

Abstract
Salt intake promotes progression of CKD by uncertain mechanisms. We hypothesized that a salt-induced reno-cerebral reflex activates a renin-angiotensin axis to promote CKD. Sham-operated and 5/6-nephrectomized rats received a normal-salt (0.4%), low-salt (0.02%), or high-salt (4%) diet for 2 weeks. High salt in 5/6-nephrectomized rats increased renal NADPH oxidase, inflammation, BP, and albuminuria. Furthermore, high salt activated the intrarenal and cerebral, but not the systemic, renin-angiotensin axes and increased the activity of renal sympathetic nerves and neurons in the forebrain of these rats. Renal fibrosis was increased 2.2-fold by high versus low salt, but intracerebroventricular tempol, losartan, or clonidine reduced this fibrosis by 65%, 69%, or 59%, respectively, and renal denervation or deafferentation reduced this fibrosis by 43% or 38%, respectively (all P<0.05). Salt-induced fibrosis persisted after normalization of BP with hydralazine. These data suggest that the renal and cerebral renin-angiotensin axes are interlinked by a reno-cerebral reflex that is activated by salt and promotes oxidative stress, fibrosis, and progression of CKD independent of BP.
Keywords: renin-angiotensin system, brain, kidney, salt, renal fibrosis
CKD is a progressive condition for which no intervention presently prevents the functional decline. Fibrosis is the final common pathway for most categories of CKD and culminates in ESRD.1,2 Fibrosis is estimated to contribute to 45% of all causes of mortality3 and is promoted by many factors present in damaged kidneys and CKD, including oxidative stress, inflammation, hypertension, hypoxia, metabolic toxins, and activation of the sympathetic nervous system (SNS).4–12
The association of hypertensive renal disease with dietary salt is well recognized.13–16 Many patients with CKD have salt-sensitive hypertension.17,18 Although restriction of dietary salt reduces albuminuria,19–22 it is not clear how salt per se accelerates the progression of renal glomerular and tubulointerstitial fibrosis.13,14,16
Several mechanisms for salt-induced renal injury have been proposed in addition to hypertension,14,23 including increased generation of Na/K-ATPase inhibitors24 and TGF-β.25 However, another potential factor is a paradoxical activation of the intrarenal renin-angiotensin system (RAS) in damaged kidneys. Thus, high salt intake enhances the intrarenal expression of angiotensinogen (AGT) in Dahl salt-sensitive (DSS) rats26 and increases urinary AGT excretion in patients with IgA nephropathy.27 Renal AGT may be rate limiting for angiotensin II (AngII) generation.26 Most organs possess a local RAS that is regulated independently and is somewhat compartmentalized from the circulation.12,28 Intracerebroventricular (ICV) AngII activates posterior hypothalamic nuclei to increase efferent renal nerve activity and BP.11 Spontaneously hypertensive rats and DSS rats have a hyperactive brain RAS,29 which contributes to hypertension.30 Thus, the brain RAS can regulate the peripheral RAS and thereby regulate the BP. However, the effect of the brain RAS on the loss of renal function in CKD has not been studied. Moreover, renal afferent nerve activity from damaged kidneys can drive hypertension via activation of the central SNS31 that itself is driven by central AngII type 1 (AT1) receptor activation.32 This suggests the intriguing possibility of extensive renal/cerebral interaction via afferent and efferent renal nerve activity in CKD with involvement of local kidney and brain RAS, but this remains speculative.
We used a 5/6-nephrectomized (5/6Nx) rat model of CKD to test the hypothesis that high salt intake contributes to progression of renal fibrosis in CKD through a neuronal interaction between the brain and the renal RAS, which we have termed the “reno-cerebral RAS axis.” Here, we demonstrate that the renal and cerebral RAS interact via changes in renal afferent and efferent sympathetic nerve activity to promote salt-induced renal fibrosis independent of BP. Activation of this RAS axis by salt may underlie progression of CKD.
Results
High Salt Induced Renal Inflammation, Fibrosis, Oxidative Stress, and Activation of the SNS in 5/6Nx Rats
Eight weeks after operation, 5/6Nx or sham rats were randomly assigned to three groups receiving a low-salt (0.02% NaCl), normal-salt (0.4% NaCl), or high-salt (4% NaCl) diet for 2 weeks. Changes in dietary salt in sham animals did not induce structural alterations in the kidneys (Figure 1, A–D) or change systolic BP (SBP) or urinary albumin excretion (UAE) (Figure 1, E and F).
Figure 1.

High salt induces renal inflammation, fibrosis, sympathetic activation, and oxidative stress in 5/6Nx rats. (A) Representative photographs of macrophage (ED-1–positive cells) infiltration and renal fibrosis (shown by PAS or Masson staining). (B–D) Quantitative analysis of macrophage infiltration (B), glomerulosclerosis index (C), and tubulointerstitial fibrosis score (D). (E) Changes in SBP. (F) Changes in UAE. (G) Concentration of NE in renal cortex. (H–I) Protein (H) and mRNA (I) levels of Noxs in renal cortex. Data from three independent experiments are expressed as the mean±SD (n=6 in each group). *P<0.05 versus normal salt in the respective group. HS, high salt; LS, low salt; NS, normal salt; PAS, periodic acid–Schiff.
By contrast, among 5/6Nx rats, high dietary salt intake evoked significant increases in tubulointerstitial macrophage infiltration (Figure 1, A and B), glomerulosclerosis index (Figure 1, A and C), and tubulointerstitial fibrosis score (Figure 1, A and D) with overexpression of fibronectin and collagen I (Supplemental Figure 1, A–D). High salt also increased SBP (Figure 1E), UAE (Figure 1F), renal NE concentration (Figure 1G), and expression of the NADPH oxidase subunits Nox2 and Nox4 (Figure 1, H and I).
High Salt Paradoxically Activated the Intrarenal RAS in 5/6Nx Rats
The components of the RAS were expressed in sham kidneys in renal tubular epithelial cells,33 except for renin that was expressed in juxtaglomerular cells (Figure 2). Salt intake in sham rats led to a graded downregulation of the expression of renal renin, AGT, angiotensin-converting enzyme (ACE)-1, AT1 receptors, and AngII in sham rats (Figure 2) with similar effects of salt on the circulating RAS (Table 1).
Figure 2.

High salt induces paradoxical activation of intrarenal RAS in 5/6Nx rats. (A) Expression of AGT: representative photographs of AGT expression (A1) and semiquantitative data of AGT expression (A2). (B) Expression of ACE-1: representative photographs of ACE-1 expression (B1) and semiquantitative data of ACE-1 expression (B2). (C) Expression of AngII: representative photographs of AngII expression (C1) and semiquantitative data of AngII expression (C2). (D) Expression of AT1 receptors: representative photographs of AT1 receptor expression (D1) and semiquantitative data of AT1 receptor expression (D2). (E) Expression of renin: representative photographs of renin expression (E1) and semiquantitative data of renin expression (E2). The semiquantitative data are expressed as the mean±SD of three independent experiments (n=6 in each group). *P<0.05 versus normal salt in the respective group. HS, high salt; LS, low salt; NS, normal salt.
Table 1.
Changes in biochemical and metabolic parameters
| Parameter | Sham Rats | 5/6Nx Rats | ||||
|---|---|---|---|---|---|---|
| Low Salt | Normal Salt | High Salt | Low Salt | Normal Salt | High Salt | |
| Body wt (g) | ||||||
| 0 wk | 453±9 | 466±10 | 468±12 | 456±10 | 461±9 | 459±11 |
| 2 wk | 491±11 | 494±13 | 495±12 | 484±12 | 485±18 | 482±15 |
| Kidney wt/body wt (mg/g) | 3.2±0.1 | 3.2±0.2 | 3.1±0.1 | 4.2±0.1a | 4.4±0.2a | 4.8±0.2a,b |
| Serum creatinine (μmol/L) | ||||||
| 0 wk | 68±4 | 71±5 | 72±3 | 107±5a | 111±6a | 112±3a |
| 2 wk | 66±4 | 72±4 | 75±8 | 110±6a | 114±6a | 117±9a |
| Serum Na+ (mmol/L) | ||||||
| 0 wk | 140±3 | 138±3 | 140±3 | 139±2 | 140±3 | 140±2 |
| 2 wk | 140±3 | 140±1 | 142±2 | 141±3 | 141±2 | 144±2a,b |
| Urine volume (ml/24 h) | ||||||
| 0 wk | 29±2 | 30±2 | 31±3 | 29±3 | 28±3 | 27±3 |
| 2 wk | 25±3b | 32±4 | 40±8b | 20±4 | 26±4 | 59±9a,b |
| Urine Na+ (mmol) | ||||||
| 0 wk | 0.83±0.16 | 0.85±0.16 | 0.83±0.19 | 0.79±0.14 | 0.78±0.19 | 0.77±0.15 |
| 2 wk | 0.13±0.08b | 0.87±0.79 | 15.32±0.19b | 0.10±0.07b | 0.75±0.13 | 10.94±0.14a,b |
| Plasma renin activity (ng AngI/ml per h) | ||||||
| 0 wk | 4.9±0.7 | 5.2±0.5 | 5.0±0.4 | 2.3±0.2a | 2.5±0.2a | 2.2±0.3a |
| 2 wk | 8.2±0.7b | 5.1±0.4 | 3.0±0.3b | 3.2±0.3a,b | 1.4±0.2a | 0.5±0.1a,b |
| Plasma AGT (ng/ml) | ||||||
| 0 wk | 3222±274 | 3149±332 | 3188±320 | 2932±245 | 3005±329 | 2878±277 |
| 2 wk | 3474±131b | 3277±154 | 3049±118b | 3256±148a,b | 2990±136a | 2712±150a,b |
| Serum ACE-1 activity (nmol/ml per min) | ||||||
| 0 wk | 86±3 | 87±4 | 89±5 | 84±3 | 85±2 | 83±4 |
| 2 wk | 90±6 | 86±4 | 88±3 | 87±2 | 86±2 | 80±3a,b |
| Plasma AngII (pg/ml) | ||||||
| 0 wk | 36±4 | 40±5 | 38±3 | 27±4a | 30±3a | 29±3a |
| 2 wk | 81±5b | 47±3 | 25±3b | 58±3a,b | 28±2a | 15±2a,b |
| Plasma NE (pg/ml) | ||||||
| 0 wk | 301±20 | 312±19 | 293±16 | 647±26a | 635±42a | 651±38a |
| 2 wk | 442±37b | 335±18 | 253±21b | 551±22a,b | 671±38a | 822±31a,b |
Data from three independent experiments are expressed as the mean±SD (n=6 in each group).
P<0.05 versus sham group fed with the same salt diet.
P<0.05 versus rats fed with normal salt in the sham or 5/6Nx group.
By contrast, the expression of the intrarenal RAS in 5/6Nx rats was paradoxically upregulated by salt loading (Figure 2). The expression of renal angiotensin peptides and enzymes (Figure 2, A–D) was increased significantly in 5/6Nx rats and increased further during high salt. This contrasts with immunodetectable renin expression that was confined to the juxtaglomerular apparatus (JGA) and was decreased significantly by high salt intake in 5/6Nx rats (Figure 2E). Thus, the salt-induced changes in the intrarenal RAS were fully dissociated from JGA renin, which is the main source of circulating renin. Indeed, although the remnant kidney of 5/6Nx rats exhibited overactivation of tissue RAS, their circulating levels were lower than that in sham rats and were reduced appropriately by salt loading (Table 1). Moreover, there was upregulation by high salt in renal homogenates of 5/6Nx rats of tissue ACE-1 activity, AngII levels, urinary AGT excretion, and expression of AGT, ACE-1, and AT1 receptors (Supplemental Figure 2). Thus, with the exception of renin, there was a complete dissociation of the intrarenal from the systemic RAS in 5/6Nx rats.
High Salt Upregulated RAS Components in Renal Tubular Cells of 5/6Nx Rats
Overexpression of AGT in high-salt–fed 5/6Nx rats was localized mainly in proximal tubular cells (Figure 3A). Expression of ACE-1 (Figure 3B), AngII (Figure 3C), and AT1 receptors (Figure 3D) was apparent in proximal tubules, thick ascending limbs of Henle, distal convoluted tubules, and collecting ducts.
Figure 3.

High-salt–induced renal RAS expression is mainly localized in tubular cells. (A) Representative photographs of AGT localization determined with double staining of antibodies against AGT and markers of renal tubular segments. (B) Location of ACE-1 determined with double staining of antibodies against ACE-1 and markers of renal tubular segments. (C) Location of AngII determined with double staining of antibodies against AngII and markers of renal tubular segments. (D) Location of AT1 receptors determined with double staining of antibodies against AT1 receptors and markers of renal tubular segments. AQP-1, aquaporin 1 (proximal tubule); AQP-2, aquaporin 2 (collecting duct); NCCT, thiazide-sensitive NaCl cotransporter (distal tubule); THP, Tamm–Horsfall protein (thick ascending limb).
High Salt Activated Central Neurons to Express RAS, Tyrosine Hydroxylase, and Noxs in 5/6Nx Rats
The brain RAS was expressed predominantly in the cardiovascular regions of the forebrain, such as the subfornical organ (SFO), the organum vasculosum laminae terminalis, the paraventricular nucleus (PVN), and the supraoptic nucleus.34 Although the expression of the brain RAS was unaffected by high salt intake in sham rats (Figure 4, A–D, Supplemental Figure 3), it was activated in 5/6Nx rats in the SFO (exposed to cerebrospinal fluid) and PVN (within the blood–brain barrier) (Figure 4, A–D) and the organum vasculosum laminae terminalis and supraoptic nucleus (Supplemental Figure 3). High salt intake in 5/6Nx rats increased the numbers of AT1 receptor–positive cells (Figure 4, A and B), AT1 receptor mRNA (Figure 4C), and AngII expression (Figure 4D) in these regions. The expression of RAS in the brain cortex (as a control) was modest and independent of salt intake in sham and 5/6Nx rats (data not shown).
Figure 4.

High salt activates central neurons to express RAS in 5/6Nx rats. (A) Representative photographs of immunohistochemistry staining of AT1 receptors and AngII in SFO and PVN. (B) Semiquantitative data of AT1 receptors. (C) Expression of AT1 receptor mRNA measured by real-time PCR. (D) Semiquantitative data of AngII. (E) Localization of central AT1 receptors or AngII determined by double staining with the antibodies against AT1 receptors or AngII (green) and the antibody-recognized NSE (red). (F) Expression of c-fos in SFO and PVN: representative photographs of immunohistochemistry staining (F1) and semiquantitative data (F2). Data are expressed as the mean±SD of three independent experiments (n=6 in each group). *P<0.05 versus sham group fed with the same salt diet. HS, high salt; LS, low salt; NS, normal salt; NSE, neuron-specific enolase.
Double immunofluorescence with antibodies recognizing the neuron-specific enolase or glial fibrillary acidic protein demonstrated that high-salt–fed 5/6Nx rats had overexpression of AT1 receptors and AngII (Figure 4E, Supplemental Figure 4) in neurons, but not glial cells, and enhanced activation of neurons, represented by increased numbers of c-fos–positive cells (Figure 4F).
Tyrosine hydroxylase (TH) is the rate-limiting enzyme for cerebral NE synthesis. Its expression was upregulated by high salt in 5/6Nx rats in c-fos–positive neurons in the rostral ventrolateral medulla (RVLM), the SFO, and the PVN (Figure 5, A and B). High salt also upregulated the expression of Nox2 and Nox4 in this model (Figure 5, C and D).
Figure 5.
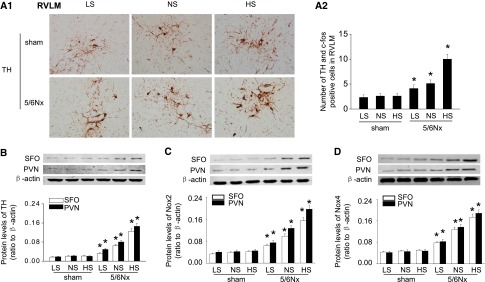
High salt intake upregulates expression of central TH and Noxs in 5/6Nx rats. (A) The number of c-fos–positive and TH-expressing neurons in RVLM, detected by double labeling of TH (brown cytoplasmic) and c-fos (red nuclear), markedly increases in high-salt–fed 5/6Nx rats: representative photographs (A1) and quantitative data (A2). (B) Expression of TH levels in SFO and PVN assessed by Western blot. (C and D) Expression of Nox2 (C) and Nox4 (D) analyzed by Western blot. Data are expressed as the mean±SD of three independent experiments (n=6 in each group). *P<0.05 versus sham group fed with the same salt diet. HS, high salt; LS, low salt; NS, normal salt.
Increased Neuronal Activation and TH Expression by High Salt in 5/6Nx Rats Was Prevented by Blockade of Central AT1 Receptors or Oxidative Stress
ICV losartan, given to 5/6Nx rats fed a high-salt diet, at 1/500 the concentration of intragastric (IG) losartan, inhibited the overexpression of AT1 receptors significantly at both protein and mRNA levels (Figure 6, A–C) and inhibited the expression of c-fos (Supplemental Figure 5) and TH (Figure 6, D and E). By contrast, ICV administration of clonidine that blocks the sympathetic outflow did not affect the expression of these systems (Figure 6, A–C, Supplemental Figure 5), indicating that the central RAS activated the SNS.
Figure 6.

Blockade of central RAS or oxidative stress inhibits salt-induced neuron activation and TH expression in 5/6Nx rats. (A–C) Central administration of losartan or tempol and RDX or SDR downregulate expression of central AT1 receptors: representative photographs of immunohistochemistry staining (A), semiquantitative data (B), and AT1 receptor mRNA level assessed by real-time PCR (C). (D and E) Central administration of losartan or tempol and RDX or SDR downregulate TH expression in SFO and PVN (D) as well as in RVLM (E). (F and G) Central administration of losartan or tempol and RDX or SDR reduce overexpression of Nox2 (F) and Nox4 (G) in brain. Data are expressed as the mean±SD of three independent experiments (n=6 in each group). *P<0.05 versus 5/6Nx rats given vehicle (0 mg/kg per day of inhibitor). Hyd, hydralazine; Los, losartan; RDX, renal denervation; SDR, selective dorsal rhizotomy.
Remarkably, blockade of sympathetic nerve traffic by renal denervation or selective blockade of renal afferent signals from the kidney by dorsal rhizotomy also alleviated high-salt–induced upregulation of brain RAS and TH (Figure 6). Thus, activation of the central RAS, and thereby activation of the central SNS, during high-salt intake in 5/6Nx rats was driven by afferent signals transmitted by sensory renal nerves. The activation of the central reactive oxygen species (ROS), RAS, and SNS was independent of hypertension because their overexpression persisted after normalization of BP with hydralazine (Figure 6).
The Activation of the Renal RAS by High Salt in 5/6Nx Rats Was Prevented by Blockade of Central RAS, ROS, or SNS
Blockade of central AT1 receptors in remnant kidneys of high-salt–fed rats by ICV losartan, at a dose of 0.2% of the effective IG dose, inhibited the overexpression of RAS at both the protein (Figure 7, A and B) and the mRNA levels (Figure 7C), and inhibited salt-induced increases in renal ACE-1 activity and urinary AGT excretion (Supplemental Figure 6). Remarkably, blockade of central sympathetic outflow by ICV clonidine or inhibition of renal afferent nerve signaling by selective dorsal rhizotomy or renal denervation also fully suppressed the activation of the intrarenal RAS by high salt (Figure 7). Moreover, both ICV and IG administration of the redox-cycling antioxidant tempol35 significantly suppressed the activation of the intrarenal RAS by high salt (Figure 7), yet the amount of tempol delivered by ICV administration was only 0.15% of that delivered by IG administration. Thus, both systemic and central oxidative stress can activate the intrarenal RAS by high salt. However, the activation of the intrarenal RAS was independent of hypertension because it persisted in 5/6Nx rats after normalization of their BP with hydralazine. Blockade of central AT1 receptors, sympathetic outflow, or oxidative stress did not affect the circulating levels of RAS in salt-fed 5/6Nx rats (Supplemental Table 1). Thus, the reno-cerebral RAS axis is regulated independently of the systemic RAS and of hypertension, and must be compartmentalized from the circulation in this model.
Figure 7.
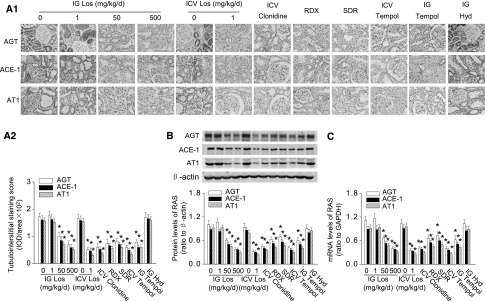
Blockade of central RAS, sympathetic signal, or oxidative stress inhibits salt-induced renal RAS activation in 5/6Nx rats. (A) Central administration of losartan or tempol and RDX or SDR downregulate overexpression of renal RAS in high-salt–fed 5/6Nx rats: representative photographs (A1) and semiquantitative analysis (A2). (B) Protein expression of intrarenal RAS in renal cortex homogenates. (C) Expression of RAS mRNA in renal cortex homogenates detected by real-time PCR. Data are expressed as the mean±SD of three independent experiments (n=6 in each group). *P<0.05 versus 5/6Nx rats given vehicle. Hyd, hydralazine; Los, losartan; RDX, renal denervation; SDR, selective dorsal rhizotomy.
Renal Inflammation, Fibrosis, and Dysfunction in 5/6Nx Rats Fed High Salt Was Attenuated by Blockade of Central RAS, SNS, or Oxidative Stress
ICV losartan decreased salt-induced renal macrophage infiltration significantly by 66% (Figure 8, A and B), decreased the glomerulosclerosis index by 68% (Figure 8, A and C), and decreased the tubulointerstitial fibrosis score by 65% (Figure 8, A and D). It also decreased extracellular matrix accumulation in renal tissue (Supplemental Figure 1, E–G), reduced renal NE levels (Figure 8E), and reduced the expression of Noxs (Figure 8F). This preservation of renal structure was associated with a reduced BP (Figure 8G), and improved UAE (Figure 8H). Blockade of sympathetic traffic by ICV clonidine, renal denervation, or selective dorsal rhizotomy or blockade of oxidative stress with ICV or IG tempol all reduced salt-induced renal injury and dysfunction in 5/6Nx rats (Figure 8), whereas normalization of BP with hydralazine was ineffective (Figure 8). Overall, renal fibrosis in the kidneys of 5/6Nx rats was increased 2.2-fold by high versus low salt, but intracerebroventricular tempol, losartan, or clonidine reduced this by 65%, 69%, or 59%, respectively, and renal denervation or deafferentation by 43% or 38%, suggesting a robust, but not exclusive, role for the brain and the renal nerves in the renal fibrosis in this model.
Figure 8.

Blockade of central RAS, sympathetic signal, or oxidative stress inhibits salt-induced renal inflammation, fibrosis, and dysfunction in 5/6Nx rats. (A–D) Central administration of losartan or tempol and RDX or SDR inhibit salt-induced renal inflammation, fibrosis, and dysfunction in 5/6Nx rats: representative photographs of macrophage infiltration and renal fibrosis (A), quantitative analysis of renal macrophage infiltration (B), glomerulosclerosis index (C), and tubulointerstitial fibrosis score (D). (E) Concentration of NE in renal cortex. (F) Protein expression of Noxs in renal cortex. (G) Changes in SBP. (H) Changes in UAE. Data are expressed as the mean±SD of three independent experiments (n=6 in each group). *P<0.05 versus 5/6Nx rats given vehicle. Hyd, hydralazine; Los, losartan; PAS, periodic acid–Schiff; RDX, renal denervation; SDR, selective dorsal rhizotomy.
Discussion
This study uncovered two new pathways in 5/6Nx rats given high salt intake. The first is that a high salt intake paradoxically activates all of the major components of the intrarenal and cerebral RAS independent of the systemic RAS or the BP. The second is that a high-salt diet promotes renal inflammation, fibrosis, and proteinuria by coactivation of the central and renal ROS and RAS via a robust intercommunication provided by the renal afferent and efferent nerves. The operation of this strong coactivation of oxidative stress and RAS in the damaged kidney and brain by high salt intake and its interlinkage via the afferent and efferent renal sympathetic nerves in a positive feedback mode is illustrated in Figure 9.
Figure 9.

Schematic diagram summarizing coactivation by salt intake of oxidative stress and RAS in the damaged kidney and brain linked via the afferent and efferent renal sympathetic nerves in a positive feedback mode.
A low salt intake normally activates the RAS in the JGA of the kidney and the circulation, as confirmed in this study. This contributes to NaCl and BP homeostasis.36,37 Apparently, there is a counter-regulation in this model of CKD in which salt loading paradoxically enhances the intrarenal epithelial RAS, despite maintenance of a suppressed circulating RAS. This extends similar findings in rat models of salt-sensitive hypertension,26,38 nephritis,39 and reduced renal mass.9 Likewise, there is evidence that high salt intake accelerates the loss of renal function in humans with CKD.16,17 AGT excretion, which mirrors intrarenal AngII during oxidative stress, is increased by salt in patients with CKD.27 Remarkably, almost all of the necessary substrates and enzymes for the local generation of AngII, exception for renin, and the receptors on which it acts, were upregulated in the kidneys of 5/6Nx rats fed a high-salt diet. The increased intrarenal AngII with salt in 5/6Nx rat kidneys likely originates in large part from increased proximal tubular AGT and ACE.40 Indeed, AGT expression is associated with fibrotic injury and can be self-sustaining because AngII itself stimulates AGT expression.41 Renovascular hypertension42 and diabetes43,44 are other examples of increased renal RAS expression and renal AngII levels that are functionally linked to hypertension and progressive loss of kidney function.
An important new finding is that activation of RAS in a remnant kidney by high salt intake leads to renal fibrosis mediated by overexpression of the central RAS. These 5/6Nx rats fed a high-salt diet had increased generation of cerebral TH, which is the rate-limiting enzyme for NE synthesis, thereby indicating increased central sympathetic drive. These rats also had increased NE levels in the kidney, indicating increased renal efferent sympathetic nerve activity. Importantly, we found a significantly increased number of TH-positive neurons in the RVLM, which is the gateway for activation of the SNS, in high-salt–fed 5/6Nx rats. Activation of RVLM neurons has been reported in salt-sensitive animals,45 such as in our study, but not in AngII-induced hypertensive rats.46 This brain response may be secondary to ROS because AngII potently activates ROS in neurons regulating cardiovascular function.47 Indeed, the activation of the central sympathetic system during high salt intake clearly was dependent on the central ROS and RAS because their blockade with ICV tempol or losartan prevented the activation of the renal RAS or the increase in renal NE levels, and lessened the development of renal fibrosis and dysfunction with high salt. In turn, these changes in kidney structure and function were dependent on increased central sympathetic outflow because its blockade with ICV clonidine also suppressed salt-induced renal RAS activation and renal injury. However, because ICV clonidine failed to affect the brain RAS activity, we conclude that the activation of the brain RAS by dietary salt is upstream from, and is a driving force for, central sympathetic activation, which provides a link between activation of the brain RAS and ROS and enhanced renal injury. Similarly, an overexpression of the brain RAS in DSS rats contributes to their hypertension,48 whereas an inhibition of brain ROS and sympathoexcitation in salt-loaded uninephrectomized rats by tempol lessens hypertension.49 However, the SNS, rather than hypertension, linked the brain and renal RAS because normalization of BP with hydralazine failed to prevent these responses to salt in 5/6Nx rats. Consistent with the hypothesis, salt-induced renal fibrosis and dysfunction were improved significantly in rats after central blockade of RAS or sympathetic outflow, in contrast with those treated with hydralazine. These data imply that salt-induced activation of the central RAS and SNS, and the progressive renal fibrosis, are associated with but not mediated by hypertension, but are susceptible to blockade of RAS or the SNS.
An unanticipated new finding is that denervation of the remnant kidney not only lessens renal injury, likely by interrupting renal efferent sympathetic activity, but also inhibits the activation of the brain-renal RAS axis, likely by interrupting renal afferent sympathetic nerve activity. Indeed, dorsal rhizotomy, which interrupts afferent nerves specifically, was as effective as renal denervation, which implies an essential role for renal afferent sympathetic nerves in the central activation of the SNS and consequent renal damage. This is consistent with observations that activation of renal afferent nerves by ROS induces neurogenic hypertension in rats50 and that central or systemic administration of tempol lowers BP in many hypertensive models in part by reduction of SNS activity.51,52 Moreover, the kidney is required to sustain hypertension and peripheral vasoconstriction.53
Central activation of RAS, ROS, and SNS enhanced renal injury by >65% only in 5/6Nx rats fed a high-salt diet. This suggests that the reno-cerebral axis may be activated selectively in states of oxidative stress or inflammation. Such a positive feedback reflex mechanism whereby renal afferent nerves from damaged or inflamed kidneys activate the brain ROS, RAS, and efferent sympathetic drive that engages renal ROS and RAS and worsens renal damage may have considerable consequences for understanding the progressive nature of renal damage and hypertension in CKD and the detrimental effects of salt intake.16 Indeed, our finding that activation of the central ROS, RAS, and SNS and renal damage are independent of hypertension provides some insight into the failure of rigorous antihypertensive therapy to slow progression of established CKD, unless there is heavy proteinuria.54,55
The upregulation of intrarenal AGT in 5/6Nx rats by salt may determine the upregulation of intrarenal AngII because AGT can be limiting for AngII production within the kidney. Remarkably, although renal NADPH oxidase activity and ROS generation in normal rats are clearly enhanced by prolonged AngII, they also are enhanced by a high-salt diet that suppresses intrarenal AngII.56,57 This was attributed to selective effects of AngII and salt on expression of NADPH oxidase components and SOD isoforms. Thus, whereas AngII increased the renal mRNA expression of NOX-1, salt increased expression of NOX-2 (as in this study). Thus, a prolonged elevation of salt intake may increase renal ROS, which may increase sympathetic nerve activity, local AGT expression, and renal AngII production, all independent of BP and circulating renin.
This study has some limitations. The mechanism of renal nerve activation by high salt intake in 5/6Nx kidneys was not established. However, endothelin-1 activates renal afferent nerves during high salt intake in normal kidneys,58 and is implicated in progression of CKD.5 Therefore, endothelin-1 may be implicated but this was not investigated in this study. The mechanism whereby efferent renal nerve activity upregulates the renal epithelial RAS was not established. However, AGT expression, which is the limiting step for intrarenal AngII generation,26 is increased by NE59 and by oxidative stress.60 This provides potential pathways by which increased efferent renal nerve activity could stimulate intrarenal AGT, and thereby stimulate intrarenal AngII generation in a state of oxidative stress such as CKD. In addition, we did not measure the concentrations of ICV infused agents in the blood or urine, and thus cannot exclude some spillover effects mediated via the systemic circulation. However, the ICV dosages of losartan (1 mg/kg per day) or tempol (4.5 μg/kg/d) were >500-fold lower than the IG doses and were chosen from preliminary studies in which intravenous injection of these doses had no detectable effects on AngII-induced increase in BP or renal NE levels. Finally, the SBP was measured under anesthesia. However, the values were similar to telemetric measurement in 5/6Nx rats.35
In summary, this study demonstrates that salt increases renal fibrosis in a model of CKD in part by activation of a reno-cerebral RAS axis interlinked by afferent and efferent sympathetic nerves. This identifies a new regulatory mechanism underlying renal damage in response to salt loading, and thereby provides insights into potential new strategies for prevention and management not only of hypertension but also of the progression of CKD by use of sympatholytic agents or interventions to interrupt renal nerve traffic.
Concise Methods
Animals
Five-week-old male Sprague–Dawley rats (Nanfang Hospital Animal Experiment Center) were maintained in a pathogen-free facility under controlled temperature (24°C±2°C) and humidity (55%±5%), with a 12-hour light/dark cycle. All animal experiments were approved by the Animal Ethics Committee of Nanfang Hospital.
Treatments
Protocol 1
Five-sixths nephrectomy or sham operation was performed at 6 weeks of age as previously described.61 After operation, all rats received a normal-salt diet for 8 weeks, and were then randomly assigned to three groups (n=6 per group) receiving a low-salt (0.02% NaCl), normal-salt (0.4% NaCl), or high-salt (4% NaCl) diet (Trophic Animal Feed High-Tech Co, Ltd) for 2 weeks.
Protocol 2
The 5/6Nx rats were fed with a high-salt (4% NaCl) diet for 2 weeks and were randomly assigned to 12 groups matched for body weight and serum creatinine level (n=6 in each group). They received the following treatments for 2 weeks: (1) IG vehicle (PBS, pH 7.4) or losartan (Sigma-Aldrich) at 1, 50, or 500 mg/kg per day (groups 1–4) as previously used in this model62; (2) ICV vehicle (artificial cerebrospinal fluid) or losartan at 1 mg/kg per day using an Alzet osmotic minipump (Durect Corp.)48 (groups 5 and 6); (3) ICV clonidine (Sigma-Aldrich) at 5.76 μg/kg per day using an osmotic minipump (group 7); (4) renal denervation as previously described63 (group 8); (5) selective dorsal rhizotomy as previously described64 (group 9); (6) ICV tempol at (4.5 μg/kg per day) using an osmotic minipump (group 10); (7) IG tempol at 30 mg/kg per day (group 11); and (8) IG hydralazine (Sigma-Aldrich) at 15 mg/kg per day (group 12).
Measurement of SBP and Renal Function
PE-50 catheters were inserted into the femoral artery under isoflurane anesthesia to measure SBP using a pressure transducer (Gould) connected to a physiologic recorder (Gilson Medical Electronics).61
Serum creatinine and sodium concentrations were measured with an automated chemistry analyzer (AU480; Beckman Coulter) and urinary albumin was measured with an ELISA kit (IMTEC Diagnostics).
Histologic Evaluation
Evaluation of Renal Fibrosis
Histologic Analyses.
The kidneys were dissected and processed for periodic acid–Schiff and Masson’s trichrome staining as previously described.61
Immunohistochemistry Analyses.
Five-micrometer-thick sections were processed10 using anti-rat fibronectin (1:200; Sigma-Aldrich) and anti-rat collagen I (1:100; EMD Millipore). Intrarenal expression of fibronectin and collagen I was semiquantitated as previously described.65
Western Blot Analyses.
The protein levels of fibronectin and collagen I in homogenates of renal cortex were determined as described.66
Real-Time PCR.
Renal mRNA levels of fibronectin and collagen I were analyzed by real-time RT-PCR.67
Evaluation of Renal Macrophage Infiltration
Kidneys were stained with a mAb (1:50) specifically recognizing macrophage marker ED-1 (Bio-Rad). Macrophage infiltration was quantified by counting the ED-1–positive cells in 20 randomly chosen glomerular profiles and in 20 (0.3×0.3 mm2) tubulointerstitial areas.
Evaluation of Cerebral Neuronal Activation
This was assessed in forebrain nuclei by using c-fos expression as a functional marker as described with modification.36 All histologic analyses were performed by two pathologists who were blinded to the treatment of the animals.
RAS Expression and Activity
Expression of RAS in Kidneys
Immunohistochemistry Analyses.
Five-micrometer-thick sections were processed10 using anti-rat AGT (1:200; IBL), anti-rat ACE-1 (1:100; Santa Cruz Biotechnology), anti-AngII (1:800; Peninsula Laboratories), anti-rat AT1 receptors (1:100; EMD Millipore), and anti-rat renin (1:50; Santa Cruz Biotechnology) antibodies and intrarenal expression was semiquantitated as described above. Intrarenal renin was assessed as previously described.68
Western Blot Analyses.
The levels of RAS components in homogenates of renal cortex were determined as previously described66 using anti-AGT (IBL), anti-ACE-1 (Santa Cruz Biotechnology), anti-AT1 receptors (EMD Millipore), and anti-β-actin (Cell Signaling Technology) antibodies.
mRNA Expression of RAS.
Renal mRNA levels of AGT, ACE-1, and AT1 receptors were analyzed by RT-PCR as previously described10 and were normalized to glyceraldehyde-3-phosphate dehydrogenase.
Localization of Intrarenal RAS.
Five-micrometer paraffin-embedded kidney sections were double stained using first primary antibodies against AGT (1:100; IBL), ACE-1 (1:100; Santa Cruz Biotechnology), AngII (1:800; Peninsula Laboratories), and AT1 receptors (1:200; EMD Millipore), separately, and second primary antibodies against the markers of tubular epithelial cells as follows: anti-aquaporin 1 (a marker of proximal tubular epithelial cells, 1:100; Abcam, Inc.), anti-Tamm–Horsfall protein (a marker of thick ascending limbs of Henle, 1:100; Santa Cruz Biotechnology), anti-thiazide-sensitive NaCl cotransporter (a marker of distal convoluted tubular epithelial cells, 1:500; EMD Millipore), anti-aquaporin 2 (a marker of collecting duct epithelial cells, 1:100; Novus) antibody, separately.
Activity of RAS in Kidney and Circulation
Level of RAS Components.
Plasma levels of AGT were assessed with an ELISA kit (IBL). Concentrations of AngII in plasma and renal cortex homogenates were assessed by a competitive ELISA (Peninsula Laboratories). Urinary AGT was assessed by ELISA (IBL).
Activity of Renin and ACE-1.
Plasma renin activity was determined by a RIA kit (SRL). Serum and renal cortex homogenates for ACE-1 activity were analyzed by a fluorometric method and normalized for protein content determined by a BCA Protein Assay Kit (Pierce).69,70
Expression of RAS in Brain
Immunohistochemistry and Real-Time RT-PCR.
The cerebral expression of AngII and AT1 receptors was quantitated as described for c-fos analysis. AT1 receptor expression was also analyzed with RT-PCR in homogenates of forebrain nuclei.
Localization of Brain RAS.
Cerebral localization of AngII and AT1 receptors was determined by double-staining immunofluorescence,65 using anti-AngII or anti-AT1 receptors as the first primary antibody, and anti-glial fibrillary acidic protein (EMD Millipore) or anti-neuron-specific enolase (EMD Millipore) as the second primary antibody.
Evaluation of Sympathetic Activity
NE Concentrations
Concentrations of NE in plasma and renal cortex homogenates were assessed with an ELISA kit (ALPCO Diagnostics)63 according to the manufacturer’s protocol.
Expression of TH in Brain
Expression of TH in forebrain nuclei was analyzed by using Western blot.36 Samples frozen in liquid nitrogen were cut into 200-μm coronal sections and specific forebrain nuclei were obtained as described above. Expression of TH in nuclei homogenates was determined by Western blot using an anti-TH antibody (Abcam, Inc.).36
The number of c-fos–positive and TH-expressing neurons in the RVLM was determined as previously described.71 Briefly, brain stem sections were double stained with antibodies against TH (EMD Millipore) and c-fos (Santa Cruz Biotechnology) and the number of labeled TH and c-fos immunopositive neurons located within the defined borders of the RVLM was assessed as delineated in the atlas of Paxinos and Watson.
Evaluation of Oxidative Stress
Expression of NADPH Oxidase Subunits in Renal Cortex
The levels of NADPH oxidase subunits Nox2 and Nox4 in homogenates of renal cortex were determined66 using anti-Nox2 and anti-Nox4 antibodies (Santa Cruz Biotechnology).
Expression of NADPH Oxidase Subunits in Brain
Brain samples frozen in liquid nitrogen were cut into 200-μm coronal sections and specific forebrain nuclei were obtained as described above. Expression of Nox2 and Nox4 was determined by Western blot using anti-Nox2 and anti-Nox4 antibodies (Santa Cruz Biotechnology).36
Statistical Analyses
All data are expressed as the mean±SD of three independent experiments. Continuous variables between groups were compared using one-way ANOVA, followed by the least significant difference test. Statistical analyses were conducted with SPSS 17.0 for Windows (SPSS). A value of P<0.05 was considered statistically significant.
Disclosures
None.
Supplementary Material
Acknowledgments
F.F.H. was supported by grants from the Major State Basic Research Development Program of China (National 973 Program; 2012CB517703) and National Natural Science Foundation of China (81430016). W.C. was supported by a National Nature and Science Young Investigator Grant (81200502). A.L. was supported by a National Nature and Science Grant (81270825 and 31201751). C.S.W. was supported by grants from the National Institutes of Health (HL-68686, DK-049870, and DK-036079) and funds from the George E. Schreiner Chair of Nephrology and the Georgetown University Center for Hypertension, Kidney, and Vascular Research.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2014050518/-/DCSupplemental.
References
- 1.Zeisberg M, Neilson EG: Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol 21: 1819–1834, 2010 [DOI] [PubMed] [Google Scholar]
- 2.Nangaku M: Mechanisms of tubulointerstitial injury in the kidney: Final common pathways to end-stage renal failure. Intern Med 43: 9–17, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Wynn TA: Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol 4: 583–594, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eddy AA, Neilson EG: Chronic kidney disease progression. J Am Soc Nephrol 17: 2964–2966, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Speed JS, Pollock DM: Endothelin, kidney disease, and hypertension. Hypertension 61: 1142–1145, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ljutić D, Kes P: The role of arterial hypertension in the progression of non-diabetic glomerular diseases. Nephrol Dial Transplant 18[Suppl 5]: v28–v30, 2003 [DOI] [PubMed] [Google Scholar]
- 7.Schlaich MP, Socratous F, Hennebry S, Eikelis N, Lambert EA, Straznicky N, Esler MD, Lambert GW: Sympathetic activation in chronic renal failure. J Am Soc Nephrol 20: 933–939, 2009 [DOI] [PubMed] [Google Scholar]
- 8.DiBona GF: Sympathetic nervous system and hypertension. Hypertension 61: 556–560, 2013 [DOI] [PubMed] [Google Scholar]
- 9.Lai EY, Luo Z, Onozato ML, Rudolph EH, Solis G, Jose PA, Wellstein A, Aslam S, Quinn MT, Griendling K, Le T, Li P, Palm F, Welch WJ, Wilcox CS: Effects of the antioxidant drug tempol on renal oxygenation in mice with reduced renal mass. Am J Physiol Renal Physiol 303: F64–F74, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao W, Xu J, Zhou ZM, Wang GB, Hou FF, Nie J: Advanced oxidation protein products activate intrarenal renin-angiotensin system via a CD36-mediated, redox-dependent pathway. Antioxid Redox Signal 18: 19–35, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campese VM, Ye S, Zhong H: Downregulation of neuronal nitric oxide synthase and interleukin-1beta mediates angiotensin II-dependent stimulation of sympathetic nerve activity. Hypertension 39: 519–524, 2002 [DOI] [PubMed] [Google Scholar]
- 12.Crowley SD, Coffman TM: Recent advances involving the renin-angiotensin system. Exp Cell Res 318: 1049–1056, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kotchen TA, Cowley AW, Jr, Frohlich ED: Salt in health and disease—a delicate balance. N Engl J Med 368: 1229–1237, 2013 [DOI] [PubMed] [Google Scholar]
- 14.Lambers Heerspink HJ, Navis G, Ritz E: Salt intake in kidney disease—a missed therapeutic opportunity? Nephrol Dial Transplant 27: 3435–3442, 2012 [DOI] [PubMed] [Google Scholar]
- 15.Krikken JA, Laverman GD, Navis G: Benefits of dietary sodium restriction in the management of chronic kidney disease. Curr Opin Nephrol Hypertens 18: 531–538, 2009 [DOI] [PubMed] [Google Scholar]
- 16.Lipkowitz MS, Wilcox CS: What level of dietary salt intake worsens renal outcomes? Am J Hypertens 27: 1243–1244, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weir MR, Fink JC: Salt intake and progression of chronic kidney disease: An overlooked modifiable exposure? A commentary. Am J Kidney Dis 45: 176–188, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Johnson RJ, Herrera-Acosta J, Schreiner GF, Rodriguez-Iturbe B: Subtle acquired renal injury as a mechanism of salt-sensitive hypertension. N Engl J Med 346: 913–923, 2002 [DOI] [PubMed] [Google Scholar]
- 19.Weir MR, Dengel DR, Behrens MT, Goldberg AP: Salt-induced increases in systolic blood pressure affect renal hemodynamics and proteinuria. Hypertension 25: 1339–1344, 1995 [DOI] [PubMed] [Google Scholar]
- 20.Vogt L, Waanders F, Boomsma F, de Zeeuw D, Navis G: Effects of dietary sodium and hydrochlorothiazide on the antiproteinuric efficacy of losartan. J Am Soc Nephrol 19: 999–1007, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bigazzi R, Bianchi S, Baldari D, Sgherri G, Baldari G, Campese VM: Microalbuminuria in salt-sensitive patients. A marker for renal and cardiovascular risk factors. Hypertension 23: 195–199, 1994 [DOI] [PubMed] [Google Scholar]
- 22.Swift PA, Markandu ND, Sagnella GA, He FJ, MacGregor GA: Modest salt reduction reduces blood pressure and urine protein excretion in black hypertensives: A randomized control trial. Hypertension 46: 308–312, 2005 [DOI] [PubMed] [Google Scholar]
- 23.Coffman TM: Under pressure: The search for the essential mechanisms of hypertension. Nat Med 17: 1402–1409, 2011 [DOI] [PubMed] [Google Scholar]
- 24.Kolmakova EV, Haller ST, Kennedy DJ, Isachkina AN, Budny GV, Frolova EV, Piecha G, Nikitina ER, Malhotra D, Fedorova OV, Shapiro JI, Bagrov AY: Endogenous cardiotonic steroids in chronic renal failure. Nephrol Dial Transplant 26: 2912–2919, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanders PW: Vascular consequences of dietary salt intake. Am J Physiol Renal Physiol 297: F237–F243, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobori H, Nishiyama A, Abe Y, Navar LG: Enhancement of intrarenal angiotensinogen in Dahl salt-sensitive rats on high salt diet. Hypertension 41: 592–597, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konishi Y, Nishiyama A, Morikawa T, Kitabayashi C, Shibata M, Hamada M, Kishida M, Hitomi H, Kiyomoto H, Miyashita T, Mori N, Urushihara M, Kobori H, Imanishi M: Relationship between urinary angiotensinogen and salt sensitivity of blood pressure in patients with IgA nephropathy. Hypertension 58: 205–211, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Velez JC: The importance of the intrarenal renin-angiotensin system. Nat Clin Pract Nephrol 5: 89–100, 2009 [DOI] [PubMed] [Google Scholar]
- 29.Veerasingham SJ, Raizada MK: Brain renin-angiotensin system dysfunction in hypertension: Recent advances and perspectives. Br J Pharmacol 139: 191–202, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hutchinson JS, Mendelsohn FA: Hypotensive effects of captopril administered centrally in intact conscious spontaneously hypertensive rats and peripherally in anephric anaesthetized spontaneously hypertensive rats. Clin Exp Pharmacol Physiol 7: 555–558, 1980 [DOI] [PubMed] [Google Scholar]
- 31.Campese VM: A new model of neurogenic hypertension caused by renal injury: Pathophysiology and therapeutic implications. Clin Exp Nephrol 7: 167–171, 2003 [DOI] [PubMed] [Google Scholar]
- 32.Ye S, Zhong H, Duong VN, Campese VM: Losartan reduces central and peripheral sympathetic nerve activity in a rat model of neurogenic hypertension. Hypertension 39: 1101–1106, 2002 [DOI] [PubMed] [Google Scholar]
- 33.Kobori H, Nangaku M, Navar LG, Nishiyama A: The intrarenal renin-angiotensin system: From physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 59: 251–287, 2007 [DOI] [PubMed] [Google Scholar]
- 34.McKinley MJ, Albiston AL, Allen AM, Mathai ML, May CN, McAllen RM, Oldfield BJ, Mendelsohn FA, Chai SY: The brain renin-angiotensin system: Location and physiological roles. Int J Biochem Cell Biol 35: 901–918, 2003 [DOI] [PubMed] [Google Scholar]
- 35.Wilcox CS, Pearlman A: Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacol Rev 60: 418–469, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jo H, Yang EK, Lee WJ, Park KY, Kim HJ, Park JS: Gene expression of central and peripheral renin-angiotensin system components upon dietary sodium intake in rats. Regul Pept 67: 115–121, 1996 [DOI] [PubMed] [Google Scholar]
- 37.Hettinger U, Lukasova M, Lewicka S, Hilgenfeldt U: Regulatory effects of salt diet on renal renin-angiotensin-aldosterone, and kallikrein-kinin systems. Int Immunopharmacol 2: 1975–1980, 2002 [DOI] [PubMed] [Google Scholar]
- 38.Chandramohan G, Bai Y, Norris K, Rodriguez-Iturbe B, Vaziri ND: Effects of dietary salt on intrarenal angiotensin system, NAD(P)H oxidase, COX-2, MCP-1 and PAI-1 expressions and NF-kappaB activity in salt-sensitive and -resistant rat kidneys. Am J Nephrol 28: 158–167, 2008 [DOI] [PubMed] [Google Scholar]
- 39.Suzuki H, Yamamoto T, Ikegaya N, Hishida A: Dietary salt intake modulates progression of antithymocyte serum nephritis through alteration of glomerular angiotensin II receptor expression. Am J Physiol Renal Physiol 286: F267–F277, 2004 [DOI] [PubMed] [Google Scholar]
- 40.Kobori H, Ozawa Y, Suzaki Y, Prieto-Carrasquero MC, Nishiyama A, Shoji T, Cohen EP, Navar LG: Young Scholars Award Lecture: Intratubular angiotensinogen in hypertension and kidney diseases. Am J Hypertens 19: 541–550, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schunkert H, Ingelfinger JR, Jacob H, Jackson B, Bouyounes B, Dzau VJ: Reciprocal feedback regulation of kidney angiotensinogen and renin mRNA expressions by angiotensin II. Am J Physiol 263: E863–E869, 1992 [DOI] [PubMed] [Google Scholar]
- 42.Navar LG, Von Thun AM, Zou L, el-Dahr SS, Mitchell KD: Enhancement of intrarenal angiotensin II levels in 2 kidney 1 clip and angiotensin II induced hypertension. Blood Press Suppl 2: 88–92, 1995 [PubMed] [Google Scholar]
- 43.Singh R, Singh AK, Leehey DJ: A novel mechanism for angiotensin II formation in streptozotocin-diabetic rat glomeruli. Am J Physiol Renal Physiol 288: F1183–F1190, 2005 [DOI] [PubMed] [Google Scholar]
- 44.Onozato ML, Tojo A, Goto A, Fujita T, Wilcox CS: Oxidative stress and nitric oxide synthase in rat diabetic nephropathy: Effects of ACEI and ARB. Kidney Int 61: 186–194, 2002 [DOI] [PubMed] [Google Scholar]
- 45.Ito S, Hiratsuka M, Komatsu K, Tsukamoto K, Kanmatsuse K, Sved AF: Ventrolateral medulla AT1 receptors support arterial pressure in Dahl salt-sensitive rats. Hypertension 41: 744–750, 2003 [DOI] [PubMed] [Google Scholar]
- 46.Pedrino GR, Calderon AS, Andrade MA, Cravo SL, Toney GM: Discharge of RVLM vasomotor neurons is not increased in anesthetized angiotensin II-salt hypertensive rats. Am J Physiol Heart Circ Physiol 305: H1781–H1789, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lob HE, Schultz D, Marvar PJ, Davisson RL, Harrison DG: Role of the NADPH oxidases in the subfornical organ in angiotensin II-induced hypertension. Hypertension 61: 382–387, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang BS, Leenen FH: Both brain angiotensin II and “ouabain” contribute to sympathoexcitation and hypertension in Dahl S rats on high salt intake. Hypertension 32: 1028–1033, 1998 [DOI] [PubMed] [Google Scholar]
- 49.Fujita M, Ando K, Kawarazaki H, Kawarasaki C, Muraoka K, Ohtsu H, Shimizu H, Fujita T: Sympathoexcitation by brain oxidative stress mediates arterial pressure elevation in salt-induced chronic kidney disease. Hypertension 59: 105–112, 2012 [DOI] [PubMed] [Google Scholar]
- 50.Ye S, Zhong H, Yanamadala S, Campese VM: Oxidative stress mediates the stimulation of sympathetic nerve activity in the phenol renal injury model of hypertension. Hypertension 48: 309–315, 2006 [DOI] [PubMed] [Google Scholar]
- 51.Xu H, Fink GD, Chen A, Watts S, Galligan JJ: Nitric oxide-independent effects of tempol on sympathetic nerve activity and blood pressure in normotensive rats. Am J Physiol Heart Circ Physiol 281: H975–H980, 2001 [DOI] [PubMed] [Google Scholar]
- 52.Patel K, Chen Y, Dennehy K, Blau J, Connors S, Mendonca M, Tarpey M, Krishna M, Mitchell JB, Welch WJ, Wilcox CS: Acute antihypertensive action of nitroxides in the spontaneously hypertensive rat. Am J Physiol Regul Integr Comp Physiol 290: R37–R43, 2006 [DOI] [PubMed] [Google Scholar]
- 53.Converse RL, Jr, Jacobsen TN, Toto RD, Jost CM, Cosentino F, Fouad-Tarazi F, Victor RG: Sympathetic overactivity in patients with chronic renal failure. N Engl J Med 327: 1912–1918, 1992 [DOI] [PubMed] [Google Scholar]
- 54.Klahr S, Levey AS, Beck GJ, Caggiula AW, Hunsicker L, Kusek JW, Striker G, Modification of Diet in Renal Disease Study Group : The effects of dietary protein restriction and blood-pressure control on the progression of chronic renal disease. N Engl J Med 330: 877–884, 1994 [DOI] [PubMed] [Google Scholar]
- 55.Wright JT, Jr, Bakris G, Greene T, Agodoa LY, Appel LJ, Charleston J, Cheek D, Douglas-Baltimore JG, Gassman J, Glassock R, Hebert L, Jamerson K, Lewis J, Phillips RA, Toto RD, Middleton JP, Rostand SG, African American Study of Kidney Disease and Hypertension Study Group : Effect of blood pressure lowering and antihypertensive drug class on progression of hypertensive kidney disease: Results from the AASK trial. JAMA 288: 2421–2431, 2002 [DOI] [PubMed] [Google Scholar]
- 56.Chabrashvili T, Kitiyakara C, Blau J, Karber A, Aslam S, Welch WJ, Wilcox CS: Effects of ANG II type 1 and 2 receptors on oxidative stress, renal NADPH oxidase, and SOD expression. Am J Physiol Regul Integr Comp Physiol 285: R117–R124, 2003 [DOI] [PubMed] [Google Scholar]
- 57.Kitiyakara C, Chabrashvili T, Chen Y, Blau J, Karber A, Aslam S, Welch WJ, Wilcox CS: Salt intake, oxidative stress, and renal expression of NADPH oxidase and superoxide dismutase. J Am Soc Nephrol 14: 2775–2782, 2003 [DOI] [PubMed] [Google Scholar]
- 58.Kopp UC, Cicha MZ, Smith LA: Differential effects of endothelin on activation of renal mechanosensory nerves: Stimulatory in high-sodium diet and inhibitory in low-sodium diet. Am J Physiol Regul Integr Comp Physiol 291: R1545–R1556, 2006 [DOI] [PubMed] [Google Scholar]
- 59.Wang TT, Wu XH, Zhang SL, Chan JS: Molecular mechanism(s) of action of norepinephrine on the expression of the angiotensinogen gene in opossum kidney cells. Kidney Int 54: 785–795, 1998 [DOI] [PubMed] [Google Scholar]
- 60.Kamiyama M, Urushihara M, Morikawa T, Konishi Y, Imanishi M, Nishiyama A, Kobori H: Oxidative stress/angiotensinogen/renin-angiotensin system axis in patients with diabetic nephropathy. Int J Mol Sci 14: 23045–23062, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li HY, Hou FF, Zhang X, Chen PY, Liu SX, Feng JX, Liu ZQ, Shan YX, Wang GB, Zhou ZM, Tian JW, Xie D: Advanced oxidation protein products accelerate renal fibrosis in a remnant kidney model. J Am Soc Nephrol 18: 528–538, 2007 [DOI] [PubMed] [Google Scholar]
- 62.Fujihara CK, Velho M, Malheiros DM, Zatz R: An extremely high dose of losartan affords superior renoprotection in the remnant model. Kidney Int 67: 1913–1924, 2005 [DOI] [PubMed] [Google Scholar]
- 63.Veelken R, Vogel EM, Hilgers K, Amann K, Hartner A, Sass G, Neuhuber W, Tiegs G: Autonomic renal denervation ameliorates experimental glomerulonephritis. J Am Soc Nephrol 19: 1371–1378, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ye S, Ozgur B, Campese VM: Renal afferent impulses, the posterior hypothalamus, and hypertension in rats with chronic renal failure. Kidney Int 51: 722–727, 1997 [DOI] [PubMed] [Google Scholar]
- 65.Xavier LL, Viola GG, Ferraz AC, Da Cunha C, Deonizio JM, Netto CA, Achaval M: A simple and fast densitometric method for the analysis of tyrosine hydroxylase immunoreactivity in the substantia nigra pars compacta and in the ventral tegmental area. Brain Res Brain Res Protoc 16: 58–64, 2005 [DOI] [PubMed] [Google Scholar]
- 66.Cao W, Zhou QG, Nie J, Wang GB, Liu Y, Zhou ZM, Hou FF: Albumin overload activates intrarenal renin-angiotensin system through protein kinase C and NADPH oxidase-dependent pathway. J Hypertens 29: 1411–1421, 2011 [DOI] [PubMed] [Google Scholar]
- 67.Xiao HB, Liu RH, Ling GH, Xiao L, Xia YC, Liu FY, Li J, Liu YH, Chen QK, Lv JL, Zhan M, Yang SK, Kanwar YS, Sun L: HSP47 regulates ECM accumulation in renal proximal tubular cells induced by TGF-β1 through ERK1/2 and JNK MAPK pathways. Am J Physiol Renal Physiol 303: F757–F765, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pupilli C, Chevalier RL, Carey RM, Gomez RA: Distribution and content of renin and renin mRNA in remnant kidney of adult rat. Am J Physiol 263: F731–F738, 1992 [DOI] [PubMed] [Google Scholar]
- 69.Friedland J, Silverstein E: A sensitive fluorimetric assay for serum angiotensin-converting enzyme. Am J Clin Pathol 66: 416–424, 1976 [DOI] [PubMed] [Google Scholar]
- 70.Fabris B, Candido R, Carraro M, Fior F, Artero M, Zennaro C, Cattin MR, Fiorotto A, Bortoletto M, Millevoi C, Bardelli M, Faccini L, Carretta R: Modulation of incipient glomerular lesions in experimental diabetic nephropathy by hypotensive and subhypotensive dosages of an ACE inhibitor. Diabetes 50: 2619–2624, 2001 [DOI] [PubMed] [Google Scholar]
- 71.Yao ST, May CN: Intra-carotid angiotensin II activates tyrosine hydroxylase-expressing rostral ventrolateral medulla neurons following blood-brain barrier disruption in rats. Neuroscience 245: 148–156, 2013 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.