

Abstract
The intestine of the human contains a dynamic population of microbes that have a symbiotic relationship with the host. In addition, there is an effect of the intestinal microbiota on metabolism and digestion. Non-alcoholic fatty liver disease (NAFLD) is a common cause worldwide of hepatic pathology and is thought to be the hepatic manifestation of the metabolic syndrome. In this review we examine the effect of the human microbiome on the components and pathogenesis of the metabolic syndrome. We are now on the threshold of therapeutic interventions on the human microbiome in order to effect human disease including NAFLD.
Keywords: Microbiome, Metabolic syndrome, Stool transplantation, Non-alcoholic fatty liver disease
Core tip: The human intestine contains more bacterial cells than mammalian cells. These have a symbiotic relationship with the host. Non-alcoholic fatty liver disease is the hepatic manifestation of the metabolic syndrome and a major cause of hepatic morbidity as a consequence of the obesity epidemic. We examine the effect of the human microbiome on the components of the metabolic syndrome and fatty liver and mention the possibility of therapeutic interventions in humans.
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is considered the hepatic manifestation of the metabolic syndrome. The metabolic syndrome is defined by clear clinical and laboratory criteria (Table 1). NAFLD encompasses a range of liver damage ranging from simple steatosis to non-alcoholic steatohepatitis (NASH), fibrosis, cirrhosis and its complications. NAFLD is present in approximately 1/3 of the United States population, who have isolated steatosis of the liver[1]. Of the patients with NAFLD, approximately 30% have NASH[2]. NASH refers to those patients who have developed liver inflammation and fibrosis. It is those patients with NASH who may develop stage 3 or 4 fibrosis (cirrhosis)[3].
Table 1.
The definitions of the metabolic syndrome
| NCEP ATP III | IDF | |
| Absolutely required | None | Central obesity (waist circumference) ≥ 94 cm in males or ≥ 80 cm in females European origin ≥ 90 cm in males or ≥ 80 cm in females |
| Criteria | Any three of the five criteria below | Central obesity plus two of the four criteria below |
| Obesity | (1) waist circumference > 40 inches in males, or > 35 inches in females | |
| Hyperglycemia | (2) fasting glucose ≥ 100 mg/dL or treated for DM | (1) fasting glucose ≥ 100 mg/dL |
| Dyslipidemia | (3) TG ≥ 150 mg/dL or treated for dyslipidemia | (2) TG ≥ 150 mg/dL or treated for dyslipidemia |
| Or (4) HDL cholesterol < 40 mg/dL in males, or < 50 mg/dL in females or under treatment | Or (3) HDL cholesterol < 40 mg/dL in males, or < 50 mg/dL in females or under treatment | |
| Hypertension | (5) > 130 mmHg systolic or > 85 mmHg diastolic or treated for HTN | (4) > 130 mmHg systolic or > 85 mmHg diastolic or treated for HTN |
NCEP ATP III: National Cholesterol and Education Program - Adult Treatment Panel III; IDF: International diabetes federation; DM: Diabetes mellitus; TG: TG: Triglycerides; HTN: Hypertension.
Many factors including diet, sedentary lifestyle and genetics have been shown to influence the progression from steatosis through NASH to cirrhosis. However, not all people who are obese develop NAFLD and neither are all patients with NAFLD obese.
GUT MICROBIOTA
The intestinal microbiome is attracting an increasing amount of attention[4]. It is becoming apparent that there is a symbiotic relationship between the intestine and its microbiota and that disturbance in this relationship can be associated with the pathogenesis of many disorders. The most striking example of such an association is Clostridium difficile infection, for which fecal transplantation from healthy donors is now an accepted treatment[5].
Distinct gut microbiota profiles are linked with specific metabolomes. Ninety-five percent of the gut microbiota of humans consists of the Firmicutes, Bacteroidetes and Actinobacteria phyla. The species level of the human microbiota, however, has higher diversity, with approximately 200 highly prevalent and up to 1000 less common bacterial species[6]. In humans as in mice, each individual has an unique bacterial species profile[7]. The gut bacteria may alter in response to a high fat diet (HFD), which could be responsible for some of the responses to an HFD.
Bacteria from human stools can be transferred to germ free (GF) mice and result in a similar microbiome in the host mice[8]. This can result in the appearance of human gut enzymatic activities in GF rodents after human fecal transplantation[9,10].
Recently, the transfer of human gut microbiome from obesity discordant twins to GF mice was shown to result in the transfer of the adiposity phenotype of the donor twin[11]. Thus, the transfer of human fecal microbiota to GF mice may result in the development of human diseases and provide an experimental study system.
INTESTINAL MICROBIOTA ARE RELATED TO OBESITY AND INSULIN RESISTANCE
The gut microbiota is now recognized as contributing to obesity and NAFLD[12]. GF mice have been found to gain less weight than conventional mice after being fed a high sugar and fat diet in spite of a higher amount of food consumption[13,14]. Furthermore, GF mice on an HFD develop an increase in insulin sensitivity[15] and GF mice colonized with conventional mouse intestinal microbiota develop an increase in body fat content[13]. There are, however, wide variations in the development of HFD-associated features[16,17], but the responsible factors are still undefined.
The insulin resistance index can be transferred by gut microbiota transplantation[18]. Gut microbiota affects both macrophage fat accumulation and systemic glucose metabolism by different mechanisms[19]. In a diet-induced obesity mouse model, administration of antibiotics improved fasting glycemia and insulin resistance independently of both food intake or adiposity[20]. Furthermore the improved insulin sensitivity correlated with less hepatic lipogenesis and steatosis in the antibiotic-treated mice[21]. Taken together, these findings suggest that the gut microbiota influences both host glucose metabolism and liver function.
A study in humans showed that transfer of intestinal microbiota from lean donors to males with the metabolic syndrome resulted in increased insulin sensitivity[21]. Dietary factors and changes in diet influence the composition of the microbiome. The intestinal microbiota of obese individuals has a different microbial diversity compared to lean persons. They have less Bacteroides and more Firmicutes[22]. Furthermore, an HFD increases the proportion of Gram-negative to Gram-positive microbes, resulting in the production of lipopolysaccharide (LPS) which is responsible for inflammation[23]. Gram-positive microbes are increased following the administration of prebiotics[24]. A prebiotic is a nondigestible food substrate which increases the growth of intestinal bacteria that can result in health benefits for the host.
The intestinal microbiome in obesity has an increased capacity to extract energy from the host diet. Bacterial enzymes extract calories from otherwise indigestible dietary polysaccharides[25]. Enteric bacteria suppress the synthesis and secretion of small intestinal fasting-induced adipocyte factor, resulting in an increased activity of lipoprotein lipase and increased liver triglyceride[13,14].
GF lean mice that were resistant to becoming obese on a fat-enriched diet had an increase of phosphorylated adenosine monophosphate-activated protein kinase (AMPK) in both the skeletal muscle and liver. AMPK phosphorylates acetyl coenzyme A (CoA) carboxylase, resulting in decreased malonyl CoA levels. Malonyl CoA controls the rate-limiting step of long-chain fatty acyl CoA entry to the mitochondria by blocking carnitine palmitoyltransferase which promotes the oxidation of fatty acid and results in a lower storage of fat[14,26].
Thus, the intestinal microbiome has an effect on both obesity and insulin resistance, as well as hepatic fat content.
GUT MICROBIOTA AND NAFLD
In view of the intimate connection between the metabolic syndrome with its concomitant insulin resistance and NAFLD, it is expected that there is an effect of the intestinal microbiome on NAFLD.
The fecal microbiota in NAFLD and NASH patients has been examined using quantitative polymerase chain reaction (PCR) and deep sequencing of a conserved region in the bacterial 16S ribosomal RNA gene[27-30]. A recent review provides a summary of the changes in the intestinal microbiota associated with NAFLD and NASH[12]. Many of these studies have variable and often contradictory findings. This may be due to differences in patient mix, methodology and documentation of liver disease.
In addition to the mixture of bacteria in the colon, patients with obesity or NAFLD have more small intestinal bacterial overgrowth[31,32]. Small intestinal bacterial overgrowth was found in 50% of patients with NASH, significantly more than that in a control population[33]. The intestinal permeability and bacterial overgrowth were shown to be related to the degree of hepatic steatosis but not inflammation or fibrosis[31].
However, it is not clear if the assessment of small bowel bacterial overgrowth by breath tests is accurate since an estimate of total fecal bacterial count by real-time PCR did not detect any difference between healthy controls and patients with NAFLD and NASH[28].
Possible mediators of the link between the enteric microbiome and the host include alcohol, choline and endotoxins. Obese animals have been shown to have higher levels of alcohol in breath tests than thin animals[34]. Alcohol reaches the liver via the portal blood and can cause triglyceride accumulation in hepatocytes[35]. In addition, alcohol may provide the “second hit” to the liver for making the transformation from steatosis to steatohepatitis[36].
Choline may also be involved in the development of NAFLD and NASH. It is well known that choline deficiency may result in chronic liver disease[37]. In animal models choline-deficient diets were utilized, but it is now known that choline deficiency can exist while there is a diet that is not deficient. HFDs produce intestinal microbiota that converts dietary choline into methylamines. This results in a reduction of serum level of phosphatidylcholine which can cause NASH[26]. Phosphatidylcholine is important for the production of very low-density lipoprotein (VLDL)[38] and thus choline deficiency secondary to the intestinal microbiome will result in lower hepatic secretion of VLDL and result in triglyceride accumulation in hepatocytes.
The products of the intestinal microbiota are also implicated in the development of NAFLD and NASH. Endotoxemia has been found in patients with NASH[39]. Toll-like receptor 4, a receptor for LPS, in hematopoietic-derived cells is necessary for the development of hepatic steatosis but not for obesity in mice[40]. Mice that are deficient in sensing pathogen-associated molecular patterns (PAMPs) or downstream signaling are resistant to NASH[41,42].
The microbial products reach the liver via the portal vein and cause inflammation. Mice that are genetically obese are more sensitive to endotoxin-induced hepatotoxicity and develop steatohepatitis after being exposed to low doses of LPS[43]. NAFLD patients have an increased intestinal permeability and changes in the intestinal tight junctions, as compared to healthy individuals[31]. The increased permeability, in combination with bacterial overgrowth, increases the hepatic exposure to endotoxins.
Alteration of the fecal microbiome by administration of probiotics has been shown to decrease the amount of intrahepatic triglyceride content in addition to a decrease in Firmicutes and an increase in Bacteroidetes[30]. A meta-analysis of the published trials of probiotics in patients with NAFLD, showed a reduction in serum transaminases, total cholesterol, tumor necrosis factor-α and an improvement in insulin resistance[44].
Dysbiosis can induce intestinal inflammation. Indeed GF mice are protected from inflammation of the small intestine[45]. Mice deficient in Nlrp3 and Nlrp6 are unable to form cytoplasmic multiprotein complexes composed of nucleotide-binding domain and leucine-rich repeat-containing proteins (NLR) family, inflammasomes. Inflammasomes are sensors of exogenous PAMPs that regulate cleavage of precursors of inflammatory cytokines including pro-interleukin 1 beta (pro-IL1β) and pro-IL18. In mice, loss of Nlrp3 and Nlrp6 inflammasomes is associated with intestinal dysbiosis and colonic inflammation via CCL5. Dysbiosis is linked to an increase in Prevotella[46]. The consequent translocation of bacteria leads to an increase in bacterial products including LPS and bacterial DNA in the portal vein. The ensuing hepatic inflammatory response promotes progression of NAFLD to NASH (Figure 1). This change in phenotype can be transmitted by co-housing wild-type and NASH-prone mice[46].
Figure 1.
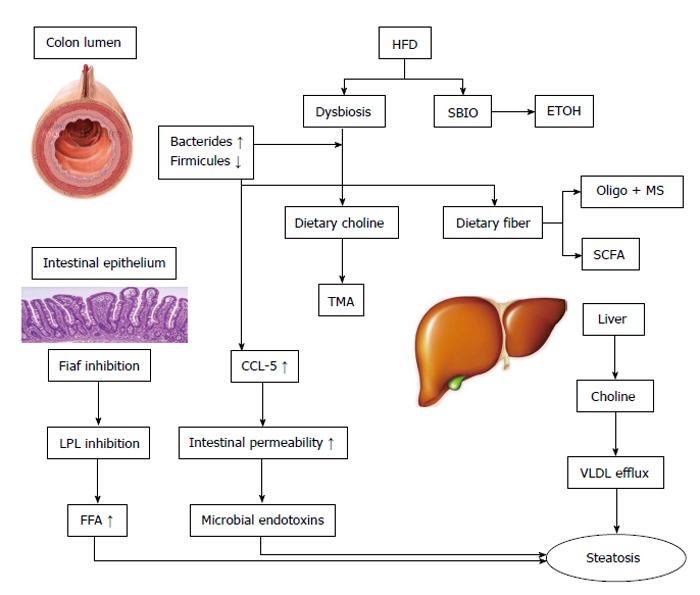
The effect of the intestinal microbiota on non-alcoholic fatty liver disease. High fat diets (HFD) produce dysbiosis and small bowel intestinal overgrowth (SBIO). There is an increase in energy extraction and fermentation of dietary fibers to oligo- and mono-saccahrides and short chain fatty acids (SCFA). There is also an increase in ethanol (ETOH) production. The microbiota metabolize choline to trimethylamine (TMA). There is a choline deficiency which decreases very low-density lipoprotein (VLDL) efflux and hepatic steatosis. In addition the intestinal microbiota suppresses the production of fasting induced adipocyte factor (Fiaf) in intestinal epithelia, which increases the activity of lipoprotein lipase and the levels of free fatty acids (FFA). Dysbiosis results in a disruption of tight junctions in the enterocytes via chemokine (C-C motif) ligand 5 (CCL-5). The resulting increase in intestinal permeability results in the translocation of microbial products to the liver and inflammation; MS: Monosaccharides; LPL: Lipoprotein lipase.
Thus, intestinal dysbiosis can induce colonic inflammation and bacterial translocation which accelerates the progression of simple steatosis to NASH. As a result of these findings attention is beginning to be directed at fecal microbiota transplantation (FMT). FMT was first used in China more than 1500 years ago[47]. In 1958, 4 cases of treatment of pseudomembranous colitis by fecal enemas were reported[48]. This is now an established treatment[49]. At present there is only one report of FMT for metabolic syndrome. Vrieze et al[21] reported 18 patients with the metabolic syndrome who underwent a stool transplant that was either autologous or from lean healthy volunteers. Six weeks following the FMT there was a significant increase in insulin sensitivity together with an increase in the levels of butyrate-producing intestinal microbiota.
In summary, there appears to be an effect of the fecal microbiome on the development of the metabolic syndrome and its hepatic manifestation NAFLD and NASH. Further investigation of this relationship will increase our understanding of this connection. There is evidence that manipulation of the fecal microbiome may result in a change in the metabolic syndrome and an improvement in the features of NAFLD. This needs to be explored further in order to investigate if there will be an improvement in clinically significant end points.
Footnotes
P- Reviewer: Abdel-Salam OME, Balaban YH, Hsieh SY, Julie NL, Rajeshwari K, Wong GLH S- Editor: Ji FF L- Editor: Wang TQ E- Editor: Liu SQ
Conflict-of-interest: The authors have no conflict of interest to declare.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: December 18, 2014
First decision: March 6, 2015
Article in press: April 20, 2015
References
- 1.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 2.Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, Landt CL, Harrison SA. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140:124–131. doi: 10.1053/j.gastro.2010.09.038. [DOI] [PubMed] [Google Scholar]
- 3.Caldwell S, Argo C. The natural history of non-alcoholic fatty liver disease. Dig Dis. 2010;28:162–168. doi: 10.1159/000282081. [DOI] [PubMed] [Google Scholar]
- 4.Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. Host-gut microbiota metabolic interactions. Science. 2012;336:1262–1267. doi: 10.1126/science.1223813. [DOI] [PubMed] [Google Scholar]
- 5.Floch MH. The power of poop: probiotics and fecal microbial transplant. J Clin Gastroenterol. 2012;46:625–626. doi: 10.1097/MCG.0b013e3182667a93. [DOI] [PubMed] [Google Scholar]
- 6.Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol. 2008;6:776–788. doi: 10.1038/nrmicro1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zoetendal EG, Akkermans AD, De Vos WM. Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Appl Environ Microbiol. 1998;64:3854–3859. doi: 10.1128/aem.64.10.3854-3859.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turnbaugh PJ, Ridaura VK, Faith JJ, Rey FE, Knight R, Gordon JI. The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci Transl Med. 2009;1:6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowey E, Adlercreutz H, Rowland I. Metabolism of isoflavones and lignans by the gut microflora: a study in germ-free and human flora associated rats. Food Chem Toxicol. 2003;41:631–636. doi: 10.1016/s0278-6915(02)00324-1. [DOI] [PubMed] [Google Scholar]
- 10.Gérard P, Béguet F, Lepercq P, Rigottier-Gois L, Rochet V, Andrieux C, Juste C. Gnotobiotic rats harboring human intestinal microbiota as a model for studying cholesterol-to-coprostanol conversion. FEMS Microbiol Ecol. 2004;47:337–343. doi: 10.1016/S0168-6496(03)00285-X. [DOI] [PubMed] [Google Scholar]
- 11.Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341:1241214. doi: 10.1126/science.1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology. 2014;146:1513–1524. doi: 10.1053/j.gastro.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci USA. 2007;104:979–984. doi: 10.1073/pnas.0605374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rabot S, Membrez M, Bruneau A, Gérard P, Harach T, Moser M, Raymond F, Mansourian R, Chou CJ. Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. FASEB J. 2010;24:4948–4959. doi: 10.1096/fj.10-164921. [DOI] [PubMed] [Google Scholar]
- 16.Jang I, Hwang D, Lee J, Chae K, Kim Y, Kang T, Kim C, Shin D, Hwang J, Huh Y, et al. Physiological difference between dietary obesity-susceptible and obesity-resistant Sprague Dawley rats in response to moderate high fat diet. Exp Anim. 2003;52:99–107. doi: 10.1538/expanim.52.99. [DOI] [PubMed] [Google Scholar]
- 17.Li H, Xie Z, Lin J, Song H, Wang Q, Wang K, Su M, Qiu Y, Zhao T, Song K, et al. Transcriptomic and metabonomic profiling of obesity-prone and obesity-resistant rats under high fat diet. J Proteome Res. 2008;7:4775–4783. doi: 10.1021/pr800352k. [DOI] [PubMed] [Google Scholar]
- 18.Le Roy T, Llopis M, Lepage P, Bruneau A, Rabot S, Bevilacqua C, Martin P, Philippe C, Walker F, Bado A, et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut. 2013;62:1787–1794. doi: 10.1136/gutjnl-2012-303816. [DOI] [PubMed] [Google Scholar]
- 19.Caesar R, Reigstad CS, Bäckhed HK, Reinhardt C, Ketonen M, Lundén GÖ, Cani PD, Bäckhed F. Gut-derived lipopolysaccharide augments adipose macrophage accumulation but is not essential for impaired glucose or insulin tolerance in mice. Gut. 2012;61:1701–1707. doi: 10.1136/gutjnl-2011-301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Membrez M, Blancher F, Jaquet M, Bibiloni R, Cani PD, Burcelin RG, Corthesy I, Macé K, Chou CJ. Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. FASEB J. 2008;22:2416–2426. doi: 10.1096/fj.07-102723. [DOI] [PubMed] [Google Scholar]
- 21.Vrieze A, Van Nood E, Holleman F, Salojärvi J, Kootte RS, Bartelsman JF, Dallinga-Thie GM, Ackermans MT, Serlie MJ, Oozeer R, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143:913–6.e7. doi: 10.1053/j.gastro.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 22.Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3:213–223. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 24.Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, Gibson GR, Delzenne NM. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia. 2007;50:2374–2383. doi: 10.1007/s00125-007-0791-0. [DOI] [PubMed] [Google Scholar]
- 25.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 26.Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A, Fearnside J, Tatoud R, Blanc V, Lindon JC, et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci USA. 2006;103:12511–12516. doi: 10.1073/pnas.0601056103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raman M, Ahmed I, Gillevet PM, Probert CS, Ratcliffe NM, Smith S, Greenwood R, Sikaroodi M, Lam V, Crotty P, et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2013;11:868–875.e1-3. doi: 10.1016/j.cgh.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 28.Mouzaki M, Comelli EM, Arendt BM, Bonengel J, Fung SK, Fischer SE, McGilvray ID, Allard JP. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology. 2013;58:120–127. doi: 10.1002/hep.26319. [DOI] [PubMed] [Google Scholar]
- 29.Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, Gill SR. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57:601–609. doi: 10.1002/hep.26093. [DOI] [PubMed] [Google Scholar]
- 30.Wong VW, Tse CH, Lam TT, Wong GL, Chim AM, Chu WC, Yeung DK, Law PT, Kwan HS, Yu J, et al. Molecular characterization of the fecal microbiota in patients with nonalcoholic steatohepatitis--a longitudinal study. PLoS One. 2013;8:e62885. doi: 10.1371/journal.pone.0062885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, Mascianà R, Forgione A, Gabrieli ML, Perotti G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–1887. doi: 10.1002/hep.22848. [DOI] [PubMed] [Google Scholar]
- 32.Sabaté JM, Jouët P, Harnois F, Mechler C, Msika S, Grossin M, Coffin B. High prevalence of small intestinal bacterial overgrowth in patients with morbid obesity: a contributor to severe hepatic steatosis. Obes Surg. 2008;18:371–377. doi: 10.1007/s11695-007-9398-2. [DOI] [PubMed] [Google Scholar]
- 33.Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut. 2001;48:206–211. doi: 10.1136/gut.48.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cope K, Risby T, Diehl AM. Increased gastrointestinal ethanol production in obese mice: implications for fatty liver disease pathogenesis. Gastroenterology. 2000;119:1340–1347. doi: 10.1053/gast.2000.19267. [DOI] [PubMed] [Google Scholar]
- 35.Hartmann P, Chen WC, Schnabl B. The intestinal microbiome and the leaky gut as therapeutic targets in alcoholic liver disease. Front Physiol. 2012;3:402. doi: 10.3389/fphys.2012.00402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Day CP. Pathogenesis of steatohepatitis. Best Pract Res Clin Gastroenterol. 2002;16:663–678. doi: 10.1053/bega.2002.0333. [DOI] [PubMed] [Google Scholar]
- 37.Blumberg H, McCollum EV. The prevention by choline of liver cirrhosis in rats on high fat, low protein diets. Science. 1941;93:598–599. doi: 10.1126/science.93.2425.598. [DOI] [PubMed] [Google Scholar]
- 38.Jiang XC, Li Z, Liu R, Yang XP, Pan M, Lagrost L, Fisher EA, Williams KJ. Phospholipid transfer protein deficiency impairs apolipoprotein-B secretion from hepatocytes by stimulating a proteolytic pathway through a relative deficiency of vitamin E and an increase in intracellular oxidants. J Biol Chem. 2005;280:18336–18340. doi: 10.1074/jbc.M500007200. [DOI] [PubMed] [Google Scholar]
- 39.Farhadi A, Gundlapalli S, Shaikh M, Frantzides C, Harrell L, Kwasny MM, Keshavarzian A. Susceptibility to gut leakiness: a possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver Int. 2008;28:1026–1033. doi: 10.1111/j.1478-3231.2008.01723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saberi M, Woods NB, de Luca C, Schenk S, Lu JC, Bandyopadhyay G, Verma IM, Olefsky JM. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009;10:419–429. doi: 10.1016/j.cmet.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. 2010;139:323–34.e7. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol. 2007;47:571–579. doi: 10.1016/j.jhep.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci USA. 1997;94:2557–2562. doi: 10.1073/pnas.94.6.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma YY, Li L, Yu CH, Shen Z, Chen LH, Li YM. Effects of probiotics on nonalcoholic fatty liver disease: a meta-analysis. World J Gastroenterol. 2013;19:6911–6918. doi: 10.3748/wjg.v19.i40.6911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ding S, Chi MM, Scull BP, Rigby R, Schwerbrock NM, Magness S, Jobin C, Lund PK. High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS One. 2010;5:e12191. doi: 10.1371/journal.pone.0012191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang F, Luo W, Shi Y, Fan Z, Ji G. Should we standardize the 1,700-year-old fecal microbiota transplantation? Am J Gastroenterol. 2012;107:1755; author reply p.1755-p.1756. doi: 10.1038/ajg.2012.251. [DOI] [PubMed] [Google Scholar]
- 48.Eiseman B, Silen W, Bascom GS, Kauvar AJ. Fecal enema as an adjunct in the treatment of pseudomembranous enterocolitis. Surgery. 1958;44:854–859. [PubMed] [Google Scholar]
- 49.van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JF, Tijssen JG, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med. 2013;368:407–415. doi: 10.1056/NEJMoa1205037. [DOI] [PubMed] [Google Scholar]