.Riser B.L., Cortes P., Zhao X., Bernstein J., Dumler F., Narins R.G. Intraglomerular pressure and mesangial stretching stimulate extracellular matrix formation in the rat. J Clin Invest. 1992;90:1932–1943. doi: 10.1172/JCI116071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yasuda T., Kondo S., Homma T., Harris R.C. Regulation of extracellular matrix by mechanical stress in rat glomerular mesangial cells. J Clin Invest. 1996;98:1991–2000. doi: 10.1172/JCI119003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J.J., Kwak S.J., Jung D.S., Kim J.J., Yoo T.H., Ryu D.R., Han S.H., Choi H.Y., Lee J.E., Moon S.J., Kim D.K., Han D.S., Kang S.W. Podocyte biology in diabetic nephropathy. Kidney Int Suppl. 2007:S36–S42. doi: 10.1038/sj.ki.5002384. [DOI] [PubMed] [Google Scholar]
- 24.Kimura K., Kawamoto K., Teranishi S., Nishida T. Role of Rac1 in fibronectin-induced adhesion and motility of human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2006;47:4323–4329. doi: 10.1167/iovs.05-1508. [DOI] [PubMed] [Google Scholar]
- 25.Brosius F.C., 3rd, Alpers C.E., Bottinger E.P., Breyer M.D., Coffman T.M., Gurley S.B., Harris R.C., Kakoki M., Kretzler M., Leiter E.H., Levi M., McIndoe R.A., Sharma K., Smithies O., Susztak K., Takahashi N., Takahashi T. Animal Models of Diabetic Complications Consortium: Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2009;20:2503–2512. doi: 10.1681/ASN.2009070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haseley L.A., Hugo C., Reidy M.A., Johnson R.J. Dissociation of mesangial cell migration and proliferation in experimental glomerulonephritis. Kidney Int. 1999;56:964–972. doi: 10.1046/j.1523-1755.1999.00641.x. [DOI] [PubMed] [Google Scholar]
- 27.Cauchi J., Alcorn D., Cancilla B., Key B., Berka J.L., Nurcombe V., Ryan G.B., Bertram J.F. Light-microscopic immunolocalization of fibroblast growth factor-1 and -2 in adult rat kidney. Cell Tissue Res. 1996;285:179–187. doi: 10.1007/s004410050635. [DOI] [PubMed] [Google Scholar]
- 28.Floege J., Hudkins K.L., Eitner F., Cui Y., Morrison R.S., Schelling M.A., Alpers C.E. Localization of fibroblast growth factor-2 (basic FGF) and FGF receptor-1 in adult human kidney. Kidney Int. 1999;56:883–897. doi: 10.1046/j.1523-1755.1999.00637.x. [DOI] [PubMed] [Google Scholar]
- 29.Strutz F., Zeisberg M., Hemmerlein B., Sattler B., Hummel K., Becker V., Müller G.A. Basic fibroblast growth factor expression is increased in human renal fibrogenesis and may mediate autocrine fibroblast proliferation. Kidney Int. 2000;57:1521–1538. doi: 10.1046/j.1523-1755.2000.00997.x. [DOI] [PubMed] [Google Scholar]
- 30.Vasko R., Koziolek M., Ikehata M., Rastaldi M.P., Jung K., Schmid H., Kretzler M., Müller G.A., Strutz F. Role of basic fibroblast growth factor (FGF-2) in diabetic nephropathy and mechanisms of its induction by hyperglycemia in human renal fibroblasts. Am J Physiol Renal Physiol. 2009;296:F1452–F1463. doi: 10.1152/ajprenal.90352.2008. [DOI] [PubMed] [Google Scholar]
- 31.Stephan C.C., Chang K.C., LeJeune W., Erichsen D., Bjercke R.J., Rege A., Biediger R.J., Kogan T.P., Brock T.A., Williamson J.R., Tilton R.G. Role for heparin-binding growth factors in glucose-induced vascular dysfunction. Diabetes. 1998;47:1771–1778. doi: 10.2337/diabetes.47.11.1771. [DOI] [PubMed] [Google Scholar]
- 32.Hamed E.A., Zakary M.M., Abdelal R.M., Abdel Moneim E.M. Vasculopathy in type 2 diabetes mellitus: role of specific angiogenic modulators. J Physiol Biochem. 2011;67:339–349. doi: 10.1007/s13105-011-0080-8. [DOI] [PubMed] [Google Scholar]
- 33.Zimering M.B., Eng J. Increased basic fibroblast growth factor-like substance in plasma from a subset of middle-aged or elderly male diabetic patients with microalbuminuria or proteinuria. J Clin Endocrinol Metab. 1996;81:4446–4452. doi: 10.1210/jcem.81.12.8954057. [DOI] [PubMed] [Google Scholar]
- 34.Shibata S., Nagase M., Yoshida S., Kawarazaki W., Kurihara H., Tanaka H., Miyoshi J., Takai Y., Fujita T. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med. 2008;14:1370–1376. doi: 10.1038/nm.1879. [DOI] [PubMed] [Google Scholar]
- 35.Togawa A., Miyoshi J., Ishizaki H., Tanaka M., Takakura A., Nishioka H., Yoshida H., Doi T., Mizoguchi A., Matsuura N., Niho Y., Nishimune Y., Nishikawa S., Takai Y. Progressive impairment of kidneys and reproductive organs in mice lacking Rho GDIalpha. Oncogene. 1999;18:5373–5380. doi: 10.1038/sj.onc.1202921. [DOI] [PubMed] [Google Scholar]
- 36.Dono R., Texido G., Dussel R., Ehmke H., Zeller R. Impaired cerebral cortex development and blood pressure regulation in FGF-2-deficient mice. EMBO J. 1998;17:4213–4225. doi: 10.1093/emboj/17.15.4213. [DOI] [PMC free article] [PubMed] [Google Scholar]