

Abstract
The molecular events that link NADPH oxidase activation and the induction of Toll-like receptor (TLR)-4 recruitment into hepatic lipid rafts in nonalcoholic steatohepatitis (NASH) are unclear. We hypothesized that in liver, NADPH oxidase activation is key in TLR4 recruitment into lipid rafts, which in turn up-regulates NF-κB translocation to the nucleus and subsequent DNA binding, leading to NASH progression. Results from confocal microscopy showed that liver from murine and human NASH had NADPH oxidase activation, which led to the formation of highly reactive peroxynitrite, as shown by 3-nitrotyrosine formation in diseased liver. Expression and recruitment of TLR4 into the lipid rafts were significantly greater in rodent and human NASH. The described phenomenon was NADPH oxidase, p47phox, and peroxynitrite dependent, as liver from p47phox-deficient mice and from mice treated with a peroxynitrite decomposition catalyst [iron(III) tetrakis(p-sulfonatophenyl)porphyrin] or a peroxynitrite scavenger (phenylboronic acid) had markedly less Tlr4 recruitment into lipid rafts. Mechanistically, peroxynitrite-induced TLR4 recruitment was linked to increased IL-1β, sinusoidal injury, and Kupffer cell activation while blocking peroxynitrite-attenuated NASH symptoms. The results strongly suggest that NADPH oxidase–mediated peroxynitrite drove TLR4 recruitment into hepatic lipid rafts and inflammation, whereas the in vivo use of the peroxynitrite scavenger phenylboronic acid, a novel synthetic molecule having high reactivity with peroxynitrite, attenuates inflammatory pathogenesis in NASH.
Nonalcoholic steatohepatitis (NASH) has been studied extensively in preclinical models and in humans. NASH manifestations range from an early sinusoidal endothelial dysfunction to inflammation followed by defective tissue repair, resulting in fibrosis.1–6 Inefficient perfusion in the fatty tissue mostly associated with fatty liver and subsequent NASH progression can result in the recruitment of other cell types, including Kupffer cells, sinusoidal endothelial cells, and circulating lymphocytes, making it a perfect microenvironment for forming inflammatory foci.7
Recent research reports have identified an emerging role of Toll-like receptor (TLR)-4 in NASH pathogenesis.8,9 Several endogenous mediators, such as gut-derived endotoxin and nuclear factor high-mobility group box 1, have been implicated in activating TLR4 signaling, leading to NASH severity.10,11 After its discovery in the 1980s, TLR signaling has been at the forefront of innate immune signaling and disease pathophysiology.12 TLR4, one of the many TLRs discovered since then, plays a pivotal role in cytokine release along with its adaptor molecules myeloid differentiation primary response 88 gene (MYD88) and TIR domain–containing adapter-inducing β-interferon. MyD88 serves as an adaptor for inflammatory signaling pathways downstream of the members of the TLR and IL-1 receptor families.13 Activation of the TLR4 pathway leads to a variety of functional outputs, including the activation of NF-κB, mitogen-activated protein kinases, and activator protein 1, making MyD88 a central node of inflammatory pathways.13 Lipopolysaccharide of Gram-negative bacterial cell walls is a strong ligand for TLR4 on the cell membrane. Myeloid differentiation 2 is associated with TLR4 on the cell surface and enables TLR4 to respond to lipopolysaccharide.14 Deficiency of Myd88 or Tlr4 in mice attenuates the development of the disease.15 Although oxidative stress via NADPH oxidase has been shown to drive NASH pathogenesis, especially fibrogenesis and Kupffer cell activation, there is no direct evidence of a single mechanism whereby a specific oxidative species can drive the recruitment of TLR4-mediated inflammation in NASH.16–18
We therefore aimed to identify a single intracellular reactive nitrative species that may play a role in developing the inflammatory pathogenesis in NASH via TLR4 recruitment into hepatic lipid rafts, an early yet distinct process in TLR4 activation and downstream signaling. One of the common sources of generation of toxic superoxide radicals may be NADPH oxidase isoforms, which can be found in both phagocytic and nonphagocytic cells.19 Liver has been shown to be a rich source of various isoforms of the family of reactive oxygen species–generating NADPH oxidases (NOX), being present in phagocytic Kupffer cells, stellate cells, and sinusoidal endothelial cells.3,17,20 Out of the various NOX isoforms, NOX2 is primarily present in Kupffer cells and stellate cells, whereas NOX1 and NOX4 have been found in other liver cell types.17 NOX2 is composed of several subunits, including glycoprotein (gp) gp91, p22 (membrane subunits), p67, p47, and Ras-related C3 botulinum toxin substrate 1 (cytosolic subunits).19 On receipt of proper signals, the cytosolic subunits align with their membrane counterparts to form an active NADPH oxidase complex that produces superoxide radicals to be released into the extracellular space.19
NOX2 is involved in NASH development.16,18,20 NOX2 stimulation by high leptin results in peroxynitrite generation, thus causing Kupffer cell activation in NASH.18 NOX2 has also been shown to facilitate the recruitment of TLR4 into lipid rafts and to help in receptor dimerization and its association with myeloid differentiation 2 in several inflammatory diseases.21 Because there is no evidence of the molecular events that follow NADPH oxidase activation concerning the type of reactive species formation and the mechanisms that link TLR4 activation, we investigated the role of peroxynitrite in TLR4 activation in NASH. Peroxynitrite causes cellular senescence and alters the immunogenicity of proteins; many functional roles of the peroxynitrite species in protein nitration in acute and chronic liver diseases have been reviewed.19,21–23 Blocking peroxynitrite in vivo with newer and more specific scavengers may provide novel therapeutic strategies for NASH complications.
Our aim was to investigate the molecular mechanisms of TLR4 induction by NADPH oxidase; the role of peroxynitrite; and the effects of peroxynitrite on sinusoidal injury, inflammation, Kupffer cell activation, and stellate cell proliferation, all significant events in NASH progression from steatosis. A high-fat diet–induced obesity (DIO) model in which hepatotoxin bromodichloromethane (BDCM) was administered to generate oxidative stress, a second hit to cause steatohepatitic lesions,24 was used. A second, widely used rodent model based on feeding with a methyl choline–deficient versus–sufficient diet (MCD and MCS, respectively) was also used. Human NASH liver and healthy human control liver were used for corroborating the results from the murine model. We used a decomposition catalyst and a scavenger of peroxynitrite to support the involvement of peroxynitrite.18,19 The results of the study, which used transgenic mice and a pharmacological approach, show for the first time a molecular basis of NADPH oxidase–mediated, peroxynitrite-driven TLR4 activation in causing sinusoidal injury, inflammation, and stellate cell proliferation in NASH.
Materials and Methods
Obese Mice
Pathogen-free, 6-week-old, customized, high-fat diet–fed adult male mice with C57BL/6J background (Jackson Laboratory, Bar Harbor, ME) were used as a model for DIO. The animals were fed a high-fat diet (60% kcal) from weeks 6 to 16. All experiments were conducted after the completion of 16 weeks. Mice with p47phox gene (alias Ncf1) knockout (KO; B6.129S2-Ncf1tm1shl N14; Taconic, Cranbury, NJ) or with Tlr4 KO (B6.B10ScN-Tlr4lps-del/JthJ; Jackson Laboratory) were fed a high-fat diet and received treatment identical to that of the DIO mice. The mice were housed one per cage in a temperature-controlled room at 23°C to 24°C with a 12-hour light/dark cycle and ad libitum access to food and water. All animals were treated in strict accordance with the NIH guideline for Humane Care and Use of Laboratory Animals and local institutional animal care and use committee standards. All experiments were approved by the Institutional Review Board at the National Institute of Environmental Health Sciences, Duke University (Durham, NC), and the University of South Carolina (Columbia, SC).
Toxin Model
Induction of Liver Injury in Obese Mice
BDCM (1.0 mmol/L per kg, diluted in corn oil) was administered through i.p. injection in 16-week-old high-fat diet–fed wild-type (DIO + BDCM) or specific gene KO mice. Two doses per week of BDCM were administered for 4 weeks. DIO mice were injected with corn oil (diluent of BDCM) and used as controls (DIO). After the treatments were completed in all mouse groups, the mice were sacrificed and liver tissue harvested for the further experiments.
Administration of Apo/FeTPPS/FBA in Obese Mice
A group of DIO mice was injected with apocynin (Apo), an NADPH oxidase inhibitor, at a dose of 10 mg/kg of body weight. A second group of DIO mice was injected with a ferric porphyrin complex that is a decomposition catalyst of peroxynitrite [iron(III) tetrakis(p-sulfonatophenyl)porphyrin; FeTPPS], at a dose of 30 mg/kg of body weight. A third group of DIO mice was injected with phenylboronic acid (FBA), a peroxynitrite scavenger, at a dose of 50 mg/kg of body weight. All of these injections were given 1 hour before each BDCM injection over a period of 4 weeks.
Human Tissues
Human liver tissue from both NASH and age- and sex-matched healthy individuals was purchased from the NIH repository at the University of Minnesota (Minneapolis, MN) and the University of Pittsburgh (Pittsburgh, PA).
Histopathology
Liver sections were collected from each animal and fixed in 10% neutral buffer formalin. These formalin-fixed, paraffin-embedded tissues were cut into 5-μm-thick sections. These sections were deparaffinized (standard protocol), stained with picrosirius red, and observed using 20× objective of light microscopy. Stained liver sections were examined for stages of fibrosis using the criteria of the NIH NASH Clinical Research Network scoring system. Stages of fibrosis were defined as 1A, mild; 1C, portal; 2, periportal; and 3, bridging. Briefly, because zone 3 perisinusoidal/pericellular fibrosis develops first and is followed by portal fibrosis, we used the fibrosis stages as described originally by Kleiner et al.25,26 Kleiner et al26 modified the fibrosis staging in the Brunt system by subclassifying fibrosis scores of stage 1 into delicate (1a); dense perisinusoidal (1b); and nonbridging, portal only (1c). Stage 2 accounts for perisinusoidal and portal/periportal fibrosis. The scoring pattern was found to be consistent with that of portal fibrosis, beginning early in the disease process and playing an important role in connection with the development of portal–central fibrous bridging, which is likely found in the most developed cases of fibrosis.
Immunofluorescence Microscopy
Formalin-fixed, paraffin-embedded tissues were cut into 5-μm-thick sections. Each section was subjected to deparaffinization and immunostaining using standard protocol. The primary antibodies anti-gp91phox, anti-p47phox, anti-TLR4, and anti-flotillin were purchased from Santa Cruz Biotechnologies (Dallas, TX) and Abcam (Cambridge, MA) and used at the recommended dilutions. Species-specific anti-IgG secondary antibodies conjugated with Alexa Fluor 488, 633, and 568 (Invitrogen, Carlsbad, CA) were used against the appropriate primary antibodies. The sections were mounted in a ProLong Gold Antifade Reagent (Life Technologies, Carlsbad, CA) with DAPI. Images were obtained under 20×/60× oil objectives using a BX73 microscope (Olympus America, Center Valley, PA). Confocal laser scanning microscopy was performed for selected images, followed by colocalization analysis using LSM image browser version 4.2.0.0121 (Carl Zeiss, Oberkochen, Germany).
IHC Analysis
Formalin-fixed, paraffin-embedded tissues were cut into 5-μm-thick sections. Each section was subjected to deparaffinization and immunostaining using standard protocol. The primary antibodies were anti–IL-1β, anti–monocyte chemotactic protein (MCP)-1, anti-CD68, and anti–α-smooth muscle actin (α-SMA). Primary antibodies were purchased from Abcam and used in recommended dilutions.
The Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA) was used for antigen-specific immunohistochemistry (IHC) analysis following the manufacturer's protocols. 3,3′-Diaminobenzidine (Sigma-Aldrich, St. Louis, MO) was used as a chromogenic substrate. Tissue sections were counterstained by Mayer's hematoxylin (Sigma-Aldrich). Sections were mounted in Simpo mount (GBI Laboratories, Mukilteo, WA) and observed under a 20× objective. Morphometric analysis was done using CellSens software version 1.12 (Olympus America).
NF-κB Transcription Factor Binding Assay
Nuclear fraction of each liver sample was isolated following the manufacturer's standard protocol (FIVEphoton Biochemicals, San Diego, CA). DNA binding activity of the transcription factor NF-κB present in those nuclear extracts was detected by a 96-well enzyme-linked immunosorbent assay method (NF-κB p65 Transcription Factor Assay Kit; Abcam) following the manufacturer's standard protocol.
Statistical Analysis
All in vivo experiments were repeated three times (n = 3 mice per group; data from each group of 3 mice were pooled). Statistical analysis was performed by analysis of variance followed by a post hoc test. Quantitative data from Western blots, as depicted by the relative intensity of the bands, were analyzed by performing a Student's t-test. P < 0.01 or P < 0.05, where applicable, was considered statistically significant.
Results
NASH Pathogenesis Is Associated with NADPH Oxidase Activation–Induced Oxidative Stress Primarily Mediated by Peroxynitrite
To study the involvement of NADPH oxidase and corresponding oxidative stress in rodent and human liver with NASH, experiments were performed to support the NADPH oxidase activation that is characterized by the association of p47phox with the membrane subunit gp91phox by immunostaining and immunofluorescence microscopy. Results showed that the DIO + BDCM group had significantly increased colocalizations of p47phox and gp91phox in NASH liver compared with those in the DIO-only group (both, P < 0.01) (Figure 1, A and C). Higher-magnification images of liver sections showed that colocalization was evident primarily in sinusoidal cells and occasionally in hepatocytes (Figure 1A). Gp91phox and p47phox colocalizations were significantly increased in MCD-model and human NASH liver (all, P < 0.05) (Supplemental Figure S1, A and C). Increased NADPH oxidase activation was associated with increased oxidative stress, as evident from the increased tyrosyl radical formation (3-nitrotyrosine immunoreactivity). Results showed that DIO + BDCM group liver had significantly increased immunoreactivity to 3-nitrotyrosine compared with that from the DIO-only group (P < 0.01) (Figure 1, B and D). Mice that had p47phox KO or that were treated with NADPH oxidase nonspecific inhibitor Apo, peroxynitrite decomposition catalyst FeTPPS, or peroxynitrite scavenger FBA showed significantly decreased 3-nitrotyrosine immunoreactivity compared with that in the DIO + BDCM group (all, P < 0.01) (Figure 1, B and D). Liver from MCD diet–fed mice and human NASH liver showed significantly increased immunoreactivity to 3-nitrotyrosine, an index of oxidative stress–mediated tyrosyl radical formation (both, P < 0.05) (Supplemental Figure S1, B and D). The results suggest that NASH pathogenesis is associated with NADPH oxidase activation primarily via NOX2 mediation and p47phox translocation, followed by peroxynitrite formation and tyrosyl radical formation. FBA scavenging of peroxynitrite also confirmed that peroxynitrite, instead of myeloperoxidase, which is also present in the injured liver, was involved in tyrosine nitration.
Figure 1.

NADPH oxidase activation and peroxynitrite-mediated tyrosine nitration. A and B: Representative images of 5-μm-thick immunostained liver sections, imaged using immunofluorescence microscopy. Nuclear stain (blue) is due to ProLong Gold Antifade Reagent (Life Technologies, Carlsbad, CA) with DAPI. A: Secondary antibodies used were Alexa Fluor 568 (Invitrogen; red) against α-gp91phox primary antibody and Alexa Fluor 488 (green) against α-p47phox primary antibody. Yellow spots/patches indicate alignment of cytoplasmic subunit of NADPH oxidase, p47phox (red), with membrane subunit glycoprotein (gp)91phox (green). B: Secondary antibody used was Alexa Fluor 633 (red) against α-3-nitrotyrosine primary antibody. C and D: Morphometry performed on three independent fields for each sample, including representative images in A for gp91phox/p47phox colocalization events per 300 cells (C) and in B for percentage of area showing positive immunoreactivity of 3-nitrotyrosine, calculated in arbitrary units (D). ∗∗P < 0.01. Apo, diet-induced obesity wild-type (DIO) mouse exposed to bromodichloromethane (BDCM) and injected with apocynin; DIO + BDCM, DIO mouse exposed to BDCM; FBA, DIO mouse exposed to BDCM and phenylboronic acid; FeTPPS, DIO mouse exposed to BDCM and iron(III) tetrakis(p-sulfonatophenyl)porphyrin; p47phox KO, high-fat diet–fed mouse with p47phox gene knockout exposed to BDCM; Tlr4 KO, high-fat diet–fed mouse with Tlr4 knockout exposed to BDCM.
NADPH Oxidase Activation–Mediated Peroxynitrite Formation and Subsequent Oxidative Stress Drives TLR4 Recruitment into Hepatic Lipid Rafts
The role of TLR4 in NASH pathogenesis has been reported recently.27,28 However, the mechanisms that drive TLR4 recruitment and subsequent signaling in NASH have been unclear. One of the primary events of TLR activation is its recruitment into the hepatic lipid rafts, after which the assembly of activation complex and ligand binding occur.29,30 Although recent studies have focused on TLR4 activation processes primarily by high-mobility group box 1, X-box 1, or bacteria from the gut, no studies have shown the mechanisms that drive TLR4 into the membrane rafts.11,31,32 To support that NADPH oxidase–mediated peroxynitrite drives TLR4 recruitment into hepatic lipid rafts, experiments were performed in mice and human liver. Results showed that DIO + BDCM group liver had a significant increase in Tlr4–flotillin (a lipid raft protein) colocalization events, as shown by immunofluorescence microscopy (sixfold increase), compared with that in the DIO-only group (P < 0.01) (Figure 2, A and B). Confocal laser scanning (Figure 2C) and analysis (Figure 2D) showed colocalization of Tlr4 and flotillin in sinusoidal cells of NASH liver (Figure 2C). Liver from p47phox gene KO mice and from mice treated with NADPH oxidase nonspecific inhibitor Apo, peroxynitrite decomposition catalyst FeTPPS, or peroxynitrite scavenger FBA had significantly decreased colocalization events of Tlr4 and flotillin (all, P < 0.05) (Figure 2, A and B, and Supplemental Figure S2, A and C). Liver from MCD diet–fed mice and human NASH liver showed significant increases in TLR4–flotillin colocalization events compared with those in respective controls (both, P < 0.05) (Supplemental Figure S2, B and D). The results suggest that NADPH oxidase activation and subsequent peroxynitrite-mediated oxidative stress drive TLR4 recruitment into hepatic lipid rafts, a significant early event of TLR4 signaling and NASH inflammatory pathogenesis.
Figure 2.

NADPH oxidase and peroxynitrite–driven Toll-like receptor (TLR)-4 recruitment into lipid rafts in hepatic cell membranes. A: Representative images of 5-μm-thick immunostained liver sections, imaged using immunofluorescence microscopy. Secondary antibodies used were Alexa Fluor 633 (Invitrogen; red) against α-Tlr4 primary antibody and Alexa Fluor 488 (green) against α-flotillin primary antibody. Nuclear stain (blue) is due to ProLong Gold Antifade Reagent (Life Technologies, Carlsbad, CA) with DAPI. Yellow spots/patches indicate alignment of Tlr4 (red), with lipid raft protein flotillin (green). B: Morphometry performed on three independent fields for each sample, including representative images in A for Tlr4–flotillin colocalization events per 300 cells. C: Confocal laser scan of Tlr4–flotillin colocalization in the membranes of liver cells with nonalcoholic steatohepatitis (NASH). D: Colocalization analysis using LSM image browser version 4.2.0.0121 (Carl Zeiss, Oberkochen, Germany). ∗∗P < 0.01. Original magnification ×60 (A). Apo, diet-induced obesity wild-type (DIO) mouse exposed to bromodichloromethane (BDCM) and apocynin; DIO + BDCM, DIO mouse exposed to BDCM; FBA, DIO mouse exposed to BDCM and phenylboronic acid; FeTPPS, DIO mouse exposed to BDCM and iron(III) tetrakis(p-sulfonatophenyl)porphyrin; p47phox KO, high-fat diet–fed mouse with p47phox gene knockout exposed to BDCM.
NF-κB Activation and DNA Binding in NASH Liver Require NADPH Oxidase–Mediated Peroxynitrite on TLR4 Recruitment into Lipid Rafts
One of the key events after TLR4 recruitment from nonrafts into lipid-enriched membrane rafts is downstream signaling, leading to NF-κB activation, translocation to the nucleus, and subsequent DNA binding.33,34 After our observations that NADPH oxidase activation and peroxynitrite generation drive TLR4 recruitment into hepatic lipid rafts, we probed the involvement of NADPH oxidase–mediated peroxynitrite in causing NF-κB activation. Results showed that the DIO + BDCM group had significant increases in NF-κB activation and binding to DNA compared with those in the DIO-only group (both, P < 0.05) (Figure 3A). p47phox gene KO mice and mice administered Apo, FeTPPS, or FBA had significantly decreased NF-κB activation and DNA binding compared with those in the DIO + BDCM group (all, P < 0.05) (Figure 3A). Liver from MCD diet–fed mice and human NASH liver had significantly increased NF-κB activation and DNA binding compared with their respective controls (all, P < 0.05) (Figure 3B). The results suggest that on TLR4 recruitment into hepatic lipid rafts after NADPH oxidase activation, downstream signaling is initiated and leads to NF-κB activation and DNA binding, significant steps toward TLR4-mediated inflammatory cytokine release and innate immune activation.
Figure 3.
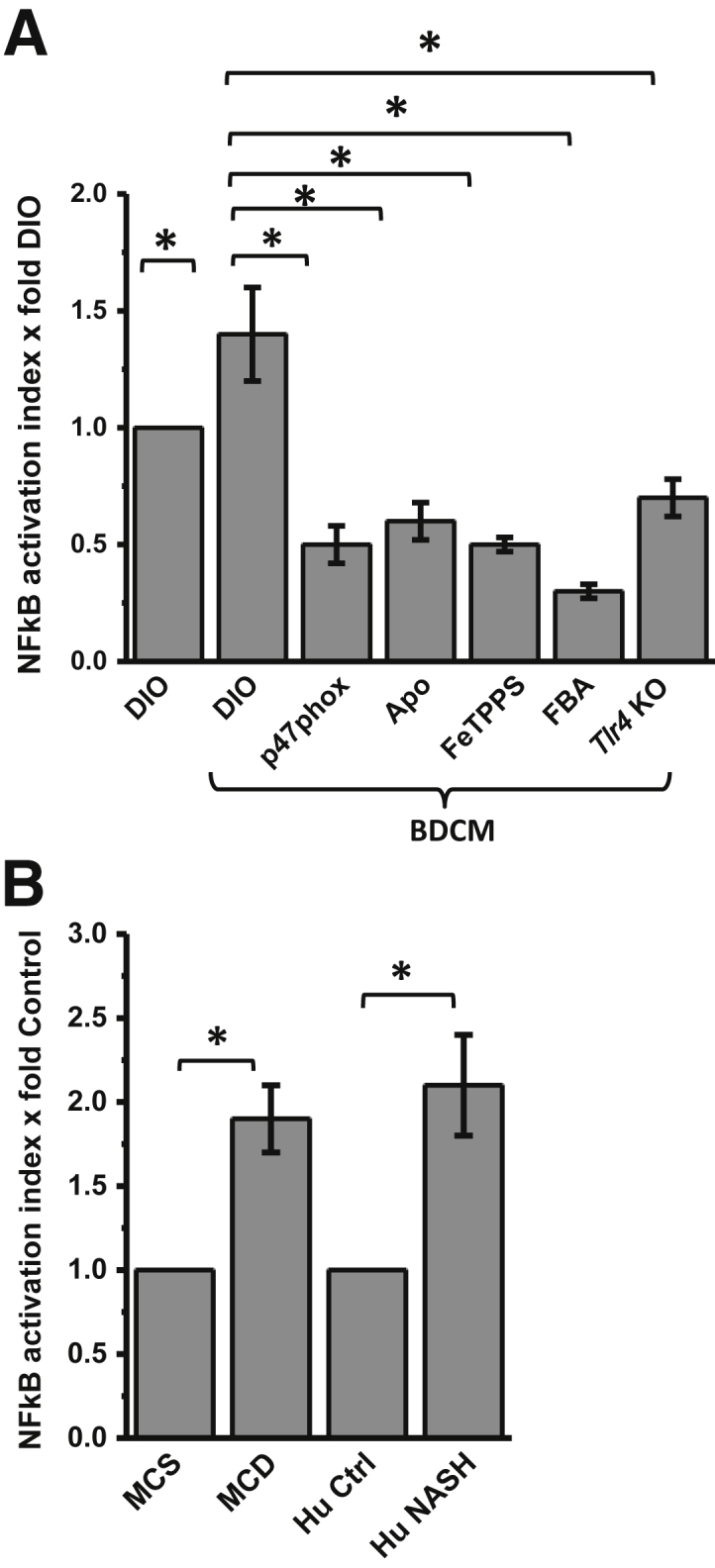
NADPH oxidase and peroxynitrite–driven NF-κB translocation and DNA-binding via Toll-like receptor (Tlr)-4 signaling. A: Results of NF-κB activation assay in murine samples of toxin-induced nonalcoholic steatohepatitis (NASH) model. NF-κB activation index (assessed from nuclear translocation and DNA binding of p65 subunit) is normalized against diet-induced obesity wild-type mouse (DIO) sample. B: Results of NF-κB activation assay in murine samples of diet-induced NASH model and human samples. NF-κB activation index is normalized against appropriate and respective control samples. ∗P < 0.05. BCDM, bromodichloromethane.
NADPH Oxidase Activation and Formation of Peroxynitrite Cause Sinusoidal Injury Primarily through Their Role in TLR4 Recruitment
One of the primary events in NASH pathogenesis is sinusoidal injury, often ascribed to insufficient perfusion and to the formation of inflammatory foci.6,7,35 Recent evidence from our laboratory and others6,36 has shown that sinusoidal injury marked by increases in intercellular adhesion molecule (ICAM)-1 and E-selectin in the liver sinusoidal endothelial cells, stellate cells, and Kupffer cells form an early basis for inflammatory pathogenesis in NASH. After such observations, it was essential that the role of NADPH oxidase-mediated formation of peroxynitrite and TLR4 recruitment into lipid rafts be shown to initiate sinusoidal injury in the NASH liver. Results showed that DIO + BDCM group liver had significant increases in immunoreactivity to Icam-1 and E-selectin compared with that in the DIO-only group (both, P < 0.01) p47phox (Figure 4). p47phox gene KO mice and those administered Apo had significantly decreased immunoreactivity to Icam-1 and E-selectin compared with those in the DIO + BDCM group (all, P < 0.01) (Figure 4, A–C), showing the involvement of NADPH oxidase activation. The use of FeTPPS or FBA further decreased the levels of Icam-1 and E-selectin in liver compared with those in the DIO + BDCM group (all, P < 0.01) (Figure 4, A and B). Mice that were deficient in Tlr4 showed significantly decreased levels of Icam-1 and E-selectin, thus showing a strong correlation between NADPH oxidase activation and the presence of Tlr4 with sinusoidal injury, as depicted by the increased levels of Icam-1 and E-selectin (both, P < 0.01) (Figure 4, A and B). MCD diet–fed mouse liver and human NASH liver showed significant increases in immunoreactivity to ICAM-1 and E-selectin compared with those in their respective controls (all, P < 0.05) (Supplemental Figure S3, A–D). Apart from the IHC analysis data, mRNA-expression profiles of ICAM-1 and E-selectin showed significant increases in murine NASH tissue and human NASH tissue, whereas mice with p47phox gene KO or Tlr4 KO and those administrated peroxynitrite scavengers had significantly decreased levels of these sinusoidal injury markers (all, P < 0.05) (Supplemental Figures S4 and S5). The results suggest that NADPH oxidase activation and the subsequent formation of peroxynitrite cause increased sinusoidal injury in NASH and are strongly associated with the presence of TLR4.
Figure 4.

NADPH oxidase and peroxynitrite–driven hepatic sinusoidal injury via Toll-like receptor (Tlr)-4 signaling. A and B: Representative images of 5-μm-thick immunostained liver sections, imaged using immunofluorescence microscopy. Secondary antibody used was Alexa Fluor 568 (Invitrogen; red) against α-intercellular adhesion molecule (Icam)-1 primary antibody (A) and against α-E-selectin primary antibody (B) (both Icam-1 and E-selectin are sinusoidal injury markers). Nuclear stain (blue) is due to ProLong Gold Antifade Reagent (Life Technologies, Carlsbad, CA) with DAPI. C and D: Morphometry performed on three independent fields for each sample including representative images in for percentage of area showing positive immunoreactivity of Icam-1 in A (C) and of E-selectin in B (D), calculated in arbitrary units. ∗∗P < 0.01. Original magnification, ×20 (A and B). Apo, diet-induced obesity wild-type (DIO) mouse exposed to bromodichloromethane (BDCM) and injected with apocynin; DIO + BDCM, DIO mouse exposed to BDCM; FBA, DIO mouse exposed to BDCM and phenylboronic acid; FeTPPS, DIO mouse exposed to BDCM and iron(III) tetrakis(p-sulfonatophenyl)porphyrin; p47phox KO, high-fat diet–fed mouse with p47phox gene knockout exposed to BDCM; Tlr4 KO, high-fat diet–fed mouse with Tlr4 knockout exposed to BDCM.
NADPH Oxidase–Mediated Peroxynitrite Generation Drives TLR4 Signaling–Induced IL-1β, Chemokine Release, and Kupffer Cell Activation in NASH
Previous experiments have shown the strong association of NADPH oxidase–mediated peroxynitrite formation and TLR4 recruitment followed by NF-κB activation, crucial processes for downstream TLR4 signaling. It is essential that TLR4-mediated IL-1β release and Kupffer cell activation, which have been shown to be mediated by TLR4 signaling, be proven to establish the significance of NADPH oxidase–driven TLR4 recruitment. Results showed that immunoreactivity to Il-1β in mouse liver slices in the DIO + BDCM group was significantly greater compared with that in the DIO-only group (P < 0.01) (Figure 5, A and D). p47phox gene KO mice and mice treated with FeTPPS showed significant decreases in Il-1β immunoreactivity compared with that in the DIO + BDCM group (both, P < 0.01) (Figure 5, A and D). Tlr4 KO mice showed a significant decrease in Il-1β immunoreactivity compared with that in the DIO + BDCM group (Figure 5, A and D), suggesting the strong involvement of TLR4 signaling.
Figure 5.

Toll-like receptor (Tlr)-4 signaling–mediated cytokine release and Kupffer cell activation. A–C: Representative images of 5-μm-thick immunostained liver sections, imaged using bright-field microscopy. Positive immunoreactivities (brown) are due to diaminobenzidine binding to respective biotinylated secondary antibodies via streptavidin–horseradish peroxidase. Nuclei (blue) counterstained with Mayer's hematoxylin solution. Immunoreactivities detected are for proinflammatory cytokines Il-1β (A), monocyte chemotactic protein (MCP)-1 (B), and Kupffer cell activation marker CD68 (C). D–F: Morphometry performed on three independent fields for each sample, including representative images for percentage of area showing positive immunoreactivity of Il-1β in A (D) and of MCP-1 in B (E) and CD68 in C (F), calculated in arbitrary units. ∗∗P < 0.01. Original magnification, ×20 (A–C). DIO, diet-induced obesity wild-type mouse; DIO + BDCM, DIO mouse exposed to bromodichloromethane (BDCM); FeTPPS, DIO mouse exposed to BDCM and iron(III) tetrakis(p-sulfonatophenyl)porphyrin; p47phox KO, high-fat diet–fed mouse with p47phox gene knockout exposed to BDCM; Tlr4 KO, high-fat diet–fed mouse with Tlr4 knockout exposed to BDCM.
MCP-1 release is considered a hallmark of Kupffer cell activation and plays a huge role in liver inflammation to attract circulating leukocytes to the inflamed liver in NASH. Results showed that the DIO + BDCM group had significantly increased MCP-1 immunoreactivity compared with that in the DIO-only group (P < 0.01) (Figure 5B). p47phox gene KO mice and mice treated with FeTPPS had significantly decreased Mcp-1 immunoreactivity compared with that in the DIO + BDCM group (both, P < 0.01) (Figure 5, B and E). Tlr4 KO mice also had a significant decrease in Mcp-1 immunoreactivity compared with that in the DIO + BDCM group (P < 0.01) (Figure 5, B and E). The inflammatory cytokines noted earlier in Results were also verified for their mRNA expression. Results showed that the mRNA expression of tumor necrosis factor-α, IL-6, lipocalin-2, serum amyloid A1 (SAA1), IL-1β, and MCP-1 were significantly increased in rodent and human NASH liver, whereas p47phox gene KO mice, Tlr4 KO mice, and mice administered FeTPPS showed significant decreases in the expression levels (all, P < 0.05) (Supplemental Figures S5–S7).
Next we explored the role of NADPH oxidase–mediated TLR recruitment in Kupffer cell activation by studying the immunoreactivity of CD68 (a Kupffer cell activation marker). Results showed that DIO + BDCM group liver had significantly increased CD68 immunoreactivity compared with that in the DIO-only group, whereas using p47phox gene KO mice or attenuating the formation of peroxynitrite significantly decreased CD68 immunoreactivity (both, P < 0.01) (Figure 5, C and F). Tlr4 KO mice had significantly decreased CD68 immunoreactivity in the liver compared with that in the DIO + BDCM group (P < 0.01) (Figure 5, C and F), suggesting a strong role of TLR4 signaling in Kupffer cell activation. The results also strongly suggest that NADPH oxidase–mediated peroxynitrite, which drove TLR4 recruitment, also correlates with TLR4 signaling, as indicated by significant attenuations in Il-1β release and Mcp-1 and CD68 protein levels when agents that block NADPH oxidase–mediated peroxynitrite were administered. The results of decreased Il-1β release and Kupffer cell activation in Tlr4 KO mice further confirmed the role of TLR4 in NASH inflammatory pathogenesis. MCD diet–fed mouse liver and human NASH liver showed significantly decreased IL-1β (both, P < 0.05) (Supplemental Figure S8, A and D), decreased MCP-1 levels (both, P < 0.05) (Supplemental Figure S8, B and E), and decreased CD68 immunoreactivity (both, P < 0.05) (Supplemental Figure S8, C and F) compared with those in their respective controls, suggesting that NADPH oxidase–driven TLR4 recruitment and subsequent NASH inflammatory pathogenesis are prevalent across different mouse models and in the human form of the disease.
A recent report suggests that the strong involvement of the TLR4/NF-κB pathway in the migration and activation of hepatic stellate cells is known to be a hallmark of NASH pathogenesis.37 To associate the phenomenon of NADPH oxidase–driven recruitment of TLR4 into hepatic lipid rafts in activating hepatic stellate cells, α-SMA immunoreactivity in liver slices was studied by IHC analysis. The DIO + BDCM group showed a significant increase in α-Sma immunoreactivity compared with that in the DIO-only group (P < 0.01) (Figure 6, A and D). p47phox gene KO mice, mice given scavenging peroxynitrite, and Tlr4 KO mice showed significant decreases in α-Sma immunoreactivity compared with that in the DIO + BDCM group (all, P < 0.01) (Figure 6, A and D). MCD diet–fed mice showed a significant increase in α-Sma immunoreactivity compared with that in the MCS group (P < 0.05) (Figure 6, B and E). Human NASH liver had a significantly increased α-SMA level, as shown by IHC analysis, compared with that in control liver from healthy subjects (P < 0.05) (Figure 6, C and F). The IHC analysis data were also verified using Western blot analysis of liver tissue homogenates (all, P < 0.05) (Supplemental Figure S9). The data strongly suggest that hepatic stellate cell proliferation depends on NADPH oxidase–mediated peroxynitrite formation and its induction of TLR4 recruitment into hepatic lipid rafts. NADPH oxidase–mediated TLR4 recruitment also strongly correlated with histological assessment of NASH, inflammation, and fibrosis, as shown by NIH NASH Clinical Research Network scores (Table 1).
Figure 6.

A–C: Representative images of 5-μm-thick immunostained liver sections, imaged using bright-field microscopy. Positive immunoreactivities (brown) are due to diaminobenzidine binding to respective biotinylated secondary antibodies via streptavidin–horseradish peroxidase. Nuclei (blue) counterstained with Mayer's hematoxylin solution. Immunoreactivities detected are for stellate cell proliferation marker α-smooth muscle actin (SMA). D–F: Morphometries performed on three independent fields for each sample including representative images for percentage of area showing positive immunoreactivity of α-SMA in A (D), B (E), and C (F), calculated in arbitrary units. ∗P < 0.05, ∗∗P < 0.01. Original magnification, ×20 (A–C). DIO, diet-induced obesity wild-type mouse; DIO + BDCM, DIO mouse exposed to bromodichloromethane (BDCM); FeTPPS, DIO mouse exposed to BDCM and iron(III) tetrakis(p-sulfonatophenyl)porphyrin; Hu Ctrl, healthy human control; Hu NASH, human nonalcoholic steatohepatitis; MCD, methionine- and choline-deficient diet–fed mouse; MCS, methionine- and choline-sufficient diet–fed mouse; p47phox KO, high-fat diet–fed mouse with p47phox gene knockout exposed to BDCM; Tlr4 KO, high-fat diet–fed mouse with Tlr4 knockout exposed to BDCM.
Table 1.
NIH NASH Clinical Research Network Scores for Rodent Model of NASH
| Symptom | DIO | DIO + BDCM | p47phox KO | FeTPPS | FBA | Tlr4 KO |
|---|---|---|---|---|---|---|
| Steatosis | 2 | 3 | 2 | 2 | 2 | 1 |
| Lobular inflammation | 1 | 3 | 1 | 2 | 0 | 0 |
| Hepatocyte ballooning | 1 | 2 | 2 | 1 | 1 | 0 |
| Stage of fibrosis∗ | 1C | 3 | 1C | 1B | 1A | 1A |
Scores were calculated after analysis of the histological features of nonalcoholic steatohepatitis (NASH) as described by Kleiner et al26 and in the guideline of the NIH NASH Clinical Research Network.
DIO, diet-induced obesity wild-type mouse; DIO + BDCM, DIO mouse exposed to bromodichloromethane (BDCM); FBA, DIO mouse exposed to BDCM and phenylboronic acid; FeTPPS, DIO mouse exposed to BDCM and iron(III) tetrakis(p-sulfonatophenyl)porphyrin; p47phox KO, high-fat diet–fed mouse with p47phox gene knockout exposed to BDCM; Tlr4 KO, high-fat diet–fed mouse with Tlr4 knockout exposed to BDCM.
Scale: 1A, mild; 1C, portal; 2, periportal; 3, bridging.
Discussion
We show that NADPH oxidase–derived peroxynitrite drives TLR4 recruitment into hepatic lipid rafts. Further, using a novel synthetic peroxynitrite scavenger, FBA, we abrogated the peroxynitrite formation and attenuated Il-1β production, sinusoidal injury, Kupffer cell activation, and stellate-cell proliferation.
NASH is clinically silent and typically has a late presentation in clinical practice, making it difficult to treat.38,39 The multiple phases of NASH pathogenesis, which include an initial lipotoxicity, macrophage activation leading to inflammation, and late-stage fibrosis, make devising a therapeutic strategy challenging. Thus, the identification of early molecular signatures of injury initiation in the fatty liver is of paramount importance. A recent report of the involvement of TLR4-mediated pathways in NASH has identified the molecular basis of inflammation that often precedes late-stage fibrosis.39
Because TLR4-mediated inflammation is a sufficiently early event in NASH pathophysiology, understanding the molecular basis of its synthesis, recruitment into lipid rafts, and downstream signaling becomes essential for resolving the disease itself.40 Our results of the strong involvement of NADPH oxidase activation in driving TLR4 into the lipid rafts show a novel mechanism of TLR4 activation and NF-κB translocation in NASH. The results also identify peroxynitrite-mediated TLR4 recruitment into lipid rafts as an early molecular event in NASH. The pharmacological approach of using a NADPH oxidase inhibitor and the use of transgenic mice that lack the p47phox subunit of NADPH oxidase confirm the role of this vital enzyme in lipid raft recruitment of TLR4.
We have reported previously that NADPH oxidase release of superoxide radicals leads to peroxynitrite formation, which in turn is instrumental in causing Kupffer cell activation in steatohepatitic injury.18 Here, we used a dual approach to study the formation of peroxynitrite via NADPH oxidase, by using a general decomposition catalyst, FeTPPS, and by helping peroxynitrite react with a novel boronic compound, FBA, in vivo.41 Modeling human NASH using rodents remains a challenge to researchers. There is no single model that illustrates the symptoms of human NASH in a clear and decisive way.42 Several concepts involving dual hits and multiple-hit paradigms have been proposed but have been the focus of debate.43,44 The lipotoxicity-based model that includes the generation of oxidative stress after the administration of hepatotoxins, a second hit, has been previously described by us and others, recognizing the fact that it does not reflect the true developmental stages of human NASH but represents an underlying condition of steatosis and insulin and leptin resistance that is found in NASH patients.24,44
Because oxidative stress is undoubtedly a major player in human NASH, our model exemplifies a logical approach to studying oxidative stress mechanisms in the progression of NASH and can be used for testing compounds that attenuate oxidative stress–mediated inflammation in this disease.
On the other hand, the MCD diet model is used widely for depicting steatohepatitic lesions coupled with fibrosis, symptoms found in more progressive stages of human NASH.42,45,46 The symptoms found in this model represent lesions of human NASH, but in a non–insulin-resistant state, although some studies have found hepatic insulin resistance in this model.47 The results reported here may be the beginning of many future studies involving NADPH oxidase activation–induced TLR4 trafficking and could be validated using other models of NASH.
The dual approach of using two different peroxynitrite blockers was significant, as FeTPPS and other metalloporphyrins have been used in various rodent models of inflammatory diseases but have low oral bioavailability and lack specificity.19 FBA, an aromatic boronate, has been shown to react specifically with peroxynitrite to yield tyrosine as a major product, with the probability of the formation of nitrotyrosine after a reaction with peroxynitrite being negligible because of the stoichiometry of the chemical reaction.48 The reaction kinetics made FBA to be a specific scavenger and blocking agent of peroxynitrite.48 FBA reacts with peroxynitrite at a rapid rate and stoichiometrically to form phenol (k = 1.6 × 106 mol per second).49 FBA is also highly specific to react with peroxynitrite and can be used for inferring the source of tyrosine nitration because it does not inhibit myeloperoxidase-dependent tyrosine nitration. We used the effectiveness of FBA as a candidate drug to attenuate peroxynitrite-mediated recruitment of TLR4 into hepatic lipid rafts. Our results of a significant attenuation of TLR4 recruitment into lipid rafts, as shown by the colocalization of Tlr4 and the lipid raft protein flotillin in NASH liver in mice, might provide a rationale for using FBA as a future small molecule to attenuate TLR4-mediated inflammation in NASH. However, caution should be exercised in interpreting the findings reported here because FBA has not been used previously in other inflammatory disease models. Further FBA treatment also abrogated Tlr4-induced NF-κB translocation and DNA binding and increased levels of sinusoidal injury markers Icam-1 and E-selectin, Kupffer cell activation marker CD68, and stellate cell proliferation. FBA-treated NASH livers had significantly less fibrosis, as assessed by NIH NASH Clinical Research Network scores.
Our results further underline an alternate mechanism of lipid raft recruitment of TLR4 via an oxidative and nitrative stress pathway, unique in NASH pathophysiology. The recruitment of TLR4 might happen as a result of nuclear respiratory factor 2 depletion, which has a direct effect on glutathione-synthesizing enzymes and lower levels of glutathione seen in NASH, although this concept remains speculative at this point.50,51 The results reported here support the role of highly reactive peroxynitrite in causing TLR4 trafficking rather than oxyradicals generated as a result of TLR4 activation, a concept that has been reported before and is widely prevalent in inflammatory diseases.52 Our data support the peroxynitrite mechanism owing to the faster-rate kinetics of formation reported for peroxynitrite (1010 mol per second), but the TLR4-induced NADPH oxidase activation in NASH cannot be ruled out owing to the complex innate immune regulations associated with the pathophysiology of the disease itself.49
In summary, we show that TLR recruitment into lipid rafts is an early event in both rodent and human NASH pathogenesis. TLR4 recruitment was initiated by NADPH oxidase, and the molecular basis of this early event was dependent on the formation of peroxynitrite. The use of FBA as a novel compound for attenuating peroxynitrite in vivo might serve as a new approach to initiating further studies involving relevant rodent models of NASH and inflammatory pathogenesis in this disease and might have significant relevance for research in drug development concerning the attenuation of NASH. The study is significant because we used two different models of NASH, elucidated the formation of peroxynitrite and TLR4 lipid raft recruitment in human NASH, and avoided in vitro cellular experimentation to support the involvement of peroxynitrite in TLR4 lipid raft recruitment, primarily because of the advanced tools for detecting peroxynitrite. However, caution should be exercised in interpreting some of the results noted in this report because we are not aware of the oral bioavailability of this compound, and future rodent-based studies need to focus more on the pharmacokinetics of FBA.
Acknowledgments
We thank the technical services of Benny Davidson (University of South Carolina School of Medicine), Dr. Gary Schools (University of South Carolina), and the Instrumentation Resource Facility (University of South Carolina) for confocal microscopy and analysis equipment usage and consulting services.
Footnotes
Supported by an NIH Pathway to Independence Award (S.C.); NIH grants R00ES019875 (S.C.), R01DK053792 (A.M.D.), P20GM103641 (S.C., M.N., and P.S.N.), and P01AT003961, R01AT006888, R01ES019313, and R01MH094755 (all to M.N. and P.S.N.); and VA Merit Award BX001357 (M.N. and P.S.N.).
Disclosures: None declared.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2015.03.024.
Supplemental Data
A and B: Representative images of 5-μm-thick immunostained liver sections, imaged using immunofluorescence microscopy. Nuclear stain (blue) is due to ProLong Gold Antifade Reagent (Life Technologies, Carlsbad, CA) with DAPI. A: Secondary antibodies used were Alexa Fluor 568 (Invitrogen; red) against α-gp91phox primary antibody and Alexa Fluor 488 (green) against α-p47phox primary antibody. Yellow spots/patches indicate alignment of cytoplasmic subunit of NADPH oxidase, p47phox (red), with membrane subunit glycoprotein 91phox (green). B: Secondary antibody used was Alexa Fluor 633 (red) against α-3-nitrotyrosine primary antibody. C and D: Morphometry performed on three independent fields for each sample, including representative images in A for gp91phox/p47phox colocalization events per 300 cells (C) and in B for percentage of area showing positive immunoreactivity of 3-nitrotyrosine (D), both calculated in arbitrary units. ∗P < 0.05. Original magnification, ×20 (A and B). Hu Ctrl, healthy human control; Hu NASH, human nonalcoholic steatohepatitis patient; MCD, methionine- and choline-deficient diet–fed mouse; MCS, methionine- and choline-sufficient diet–fed mouse.
A and B: Representative images of 5-μm-thick immunostained liver sections, imaged using immunofluorescence microscopy. Nuclear stain (blue) is due to ProLong Gold Antifade Reagent (Life Technologies, Carlsbad, CA) with DAPI; yellow spots/patches indicate alignment of Toll-like receptor 4 (red), with lipid raft protein flotillin (green). Secondary antibodies used were Alexa Fluor 633 (Invitrogen; red) against α-TLR4 primary antibody, Alexa Fluor 488 (green) against α-flotillin primary antibody. C and D: Morphometry performed on three independent fields for each sample, including representative images in A (C) and B (D) for TLR4–flotillin colocalization events per 300 cells. ∗P < 0.05. Apo, diet-induced obesity wild-type (DIO) mouse exposed to bromodichloromethane (BDCM) and injected with apocynin; DIO, diet-induced obesity wild-type mouse; DIO + BDCM, DIO mouse exposed to BDCM; FBA, DIO mouse exposed to BDCM and phenylboronic acid; FeTPPS, DIO mouse exposed to BDCM and iron(III) tetrakis(p-sulfonatophenyl)porphyrin; Hu Ctrl, healthy human control; Hu NASH, human nonalcoholic steatohepatitis; MCD, methionine- and choline-deficient diet–fed mouse; MCS, methionine- and choline-sufficient diet–fed mouse; p47phox KO, high-fat diet–fed mouse with p47phox gene knockout exposed to BDCM.
A and C: Representative images of 5-μm-thick immunostained liver sections, imaged using immunofluorescence microscopy. Secondary antibody used in A was Alexa Fluor 568 (Invitrogen; red) against α-intercellular adhesion molecule (ICAM)-1 primary antibody (A) and against α-E-selectin primary antibody (C; both ICAM-1 and E-selectin are sinusoidal injury markers). Nuclear stain (blue) is due to ProLong Gold Antifade Reagent (Life Technologies, Carlsbad, CA) with DAPI. B and D: Morphometry performed on three independent fields for each sample, including representative images for percentage of area showing positive immunoreactivity of ICAM-1 in A (B) and of E-selectin in C (D), both calculated in arbitrary units. ∗P < 0.05. Original magnification, ×20 (A and C). Hu Ctrl, healthy human control; Hu NASH, human nonalcoholic steatohepatitis; MCD, methionine- and choline-deficient diet–fed mouse; MCS, methionine- and choline-sufficient diet–fed mouse.
Real-time quantitative PCR analyses of mRNA levels of intercellular adhesion molecule (Icam)-1 and E-selectin in murine nonalcoholic steatohepatitis (NASH) models. A and B: Icam-1 in toxin (A) and in methionine- and choline-sufficient and -deficient diet–fed (MCS and MCD, respectively) (B) mouse models of NASH. C and D: E-selectin in toxin model of NASH (C) and in MCS and MCD models of NASH (D). ∗P < 0.05. BDCM, bromodichloromethane.
Real-time quantitative PCR analyses of mRNA levels of proinflammatory mediators in human control and human nonalcoholic steatohepatitic liver. Tumor necrosis factor (TNF)-α (A), IL-6 (B), lipocalin(LCN)-2 (C), serum amyloid A1 (SAA1) (D), IL-1β (E), monocyte chemotactic protein (MCP)-1 (F), intercellular adhesion molecule (ICAM)-1 (G), and E-selectin (H). ∗P < 0.05.
Real-time quantitative PCR analyses of mRNA levels of tumor necrosis factor (Tnf)-α, Il-6, and lipocalin (Lcn)-2 in murine nonalcoholic steatohepatitis (NASH) models. Tnf-α in the toxin (A) and methionine- and choline-sufficient and -deficient diet–fed (MCS and MCD, respectively) (B) models of NASH. Il-6 in the toxin (C) and MCS/MCD (D) models of NASH. Lcn-2 in the toxin (E) and MCS/MCD (F) models of NASH. ∗P < 0.05. BDCM, bromodichloromethane.
Real-time quantitative PCR analyses of mRNA levels of serum amyloid A1 (Saa1), Il-1β, and monocyte chemotactic protein (MCP)-1 in murine models of nonalcoholic steatohepatitis (NASH). Saa1 in the toxin (A) and methionine- and choline-sufficient and -deficient diet–fed (MCS and MCD, respectively) (B) models of NASH. Il-1β in the toxin (C) and MCS/MCD (D) models of NASH. MCP-1 in the toxin (E) and MCS/MCD (F) models of NASH. ∗P < 0.05. BDCM, bromodichloromethane.
Representative images of 5-μm-thick immunostained liver sections, imaged using bright-field microscopy. A–C: Positive immunoreactivities (brown) for the proinflammatory cytokines IL-1β (A), monocyte chemotactic protein (MCP)-1 (B), and Kupffer cell activation marker CD68 (C) are due to diaminobenzidine binding to respective biotinylated secondary antibodies via streptavidin–horseradish peroxidase. Nuclei (blue) counterstained with Mayer's hematoxylin solution. D–F: Morphometry performed on three independent fields for each sample, including representative images for percentage of area showing positive immunoreactivity of IL-1β in A (D), MCP-1 in B (E), and CD68 in C (F), all calculated in arbitrary units. ∗P < 0.05. Original magnification, ×20 (A–C). Hu Ctrl, healthy human control; Hu NASH, human nonalcoholic steatohepatitis; MCD, methionine- and choline-deficient diet–fed mouse; MCS, methionine- and choline-sufficient diet–fed mouse.
Western blot analysis of α-smooth muscle actin (SMA) in liver homogenates from rodent models of nonalcoholic steatohepatitis (NASH) and human NASH liver. A: Immunoblot images for the immunoreactive bands of α-SMA and β-actin. B: Band analysis after normalization against β-actin. ∗P < 0.05. Apo, diet-induced obesity wild-type (DIO) mouse exposed to bromodichloromethane (BDCM) and injected with apocynin; FeTPPS, DIO mouse exposed to BDCM and iron(III) tetrakis(p-sulfonatophenyl)porphyrin; Hu Ctrl, healthy human control; Hu NASH, human nonalcoholic steatohepatitis; MCD, methionine- and choline-deficient diet–fed mouse; MCS, methionine- and choline-sufficient diet–fed mouse; p47phox, high-fat diet–fed mouse with p47phox gene knockout exposed to BDCM; Tlr4 KO, high-fat diet–fed mouse with Tlr4 knockout exposed to BDCM.
References
- 1.Bohinc B.N., Diehl A.M. Mechanisms of disease progression in NASH: new paradigms. Clin Liver Dis. 2012;16:549–565. doi: 10.1016/j.cld.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Bosch J., Abraldes J.G., Fernandez M., Garcia-Pagan J.C. Hepatic endothelial dysfunction and abnormal angiogenesis: new targets in the treatment of portal hypertension. J Hepatol. 2010;53:558–567. doi: 10.1016/j.jhep.2010.03.021. [DOI] [PubMed] [Google Scholar]
- 3.Diehl A.M. Nonalcoholic steatohepatitis. Semin Liver Dis. 1999;19:221–229. doi: 10.1055/s-2007-1007111. [DOI] [PubMed] [Google Scholar]
- 4.Farrell G.C., van Rooyen D., Gan L., Chitturi S. NASH is an Inflammatory Disorder: pathogenic, Prognostic and Therapeutic Implications. Gut Liver. 2012;6:149–171. doi: 10.5009/gnl.2012.6.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marra F., Aleffi S., Bertolani C., Petrai I., Vizzutti F. Adipokines and liver fibrosis. Eur Rev Med Pharmacol Sci. 2005;9:279–284. [PubMed] [Google Scholar]
- 6.Pasarin M., La Mura V., Gracia-Sancho J., Garcia-Caldero H., Rodriguez-Vilarrupla A., Garcia-Pagan J.C., Bosch J., Abraldes J.G. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PLoS One. 2012;7:e32785. doi: 10.1371/journal.pone.0032785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tilg H., Diehl A.M. Cytokines in alcoholic and nonalcoholic steatohepatitis. N Engl J Med. 2000;343:1467–1476. doi: 10.1056/NEJM200011163432007. [DOI] [PubMed] [Google Scholar]
- 8.Clark J.M., Brancati F.L., Diehl A.M. Nonalcoholic fatty liver disease. Gastroenterology. 2002;122:1649–1657. doi: 10.1053/gast.2002.33573. [DOI] [PubMed] [Google Scholar]
- 9.Li Z., Clark J., Diehl A.M. The liver in obesity and type 2 diabetes mellitus. Clin Liver Dis. 2002;6:867–877. doi: 10.1016/s1089-3261(02)00060-0. [DOI] [PubMed] [Google Scholar]
- 10.Diehl A.M., Li Z.P., Lin H.Z., Yang S.Q. Cytokines and the pathogenesis of non-alcoholic steatohepatitis. Gut. 2005;54:303–306. doi: 10.1136/gut.2003.024935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abdelmalek M.F., Diehl A.M. Nonalcoholic fatty liver disease as a complication of insulin resistance. Med Clin North Am. 2007;91:1125–1149. doi: 10.1016/j.mcna.2007.06.001. ix. [DOI] [PubMed] [Google Scholar]
- 12.Gay N.J., Symmons M.F., Gangloff M., Bryant C.E. Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol. 2014;14:546–558. doi: 10.1038/nri3713. [DOI] [PubMed] [Google Scholar]
- 13.Deguine J., Barton G.M. MyD88: a central player in innate immune signaling. F1000Prime Rep. 2014;6:97. doi: 10.12703/P6-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dziarski R., Wang Q., Miyake K., Kirschning C.J., Gupta D. MD-2 enables Toll-like receptor 2 (TLR2)-mediated responses to lipopolysaccharide and enhances TLR2-mediated responses to Gram-positive and Gram-negative bacteria and their cell wall components. J Immunol. 2001;166:1938–1944. doi: 10.4049/jimmunol.166.3.1938. [DOI] [PubMed] [Google Scholar]
- 15.Choi S., Diehl A.M. Role of inflammation in nonalcoholic steatohepatitis. Curr Opin Gastroenterol. 2005;21:702–707. doi: 10.1097/01.mog.0000182863.96421.47. [DOI] [PubMed] [Google Scholar]
- 16.De Minicis S., Seki E., Oesterreicher C., Schnabl B., Schwabe R.F., Brenner D.A. Reduced nicotinamide adenine dinucleotide phosphate oxidase mediates fibrotic and inflammatory effects of leptin on hepatic stellate cells. Hepatology. 2008;48:2016–2026. doi: 10.1002/hep.22560. [DOI] [PubMed] [Google Scholar]
- 17.Paik Y.H., Kim J., Aoyama T., De Minicis S., Bataller R., Brenner D.A. Role of NADPH oxidases in liver fibrosis. Antioxid Redox Signal. 2014;20:2854–2872. doi: 10.1089/ars.2013.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chatterjee S., Ganini D., Tokar E.J., Kumar A., Das S., Corbett J., Kadiiska M., Waalkes M., Diehl A.M., Mason R.P. Leptin is key to peroxynitrite-mediated oxidative stress and Kupffer cell activation in experimental nonalcoholic steatohepatitis. J Hepatol. 2013;58:778–784. doi: 10.1016/j.jhep.2012.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang L., Li P., Fu S., Calay E.S., Hotamisligil G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467–478. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang S.Q., Lin H.Z., Lane M.D., Clemens M., Diehl A.M. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci U S A. 1997;94:2557–2562. doi: 10.1073/pnas.94.6.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hotamisligil G.S., Shargill N.S., Spiegelman B.M. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 22.Reaven G. Metabolic syndrome: pathophysiology and implications for management of cardiovascular disease. Circulation. 2002;106:286–288. doi: 10.1161/01.cir.0000019884.36724.d9. [DOI] [PubMed] [Google Scholar]
- 23.Gallagher E.J., Leroith D., Karnieli E. Insulin resistance in obesity as the underlying cause for the metabolic syndrome. Mt Sinai J Med. 2010;77:511–523. doi: 10.1002/msj.20212. [DOI] [PubMed] [Google Scholar]
- 24.Seth R.K., Kumar A., Das S., Kadiiska M.B., Michelotti G., Diehl A.M., Chatterjee S. Environmental toxin-linked nonalcoholic steatohepatitis and hepatic metabolic reprogramming in obese mice. Toxicol Sci. 2013;134:291–303. doi: 10.1093/toxsci/kft104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brunt E.M., Janney C.G., Di Bisceglie A.M., Neuschwander-Tetri B.A., Bacon B.R. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467–2474. doi: 10.1111/j.1572-0241.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 26.Kleiner D.E., Brunt E.M., Van Natta M., Behling C., Contos M.J., Cummings O.W., Ferrell L.D., Liu Y.C., Torbenson M.S., Unalp-Arida A., Yeh M., McCullough A.J., Sanyal A.J. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 27.Chen S.H., He F., Zhou H.L., Wu H.R., Xia C., Li Y.M. Relationship between nonalcoholic fatty liver disease and metabolic syndrome. J Dig Dis. 2011;12:125–130. doi: 10.1111/j.1751-2980.2011.00487.x. [DOI] [PubMed] [Google Scholar]
- 28.Kelley C.E., Brown A.J., Diehl A.M., Setji T.L. Review of nonalcoholic fatty liver disease in women with polycystic ovary syndrome. World J Gastroenterol. 2014;20:14172–14184. doi: 10.3748/wjg.v20.i39.14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Angulo P. Treatment of nonalcoholic fatty liver disease. Ann Hepatol. 2002;1:12–19. [PubMed] [Google Scholar]
- 30.Chalasani N., Younossi Z., Lavine J.E., Diehl A.M., Brunt E.M., Cusi K., Charlton M., Sanyal A.J. The diagnosis and management of non-alcoholic fatty liver disease: practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55:2005–2023. doi: 10.1002/hep.25762. [DOI] [PubMed] [Google Scholar]
- 31.Rosso N., Chavez-Tapia N.C., Tiribelli C., Bellentani S. Translational approaches: from fatty liver to non-alcoholic steatohepatitis. World J Gastroenterol. 2014;20:9038–9049. doi: 10.3748/wjg.v20.i27.9038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vernon G., Baranova A., Younossi Z.M. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–285. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- 33.Bugianesi E., Moscatiello S., Ciaravella M.F., Marchesini G. Insulin resistance in nonalcoholic fatty liver disease. Curr Pharm Des. 2010;16:1941–1951. doi: 10.2174/138161210791208875. [DOI] [PubMed] [Google Scholar]
- 34.Lu Y.C., Yeh W.C., Ohashi P.S. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42:145–151. doi: 10.1016/j.cyto.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 35.Farrell G.C., Teoh N.C., McCuskey R.S. Hepatic microcirculation in fatty liver disease. Anat Rec (Hoboken) 2008;291:684–692. doi: 10.1002/ar.20715. [DOI] [PubMed] [Google Scholar]
- 36.Pourhoseini S., Seth R.K., Das S., Dattaroy D., Kadiiska M.B., Xie G., Michelotti G.A., Nagarkatti M., Diehl A.M., Chatterjee S. Upregulation of miR21 and repression of Grhl3 by leptin mediates sinusoidal endothelial injury in experimental nonalcoholic steatohepatitis. PLoS One. 2015;10:e0116780. doi: 10.1371/journal.pone.0116780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sumida Y., Niki E., Naito Y., Yoshikawa T. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free Radic Res. 2013;47:869–880. doi: 10.3109/10715762.2013.837577. [DOI] [PubMed] [Google Scholar]
- 38.Ramesh S., Sanyal A.J. Evaluation and management of non-alcoholic steatohepatitis. J Hepatol. 2005;(42 Suppl):S2–S12. doi: 10.1016/j.jhep.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 39.Michelotti G.A., Machado M.V., Diehl A.M. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol. 2013;10:656–665. doi: 10.1038/nrgastro.2013.183. [DOI] [PubMed] [Google Scholar]
- 40.Petrasek J., Csak T., Szabo G. Toll-like receptors in liver disease. Adv Clin Chem. 2013;59:155–201. doi: 10.1016/b978-0-12-405211-6.00006-1. [DOI] [PubMed] [Google Scholar]
- 41.Aubert J., Begriche K., Knockaert L., Robin M.A., Fromenty B. Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: mechanisms and pathophysiological role. Clin Res Hepatol Gastroenterol. 2011;35:630–637. doi: 10.1016/j.clinre.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 42.Nakamura A., Terauchi Y. Lessons from mouse models of high-fat diet-induced NAFLD. Int J Mol Sci. 2013;14:21240–21257. doi: 10.3390/ijms141121240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Day C.P., James O.F. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 44.Takaki A., Kawai D., Yamamoto K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH) Int J Mol Sci. 2013;14:20704–20728. doi: 10.3390/ijms141020704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koppe S.W., Sahai A., Malladi P., Whitington P.F., Green R.M. Pentoxifylline attenuates steatohepatitis induced by the methionine choline deficient diet. J Hepatol. 2004;41:592–598. doi: 10.1016/j.jhep.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 46.Oz H.S., Chen T.S., Neuman M. Methionine deficiency and hepatic injury in a dietary steatohepatitis model. Dig Dis Sci. 2008;53:767–776. doi: 10.1007/s10620-007-9900-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ota T., Takamura T., Kurita S., Matsuzawa N., Kita Y., Uno M., Akahori H., Misu H., Sakurai M., Zen Y., Nakanuma Y., Kaneko S. Insulin resistance accelerates a dietary rat model of nonalcoholic steatohepatitis. Gastroenterology. 2007;132:282–293. doi: 10.1053/j.gastro.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 48.Gawrieh S., Opara E.C., Koch T.R. Oxidative stress in nonalcoholic fatty liver disease: pathogenesis and antioxidant therapies. J Investig Med. 2004;52:506–514. doi: 10.1136/jim-52-08-22. [DOI] [PubMed] [Google Scholar]
- 49.Sikora A., Zielonka J., Lopez M., Joseph J., Kalyanaraman B. Direct oxidation of boronates by peroxynitrite: mechanism and implications in fluorescence imaging of peroxynitrite. Free Radic Biol Med. 2009;47:1401–1407. doi: 10.1016/j.freeradbiomed.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Skamarauskas J.T., Oakley F., Smith F.E., Bawn C., Dunn M., Vidler D.S., Clemence M., Blain P.G., Taylor R., Gamcsik M.P., Thelwall P.E. Noninvasive in vivo magnetic resonance measures of glutathione synthesis in human and rat liver as an oxidative stress biomarker. Hepatology. 2014;59:2321–2330. doi: 10.1002/hep.26925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kong X., Thimmulappa R., Kombairaju P., Biswal S. NADPH oxidase-dependent reactive oxygen species mediate amplified TLR4 signaling and sepsis-induced mortality in Nrf2-deficient mice. J Immunol. 2010;185:569–577. doi: 10.4049/jimmunol.0902315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park H.S., Jung H.Y., Park E.Y., Kim J., Lee W.J., Bae Y.S. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J Immunol. 2004;173:3589–3593. doi: 10.4049/jimmunol.173.6.3589. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A and B: Representative images of 5-μm-thick immunostained liver sections, imaged using immunofluorescence microscopy. Nuclear stain (blue) is due to ProLong Gold Antifade Reagent (Life Technologies, Carlsbad, CA) with DAPI. A: Secondary antibodies used were Alexa Fluor 568 (Invitrogen; red) against α-gp91phox primary antibody and Alexa Fluor 488 (green) against α-p47phox primary antibody. Yellow spots/patches indicate alignment of cytoplasmic subunit of NADPH oxidase, p47phox (red), with membrane subunit glycoprotein 91phox (green). B: Secondary antibody used was Alexa Fluor 633 (red) against α-3-nitrotyrosine primary antibody. C and D: Morphometry performed on three independent fields for each sample, including representative images in A for gp91phox/p47phox colocalization events per 300 cells (C) and in B for percentage of area showing positive immunoreactivity of 3-nitrotyrosine (D), both calculated in arbitrary units. ∗P < 0.05. Original magnification, ×20 (A and B). Hu Ctrl, healthy human control; Hu NASH, human nonalcoholic steatohepatitis patient; MCD, methionine- and choline-deficient diet–fed mouse; MCS, methionine- and choline-sufficient diet–fed mouse.
A and B: Representative images of 5-μm-thick immunostained liver sections, imaged using immunofluorescence microscopy. Nuclear stain (blue) is due to ProLong Gold Antifade Reagent (Life Technologies, Carlsbad, CA) with DAPI; yellow spots/patches indicate alignment of Toll-like receptor 4 (red), with lipid raft protein flotillin (green). Secondary antibodies used were Alexa Fluor 633 (Invitrogen; red) against α-TLR4 primary antibody, Alexa Fluor 488 (green) against α-flotillin primary antibody. C and D: Morphometry performed on three independent fields for each sample, including representative images in A (C) and B (D) for TLR4–flotillin colocalization events per 300 cells. ∗P < 0.05. Apo, diet-induced obesity wild-type (DIO) mouse exposed to bromodichloromethane (BDCM) and injected with apocynin; DIO, diet-induced obesity wild-type mouse; DIO + BDCM, DIO mouse exposed to BDCM; FBA, DIO mouse exposed to BDCM and phenylboronic acid; FeTPPS, DIO mouse exposed to BDCM and iron(III) tetrakis(p-sulfonatophenyl)porphyrin; Hu Ctrl, healthy human control; Hu NASH, human nonalcoholic steatohepatitis; MCD, methionine- and choline-deficient diet–fed mouse; MCS, methionine- and choline-sufficient diet–fed mouse; p47phox KO, high-fat diet–fed mouse with p47phox gene knockout exposed to BDCM.
A and C: Representative images of 5-μm-thick immunostained liver sections, imaged using immunofluorescence microscopy. Secondary antibody used in A was Alexa Fluor 568 (Invitrogen; red) against α-intercellular adhesion molecule (ICAM)-1 primary antibody (A) and against α-E-selectin primary antibody (C; both ICAM-1 and E-selectin are sinusoidal injury markers). Nuclear stain (blue) is due to ProLong Gold Antifade Reagent (Life Technologies, Carlsbad, CA) with DAPI. B and D: Morphometry performed on three independent fields for each sample, including representative images for percentage of area showing positive immunoreactivity of ICAM-1 in A (B) and of E-selectin in C (D), both calculated in arbitrary units. ∗P < 0.05. Original magnification, ×20 (A and C). Hu Ctrl, healthy human control; Hu NASH, human nonalcoholic steatohepatitis; MCD, methionine- and choline-deficient diet–fed mouse; MCS, methionine- and choline-sufficient diet–fed mouse.
Real-time quantitative PCR analyses of mRNA levels of intercellular adhesion molecule (Icam)-1 and E-selectin in murine nonalcoholic steatohepatitis (NASH) models. A and B: Icam-1 in toxin (A) and in methionine- and choline-sufficient and -deficient diet–fed (MCS and MCD, respectively) (B) mouse models of NASH. C and D: E-selectin in toxin model of NASH (C) and in MCS and MCD models of NASH (D). ∗P < 0.05. BDCM, bromodichloromethane.
Real-time quantitative PCR analyses of mRNA levels of proinflammatory mediators in human control and human nonalcoholic steatohepatitic liver. Tumor necrosis factor (TNF)-α (A), IL-6 (B), lipocalin(LCN)-2 (C), serum amyloid A1 (SAA1) (D), IL-1β (E), monocyte chemotactic protein (MCP)-1 (F), intercellular adhesion molecule (ICAM)-1 (G), and E-selectin (H). ∗P < 0.05.
Real-time quantitative PCR analyses of mRNA levels of tumor necrosis factor (Tnf)-α, Il-6, and lipocalin (Lcn)-2 in murine nonalcoholic steatohepatitis (NASH) models. Tnf-α in the toxin (A) and methionine- and choline-sufficient and -deficient diet–fed (MCS and MCD, respectively) (B) models of NASH. Il-6 in the toxin (C) and MCS/MCD (D) models of NASH. Lcn-2 in the toxin (E) and MCS/MCD (F) models of NASH. ∗P < 0.05. BDCM, bromodichloromethane.
Real-time quantitative PCR analyses of mRNA levels of serum amyloid A1 (Saa1), Il-1β, and monocyte chemotactic protein (MCP)-1 in murine models of nonalcoholic steatohepatitis (NASH). Saa1 in the toxin (A) and methionine- and choline-sufficient and -deficient diet–fed (MCS and MCD, respectively) (B) models of NASH. Il-1β in the toxin (C) and MCS/MCD (D) models of NASH. MCP-1 in the toxin (E) and MCS/MCD (F) models of NASH. ∗P < 0.05. BDCM, bromodichloromethane.
Representative images of 5-μm-thick immunostained liver sections, imaged using bright-field microscopy. A–C: Positive immunoreactivities (brown) for the proinflammatory cytokines IL-1β (A), monocyte chemotactic protein (MCP)-1 (B), and Kupffer cell activation marker CD68 (C) are due to diaminobenzidine binding to respective biotinylated secondary antibodies via streptavidin–horseradish peroxidase. Nuclei (blue) counterstained with Mayer's hematoxylin solution. D–F: Morphometry performed on three independent fields for each sample, including representative images for percentage of area showing positive immunoreactivity of IL-1β in A (D), MCP-1 in B (E), and CD68 in C (F), all calculated in arbitrary units. ∗P < 0.05. Original magnification, ×20 (A–C). Hu Ctrl, healthy human control; Hu NASH, human nonalcoholic steatohepatitis; MCD, methionine- and choline-deficient diet–fed mouse; MCS, methionine- and choline-sufficient diet–fed mouse.
Western blot analysis of α-smooth muscle actin (SMA) in liver homogenates from rodent models of nonalcoholic steatohepatitis (NASH) and human NASH liver. A: Immunoblot images for the immunoreactive bands of α-SMA and β-actin. B: Band analysis after normalization against β-actin. ∗P < 0.05. Apo, diet-induced obesity wild-type (DIO) mouse exposed to bromodichloromethane (BDCM) and injected with apocynin; FeTPPS, DIO mouse exposed to BDCM and iron(III) tetrakis(p-sulfonatophenyl)porphyrin; Hu Ctrl, healthy human control; Hu NASH, human nonalcoholic steatohepatitis; MCD, methionine- and choline-deficient diet–fed mouse; MCS, methionine- and choline-sufficient diet–fed mouse; p47phox, high-fat diet–fed mouse with p47phox gene knockout exposed to BDCM; Tlr4 KO, high-fat diet–fed mouse with Tlr4 knockout exposed to BDCM.