

Abstract
Upon activation by with-no-lysine kinases, STE20/SPS1-related proline–alanine-rich protein kinase (SPAK) phosphorylates and activates SLC12A transporters such as the Na+-Cl− cotransporter (NCC) and Na+-K+-2Cl− cotransporter type 1 (NKCC1) and type 2 (NKCC2); these transporters have important roles in regulating BP through NaCl reabsorption and vasoconstriction. SPAK knockout mice are viable and display hypotension with decreased activity (phosphorylation) of NCC and NKCC1 in the kidneys and aorta, respectively. Therefore, agents that inhibit SPAK activity could be a new class of antihypertensive drugs with dual actions (i.e., NaCl diuresis and vasodilation). In this study, we developed a new ELISA-based screening system to find novel SPAK inhibitors and screened >20,000 small-molecule compounds. Furthermore, we used a drug repositioning strategy to identify existing drugs that inhibit SPAK activity. As a result, we discovered one small-molecule compound (Stock 1S-14279) and an antiparasitic agent (Closantel) that inhibited SPAK-regulated phosphorylation and activation of NCC and NKCC1 in vitro and in mice. Notably, these compounds had structural similarity and inhibited SPAK in an ATP-insensitive manner. We propose that the two compounds found in this study may have great potential as novel antihypertensive drugs.
Keywords: cell physiology, transport physiology, cell signaling, hypertension
Hypertension is one of the most important health problems worldwide because it may cause heart attack and stroke, which occur frequently at present. New insight into the mechanisms of BP regulation was recently provided by studies on monogenic hypertensive diseases such as Liddle’s syndrome and pseudohypoaldosteronism type II (PHAII).1,2 PHAII is an autosomal dominant disease characterized by hypertension, hyperkalemia, and metabolic acidosis. With-no-lysine kinase WNK1 and WNK4 mutations were identified as the genes responsible for PHAII.3 We previously generated PHAII model mice (the Wnk4D561A/+ knock-in mouse) that carried the same mutation as patients with PHAII and discovered that the constitutive activation of the WNK-Oxidative stress-responsive 1 (OSR1) and STE20/SPS1-related proline–alanine-rich protein kinase (SPAK)-NaCl cotransporter (NCC) signal cascade is the major pathogenic mechanism of PHAII.4,5 The increased phosphorylation and activation of NCC induces excessive NaCl reabsorption in the kidney and causes salt-sensitive hypertension. OSR1/SPAK kinases phosphorylate and activate not only NCC but also other Slc12a transporters such as Na+-K+-2Cl− cotransporter type 1 (NKCC1) and type 2 (NKCC2).6 Recent studies demonstrated the notion that WNK-OSR1/SPAK-NKCC1 phosphorylation and activation in vascular smooth muscle cells can play a pivotal role in the regulation of vascular tonus.7–10 Moreover, WNK signaling is positively controlled by aldosterone, angiotensin II, and insulin, which may contribute to hypertension in patients with hyperaldosteronism and hyperinsulinemia.11–15 Therefore, drugs that inhibit this signal cascade are expected to treat hypertension through dual actions (i.e., NaCl diuresis and vasodilation), and may be particularly effective in patients with hyperaldosteronism or hyperinsulinemia. In this study, we focused on the SPAK kinase because SPAK knockout mice were not fatal and displayed hypotension with low NCC and NKCC1 phosphorylation in mouse kidney and aorta, respectively.16,17 The purposes of this study were to develop a new high-throughput screening system using ELISA and to discover novel SPAK inhibitors from libraries of small-molecule compounds and existing drugs.
Results
Development of an ELISA System for the Detection of SPAK-Regulated NKCC2 Phosphorylation
To find novel inhibitors of the SPAK kinase, we developed a new screening system using ELISA. Previous studies have shown that SPAK possessed very low kinase activity in vitro, which hampered a high-throughput screening of the inhibitors.18 In this study, we adopted a combination strategy that included the recombinant SPAK [T233E] protein, in which the T-loop Thr residue phosphorylated by WNK isoforms was mutated to Glu to mimic phosphorylation, and the Mouse protein 25α (MO25α) to detect the SPAK-regulated phosphorylation more sensitively.18–20 MO25α is an enhancer of SPAK kinase. We used a fragment of human NKCC2 (residues 1–174) including SPAK phosphorylation sites as a substrate for SPAK because NKCC2 phosphorylation has been known to be the most detectable during in vitro experiments.18 They were all prepared as glutathione S-transferase (GST) fusion proteins and purified.
First, to determine whether the kinase reaction functions properly in vitro, the GST-NKCC2 phosphorylation reaction was confirmed by the immunoblotting of three different anti-phospho-NKCC2 antibodies. As shown in Figure 1, the most dramatic increase in ATP-dependent NKCC2 phosphorylation was observed in the presence of GST-MO25α. Next, we tested the utility of these anti-phospho-NKCC2 antibodies in the ELISA system. First, the ELISA plates were coated with 5 pmol of GST-NKCC2 [1–174] per well. After this, the kinase reaction was induced on the ELISA plate using GST-SPAK [T233E] with GST-MO25α in the presence of ATP. Finally, the phosphorylation of GST-NKCC2 was detected with each anti-phospho-NKCC2 antibody. As shown in Figure 2, two of the three anti-phospho-NKCC2 antibodies, pT2 and pNKCC2 (pThr100/105), succeeded in detecting NKCC2 phosphorylation. Finally, we adopted the anti-phospho-NKCC2 (pThr100/105) antibody as a primary antibody. To determine the dose-dependent kinetics, we incubated 0.5 pmol of GST-SPAK [T233E] in the presence of different concentrations of substrate, GST-NKCC2, and ATP. In vitro GST-NKCC2 phosphorylation increased according to the amount of coated GST-NKCC2 (Supplemental Figure 1A) and ATP concentrations (Supplemental Figure 1B). On the basis of these results, we determined that the optimum amounts of GST-NKCC2 and ATP were 5 pmol/well and 0.1 mM, respectively, in this screening.
Figure 1.
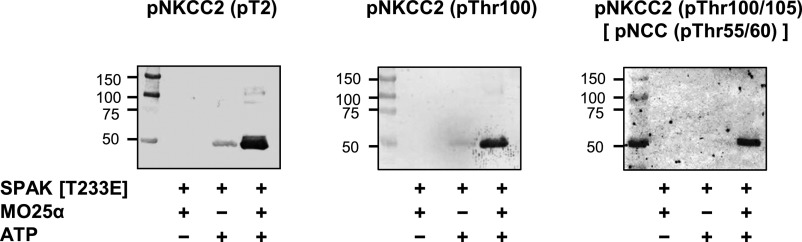
Confirmation of the in vitro phosphorylation reaction of GST-NKCC2 using three different anti-phospho-NKCC2 antibodies. GST-NKCC2 is incubated with GST-SPAK [T233E] in the absence or presence of MO25α. Subsequently, the GST-NKCC2 phosphorylation reaction is confirmed by the immunoblotting by using three different anti-phospho-NKCC2 antibodies: anti-pNKCC2 (pT2), pNKCC2 (pThr100), and pNKCC2 (pThr100/105)[pNCC (pThr50/55)]. The most dramatic increase in ATP-dependent NKCC2 phosphorylation is observed in the presence of GST-MO25α.
Figure 2.

Kinase assay and detection of phosphorylated NKCC2 in the ELISA system. GST-NKCC2 [1–174] is coated to each well of the ELISA plate and incubated with GST-SPAK [T233E] in the presence of GST-MO25α. Anti-p-NKCC2 (pT2) and p-NKCC2 (pThr100/105) [p-NCC (pTh50/55)] antibodies succeed in detecting NKCC2 phosphorylation. Anti-p-NKCC2 (pThr100) antibody is not appropriate for ELISA, because a nonspecific blue color change also develops in negative control wells.
ELISA Screening of a Small-Molecule Compound Library for SPAK Inhibitors
We applied this newly developed indirect ELISA system for screening approximately 20,000 small-molecule compounds owned by the Tokyo Medical and Dental University Chemical Biology Screening Center. We actually performed the screening by adding each compound at 50 μM into the kinase reaction buffer and obtained the signals at 620 nm. Representative results of the initial screening are shown in Figure 3. As a result of the initial screening of 21,727 compounds, we found 757 positive compounds. In the secondary assay of these positive compounds, the inhibitory effects were almost the same as those observed in the primary assay (reproducibility was >80%), indicating that our system was solid (Supplemental Figure 2). Subsequently, we carefully narrowed down the candidates by gradually decreasing the final concentrations of the compounds (50–10 μM, data not shown). Finally, we discovered Stock 1S-14279 (1S-14279; PubChem CID 01676700) that showed the highest SPAK inhibitory effect. Figure 4A shows the chemical structure of 1S-14279. The median inhibitory concentration (IC50) value of 1S-14279 for SPAK was 0.26 μM (Figure 4B).
Figure 3.
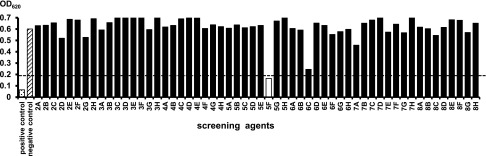
Representative figure of primary screening of SPAK inhibitors in the newly developed ELISA system. Each compound is added in the kinase reaction at a final concentration of 50 μM. As a positive control indicating nonphosphorylation of NKCC2, ATP-free kinase buffer is used. A kinase reaction without any compounds is used as a negative control for screening. The arbitrary cut-off value of OD620 is set to 0.2. The compound shown as a white bar is regarded as a positive compound having a strong inhibitory effect on SPAK-regulated NKCC2 phosphorylation.
Figure 4.

A hit compound obtained from the small-molecule compound library. (A) Chemical structure and substance name of a hit compound. (B) Confirmation of the inhibitory effect of 1S-14279 in the ELISA system. 1S-14279 is added at various concentrations (0.005–50 μM). The IC50 value of 1S-14279 is 0.26 μM (n=3, mean±SEM). (C) Surface plasmon resonance analysis of 1S-14279 binding to SPAK [T233E]. 1S-14279 is applied at the indicated concentrations. Nonspecific binding to the reference cell is subtracted from each sensorgram to obtain the specific-binding responses. 1S-14279 interacts with SPAK in a concentration-dependent manner. RU, resonance unit.
Surface Plasmon Resonance Analyses of the Binding Interactions between 1S-14279 and SPAK Kinase
With our ELISA screening system, it was unclear whether 1S-14279 directly interacts with SPAK. To determine the interaction of 1S-14279 and SPAK, we performed a binding assay using the Biacore system. Gradient concentrations (1.6–12.5 μM) of 1S-14279 were injected into GST-SPAK–immobilized flow cells. As shown in Figure 4C, 1S-14279 directly bound to SPAK in a concentration-dependent manner. When a simple 1:1 binding model was fitted, the equilibrium constant KD was 7.0E-6 (M).
Inhibitory Effect of 1S-14279 on SPAK Activity Was Independent of ATP Concentrations
Next, we clarified whether the kinase inhibitory effect of 1S-14279 was due to the competition for ATP bound to SPAK because many kinase inhibitors were ATP competitive. To clarify this issue, we tested the inhibitory effect of 1S-14279 and staurosporine (a nonspecific kinase inhibitor) at different ATP concentrations. Staurosporine interacts with up to 253 human protein kinases with high affinity and inhibits SPAK activity.21,22 As shown in Figure 5A, the IC50 values of staurosporine as an ATP-competitive inhibitor varied 30-fold (0.05 μM and 1.5 μM at 0.01 mM and 2 mM ATP, respectively). By contrast, the IC50 values of 1S-14279 did not alter with the ATP concentration tested (0.21 μM and 0.26 μM at 0.01 mM and 2 mM ATP, respectively) (Figure 5B). These results clearly suggested that 1S-14279 acts as a non-ATP–competitive inhibitor for SPAK because the inhibitory effect was independent of ATP concentration.23
Figure 5.

Effect of increasing concentrations of staurosporine and 1S-14279 on SPAK activity at different ATP concentrations in the ELISA system. (A and B) Kinetic data determined for staurosporine (A) and 1S-14279 (B). ATP concentrations in the reaction mixture vary from 0.01 to 2 mM. 1S-14279 and staurosporine are added at various concentrations (from 0.01 to 50 and from 1E-4 to 100 μM, respectively). Data points are the average of three determinations, and the error bars are ±SEM. The inner panels show IC50 values, determined for each ATP concentrations. IC50 results are 0.05, 0.27, and 1.6 μM for staurosporine at 0.01, 0.1, and 2 mM ATP, respectively. IC50 values of 1S-14279 are not affected by ATP concentration (0.21, 0.13, and 0.26 μM at 0.01, 0.1, and 2 mM ATP, respectively).
Drug Repositioning Strategy to Identify Existing Drugs That Inhibit SPAK Kinase Activity
We further screened a library of 840 existing drugs prepared by our coauthors at Keio University to more efficiently identify clinically applicable SPAK inhibitors. We found that Closantel (N-[5-chloor-4-[(4-chloorfenyl)-cyanomethyl]-2-methylfenyl]-2-hydroxy-3,5-di-joodbenzamide), an anthelmintic drug, efficiently inhibited SPAK-regulated NKCC2 phosphorylation in vitro. Figure 6A shows the chemical structure of Closantel, which is similar in structure to 1S-14279. Figure 6B shows the results of the concentration-dependent inhibitory effect of Closantel on ELISA. The IC50 value of Closantel was 0.77 μM. Furthermore, we tested the inhibitory effect of Closantel with different ATP concentrations. There was little change in the IC50 values (0.65 μM and 0.81 μM at 0.01 mM and 2 mM ATP, respectively, Figure 6C); this behavior was also similar to that of 1S-14279. In the Biacore systems, Closantel directly bound to SPAK and showed relatively faster binding and dissociation than 1S-14279.
Figure 6.

A hit compound obtained from a library of existing drugs. (A) Chemical structure of Closantel and its comparison with 1S-14279. (B) Confirmation of inhibitory effects of Closantel in the ELISA system. The compound is added at various concentrations (0.005–100 μM). The IC50 value of Closantel is 0.77 μM (n=3, mean±SEM). (C) Inhibitory effect of increasing concentrations of Closantel on SPAK activity at different ATP concentrations. Closantel is added at various concentrations (0.01–50 μM). Data points are the average of three determinations, and error bars are ±SEM. The inner panel shows IC50 values determined for each ATP concentrations. IC50 values for Closantel are not affected by ATP concentration (0.65, 0.77, and 0.81 μM at 0.01, 0.1, and 2 mM ATP, respectively). (D) Surface plasmon resonance analysis of Closantel binding to SPAK [T233E]. Closantel is applied at the indicated concentrations. Nonspecific binding to the reference cell is subtracted from each sensorgram to obtain the specific-binding responses. Closantel interacts with SPAK in a concentration-dependent manner. RU, resonance unit.
SPAK Specificity of 1S-14279 and Closantel
To investigate the specificity of these compounds as kinase inhibitors, we conducted a profiling study in the RapidKinase48 panel. The compounds were tested at a final concentration of 1.0E-5 (M), and the kinase profiling revealed that they worked on a few serine/threonine kinases such as Aurora A kinase and Proviral Integration of Moloney virus 2 kinase, although the inhibition did not exceed 80% (Figure 7). Although SPAK kinase was not included in this panel, our ELISA assay showed an IC50 of Closantel and 1S-14279 for SPAK in the range of 1.0E-7 (M), suggesting that both compounds have a potential to behave as specific SPAK inhibitors at lower concentrations.
Figure 7.

RapidKinaseTM48 panel test of 1S-14279 and Closantel. The percent inhibition of kinase activity is tested at 1.0E-05 M. Mean percent inhibition data are an average of duplicate measurements. AurA, Aurora A kinase; PIM2, Proviral Integrations of Moloney virus 2 kinase.
1S-14279 and Closantel Showed Inhibitory Effects on Hypotonicity-Induced WNK-SPAK-NCC/NKCC Signaling in Mouse Renal Distal Tubule-Derived Cells and Vascular Smooth Muscle Cells
To test whether these compounds possess an in vivo inhibitory effect against SPAK, we used mouse renal distal tubule–derived (mpkDCT) cells and mouse vascular smooth muscle (MOVAS) cells, which endogenously express NCC and NKCC1, and performed cell-based inhibitory assays.6,24 We used 30-minute hypotonic shock (170 mOsm/g H2O) to activate WNK-SPAK-NCC/NKCC signaling.25 Both 1S-14279 and Closantel demonstrated a dose-dependent inhibitory effect of phosphorylation of endogenous NCC (pThr53) in mpkDCT cells (Figures 8A and 9A) and of NKCC1 (pThr206) in MOVAS cells (Figures 8B and 9B). To exclude the possibility that the decrease in phosphorylation was due to nonspecific effects, we evaluated the effect of these compounds on phospho-p38 MAPK expression, which is an isolated phosphorylation event from WNK-SPAK signaling.26 As shown in Figures 8 and 9, even with the high concentration of these compounds, the phosphorylation of p38 expression was not reduced but was slightly increased. These data support the specificity of the inhibitory effect of 1S-14279 and Closantel on SPAK activity.
Figure 8.

Inhibitory effect of 1S-14279 on WNK-SPAK-NCC/NKCC1 signaling in mpkDCT and MOVAS cells. (A) The left panel shows the inhibitory effect of 1S-14279 in mpkDCT cells. The phosphorylation of NCC in mpkDCT cells is drastically and dose-dependently reduced by 1S-14279 (1.6–25 μM). The right panel shows quantification of the results of the blots (n=4, mean±SEM). (B) The left panel shows the inhibitory effect of 1S-14279 in MOVAS cells. The phosphorylation of NKCC1 is drastically and dose-dependently reduced by 1S-14279 (1.6–25 μM). The right panel shows quantification of the results of the blots (n=4, mean±SEM). *P<0.05; **P<0.01. Cont/ctrl, control.
Figure 9.

Inhibitory effect of Closantel on WNK-SPAK-NCC/NKCC1signaling in mpkDCT and MOVAS cells. (A) The left panel shows the inhibitory effect of Closantel in mpkDCT cells. The phosphorylation of NCC in mpkDCT cells is drastically and dose-dependently reduced by Closantel (1.6–25 μM). The right panel shows quantification of the results of the blots (n=4, mean±SEM). (B) The left panel shows the inhibitory effect of Closantel in MOVAS cells. The phosphorylation of NKCC1 is drastically and dose-dependently reduced by Closantel (1.6–25 μM). The right panel shows quantification of the results of the blots (n=4, mean±SEM). *P<0.05; **P<0.01. Cont/ctrl, control.
Acute Effects of 1S-14279 and Closantel in Mice
To define whether these compounds are effective in animals, we examined NCC and NKCC2 phosphorylation in the kidney and NKCC1 phosphorylation in the aorta of mice intraperitoneally injected with 20 mg/kg of 1S-14279 or Closantel. As shown in Figures 10A and 11A, markedly reduced NCC phosphorylation (pSer71) with total NCC reduction was observed in the kidneys after 30 minutes of drug administration. On the other hand, NKCC2 phosphorylation (pThr96), which was mainly regulated by OSR1 but not SPAK,27 was not affected. To exclude the nonspecific effects of the compounds, we also evaluated the effect of these compounds on phospho-p38 MAPK expression, as mentioned in the cell-based inhibitory assay. Even with the administration of these compounds, the expression of phospho-p38 was not reduced and was slightly increased, similar to that observed in the cell-based study. Similarly, NKCC1 phosphorylation (pThr206) at the SPAK phosphorylation site was reduced in the aorta. We also followed the time course of the inhibitory effect after a single injection of 1S-14279 and Closantel. After 30 minutes of injection, the level of phosphorylated NCC rapidly decreased; however, it entirely recovered after 120 minutes (Supplemental Figure 3). The SPAK inhibitory effects of 1S-14279 and Closantel were short acting and reversible. We then chose Closantel for BP measurement because preliminary experiments revealed that mice typically died after repeated injection of 1S-14279, and Closantel showed no lethal effects. As shown in Figure 12, the acute injection of Closantel induced marked hypotension from baseline, which was greatest immediately after injection. The heart rate also decreased just after the hypotensive reaction and recovered earlier than the BP.
Figure 10.

Inhibitory effect of 1S-14279 on WNK-SPAK-NCC/NKCC1 signaling in mouse kidney and aorta. (A) Representative immunoblots of total- and p-NCC, NKCC2, and p38 of the kidney at 30 minutes after infusion of 1S-14279. The NCC phosphorylation is drastically reduced by 1S-14279. No significant increases in pNKCC2 and p-p38 levels are observed. The lower panel shows quantification of the results of the blots (n=4, mean±SEM). (B) Representative immunoblots of total- and p-NKCC1 of the aorta at 30 minutes after infusion of 1S-14279. The expression of p-NKCC1 is markedly reduced. No significant difference in total NKCC1 abundance. The lower panel shows quantification of the results of the blots (n=4, mean±SEM). *P<0.05; **P<0.01. Ctrl, control.
Figure 11.

Inhibitory effect of Closantel on WNK-SPAK-NCC/NKCC1 signaling in mouse kidney and aorta. (A) Representative immunoblots of total- and p-NCC, NKCC2 and p38 of the kidney at 30 minutes after infusion of Closantel. The NCC phosphorylation is drastically reduced by Closantel. No significant increases in pNKCC2 and p-p38 levels are observed. The lower panel shows quantification of the results of the blots (n=3, mean±SEM). (B) Representative immunoblots of total- and p-NKCC1 of the aorta at 30 minutes after infusion of Closantel. The expression of p-NKCC1 is markedly reduced. No significant difference in total NKCC1 abundance. The lower panel shows quantification of the results of the blots (n=4, mean±SEM). *P<0.05; **P<0.01. Ctrl, control.
Figure 12.

Acute Closantel administration-induced hypotension in mice. The line graphs represent the systolic arterial BP (black triangles; n=5) and heart rate (gray diamonds; n=5) before and after vehicle (40% DMSO) and Closantel (20 mg/kg in 40% DMSO) administration. The dashed vertical lines indicate the beginning of drug administration. The shaded area represents the period under analysis. The bar graphs show the average decrease in systolic arterial pressure during the 30- to 60-minute postinjection period (*P<0.05; n=5, mean±SEM). Closantel induces a marked reduction in the BP, which appears to precede the decrease in the heart rate, and persists longer. HR, heart rate; IP, intraperitoneal; Ab, antibody.
Effect of Chronic Closantel Treatment in Mice
Next, we examined the effect of chronic treatment with oral Closantel. C57BL/6 mice were fed the mouse chow containing Closantel at a dosage of 300 mg/kg per day. Mice appeared to be healthy during the Closantel treatment. As shown in Figure 13, by day 7 of treatment, NCC and NKCC2 phosphorylation were markedly decreased in the kidneys, and NKCC1 phosphorylation was reduced in the aorta. However, BP was not decreased by Closantel, and we observed no significant differences in serum and urine electrolytes between the Closantel and control groups (Supplemental Figure 4, Supplemental Table 1).
Figure 13.

Inhibitory effect of chronic Closantel treatment on WNK-SPAK-NCC/NKCC1 signaling in mouse kidney and aorta. The dosage of Closantel added to the mouse chow is 300 mg/kg per day. (A) Representative immunoblots of total- and p-NCC and NKCC2 after 7 days treatment of Closantel. NCC and NKCC2 phosphorylation are markedly reduced by Closantel. The lower panel shows the quantification of the results of the blots (n=4, mean±SEM). (B) Representative immunoblots for the total- and p-NKCC1 of the aorta after 7 days Closantel administration. The expression of p-NKCC1 is markedly reduced. The lower panel shows the quantification of the results of the blots (n=4, mean±SEM). *P<0.05; **P<0.01.
Discussion
We previously reported that the constitutive activation of the WNK-OSR1/SPAK-NCC signal cascade is the major pathogenic mechanism of PHAII.28–30 The increased level of NCC phosphorylation induces excessive NaCl reabsorption and causes salt-sensitive hypertension. WNK-OSR1/SPAK kinase also phosphorylates and activates NKCC1 in vascular smooth muscle cells and leads to vasoconstriction.7–10 Furthermore, we revealed that this cascade was activated not only in PHAII but also in hyperinsulinemic conditions and could also cause salt-sensitive hypertension.13 Therefore, drugs that inhibit this signal cascade could become new antihypertensive drugs that have dual effects as a diuretic and vasodilator and could be particularly beneficial for patients with hyperinsulinemia (e.g., metabolic syndrome and obesity). However, directly inhibiting WNK kinases could have a risk of unexpected adverse reactions because homozygous WNK1 and OSR1 knockout mice are embryonically lethal. On the other hand, it was previously reported that SPAK knockout mice were not fatal and displayed hypotension with low NCC and NKCC1 phosphorylation in the kidney and aorta, respectively.16,17 Thus, the SPAK kinase may become a prime target for drug development with the inhibition this signal cascade.
In this study, we sought to develop a new screening system using ELISA that could demonstrate SPAK-regulated phosphorylation in vitro. As a result of our screening, we discovered two hit compounds: 1S-14279 from a small-molecule compound library and Closantel from a library of existing drugs. Surprisingly, these two compounds had quite similar constitutions and both possessed strong SPAK inhibitory effects not only in vitro but also in cultured cell lines and in mice. Closantel is widely used as an antiparasitic agent in livestock either parenterally or orally at a single dose of 5–10 mg/kg. Some observations in humans for the treatment of liver fluke disease have been reported (E. Bernardiner, unpublished data). In recent years, the so-called drug repositioning strategy, in which an existing drug currently used for a specific disease is applied to another disease, has gained increasing attention from both academia and industry.31 An advantage of this strategy is that existing drugs have already passed several stages of clinical development, which could reduce the development risk and costs. In this study, we identified a novel function of Closantel, which appeared to be less toxic than 1S-14279 because mice tolerated chronic treatment with Closantel, but not with 1S-14279. Closantel is a salicylanilide derivative that acts as an uncoupler of the oxidative phosphorylation in the cell mitochondria, which disturbs ATP production. However, in our study, another salicylanilide, oxyclozanide, had no SPAK inhibitory effect (data not shown). The compound structure of Closantel was quite important in the exertion of inhibitory actions on SPAK and the constitutional similarity of 1S-14279; Closantel may give us clues to the synthesis of compounds that have higher efficacy and less toxicity through further structural three-dimensional analysis.
It is noteworthy that both Closantel and 1S-14279 acted as non-ATP–competitive SPAK inhibitors. Most protein kinase inhibitors show an ATP-competitive type of kinase inhibition, which makes it difficult to determine high specificity because the human kinome is composed of >500 protein kinases that share a high degree of identity in the ATP binding pocket. Moreover, ATP-competitive inhibitors must compete with high intracellular ATP levels, leading to a discrepancy between IC50 values measured during an in vitro study versus those measured during an in vivo study.32 Thus, the ATP intensiveness of Closantel and 1S-14279 would be a clear advantage in generating highly specific inhibitors for SPAK.
In this study, we demonstrated that acute and chronic Closantel administration successfully decreased the expression of both phospho-NCC in the kidney and phospho-NKCC1 in the aorta. Similar results were observed in our previous studies with SPAK knockout mice.17 After acute Closantel administration, the BP decreased very rapidly, suggesting that at least in the acute phase, the vasorelaxing effect of SPAK inhibition might be dominant over its natriuretic effect. Although heart rate also decreased, whether this resulted from specific SPAK inhibition and contributed to the decrease in BP requires further investigation. The involvement of SPAK in regulating intracellular Na+ and Cl+ in cardiomyocytes has been reported,33,34 and an unidentified role of SPAK in the heart could be clarified by the discovery of SPAK inhibitors. Unfortunately, the natriuretic action of acute Closantel treatment could not be confirmed because we could not reproducibly collect urine samples in the short experimental period (the duration of activity of Closantel was too short to exert a diuretic effect; Supplemental Figure 3). In addition, we observed no significant increases in sodium excretion under chronic Closantel administration, although this may not be surprising given that SPAK knockout mice showed no such increase.17 Experiments using larger animals and ureteric cannulation are necessary for detailed analysis of Closantel on kidney functions. Furthermore, hypertensive models are needed to assess the antihypertensive effect of Closantel, which may not have an effect on baseline BP like other antihypertensive drugs. Nevertheless, this study could clearly demonstrate that chronic Closantel treatment could inhibit SPAK. Furthermore, OSR1 could also be inhibited by chronic Closantel treatment based on the observation that NKCC2 phosphorylation, which is mainly regulated by OSR1,17 was decreased. This is plausible given that SPAK and OSR1 share high homology in their catalytic and regulatory domains.17
In conclusion, we found two hit compounds that inhibit SPAK kinase both in vitro and in vivo. Further pharmacologic modification of these compounds and their in vivo validation in other animal species and disease models could lead to the development of a novel antihypertensive drug.
Concise Methods
Molecular Cloning and Plasmid Construction
The pRK5 expression plasmid containing T7-tagged whole SPAK was kindly provided by T. Moriguchi and H. Shibuya (Tokyo Medical and Dental University). The active SPAK [T233E] mutation18 was introduced using the QuickChange Site-Directed Mutagenesis Kit (Agilent Technologies Inc., Santa Clara, CA) and cloned into the pGEX6p bacterial expression vector (GE Healthcare UK Ltd.). To clone human NKCC2 [1–174], RT-PCR was carried out using human kidney mRNA as a template, and the resulting PCR product was cloned into the pGEX6p bacterial expression vector. MO25α cDNA was generated by RT-PCR using mouse kidney mRNA as a template, and cloned into the pGEX6p expression vector.
Expression of GST-Tagged Fusion Proteins in Escherichia coli
All recombinant GST fusion proteins were transformed into BL21 E. coli cells. One-liter cultures of the transformed E. coli were grown at 37°C in 2-YT broth containing 100 μg/ml ampicillin until the absorbance value at 600 nm reached 0.5. Isopropyl-d-galactosidase (0.2 mM) was then added to induce protein expression, and the cells were cultured for an additional 16 hours at 28°C. The cells were then collected by centrifugation at 4°C, lysed by sonication in 40 ml of ice-cold 1× PBS buffer containing Complete Protease Inhibitors (Roche Diagnostics), and incubated with 1% Triton X-100 and 0.5% salcosyl. The GST-tagged proteins were purified from the lysates using 1.2 ml of glutathione-Sepharose beads and eluted in an elution buffer containing 83 mM Tris-HCl, 150 mM KOH, and 30 mM glutathione.26
Antibodies and Immunoblotting Analyses
Quantitative immunoblotting was performed as previously described.4 Blots were probed with the following primary antibodies: anti-total NCC,35 anti-phosphorylated NCC (pThr53 and pSer71 in mouse NCC),11 anti-total NKCC1 (T4) (Hybridoma Bank, University of Iowa, Iowa City, IA), anti-phosphorylated NKCC1 (pThr206 in mouse NKCC1),27 anti-total NKCC2 (Alpha Diagnostic, San Antonio, TX), anti-phosphorylated NKCC2 (pThr100 in human NKCC2),36 anti-phosphorylated NKCC2 (termed pT2 antibody; kindly provided by K. Mutig),37 anti-phosphorylated p38, anti-total p38 (Cell Signaling Technology, Danvers, MA), and anti-actin (Cytoskeleton, Denver, CO). Rabbit antiserum against phosphorylated NCC [residues 49–64 of mouse NCC phosphorylated at Thr53/58; Cys+LYMR(pT)FGYN(pT)IDVVPA] was generated and affinity purified. This antibody could be used to detect the phosphorylation of human NKCC2 at residues Thr100/105, which are equivalent to Thr55/60 in human NCC and Thr53/58 in mouse NCC. Alkaline phosphatase–conjugated anti-IgG antibodies (Promega, Madison, WI) were used as secondary antibodies and Western Blue (Promega) was used as a substrate for signal detection. The intensity of the bands was analyzed and quantified using ImageJ software (National Institutes of Health, Bethesda, MD).
In Vitro Kinase Assays
GST-NKCC1 [1–174] protein (7 μg) purified from E. coli was incubated with 1 μg of GST-SPAK [T233E] and 5 μg of GST-MO25α. Kinase assay reactions were performed for 60 minutes at 30°C in a 25 μl of kinase reaction buffer (50 mM Tris-HCl [pH 7.5], 1 mM dithiothreitol, 1 mM EGTA) containing 10 mM MgCl2 and 0.1 mM ATP as previously described.6 The reactions were stopped by the addition of SDS sample buffer (Cosmo Bio Inc., Tokyo, Japan) followed by denaturation for 20 minutes at 60°C. The reaction products were analyzed by SDS-PAGE.
Chemical Screening Using an Indirect ELISA System
GST-NKCC2 [1–174] (5 pmol/well) in a bicarbonate buffer (15 mM Na2CO3, 35 mM NaHCO3) was applied to 96-well ELISA plates (Iwaki & Co., Ltd., Tokyo, Japan) and incubated overnight at 4°C for immobilization. After blocking with 1.5% (w/v) albumin in tris-buffered saline (TBS)-0.02% Tween 20 for 30 minutes at room temperature, kinase reactions were performed in 100 μl of kinase reaction buffer containing 10 mM MgCl2, 0.1 mM ATP, 0.5 pmol of GST-SPAK [T233E], and 5 pmol of GST-MO25α. After a 1-hour incubation at 30°C under gentle shaking, the plates were washed five times with wash buffer (TBS-0.02% Tween 20) using a Microplate Washer (ImmunoWash 1575; Bio-Rad, Mississauga, ON, Canada). Subsequently, 100 μl of 1.5 μg/ml rabbit anti-pNKCC2 antibody (pThr100/105; NCC pThr55/60) was applied to each well for the pNKCC2 assay and incubated overnight at 4°C. After washing, 100 μl of alkaline phosphatase–labeled anti-rabbit secondary antibody was added to each well and the plates were incubated for 1 hour at 37°C. After washing, 100 μl of BluePhos (KPL Inc., Gaithersburg, MD) was applied to each well, and the blue color developed was measured by reading the absorbance at 620 nm in a microplate reader (Spectrafluor; Tecan Japan Co., Ltd., Kanagawa, Japan). For chemical library screening, each compound was added to the kinase reaction at a final concentration of 50 μM. For inhibition studies, increasing concentrations of each compound were tested in triplicate. Curve fitting was performed using ORIGIN8.1 data analysis and graphing software (OriginLab Co., Ltd., Northampton, MA), and data are presented as the mean±SEM. Stock solutions of compounds, in 100% DMSO, were diluted with water to a final DMSO concentration of 1%. Controls without inhibitors were also incubated in the presence of 1% DMSO.
Kinetic Binding Analyses Using a Biacore Biosensor
The binding kinetics between SPAK and the various compounds were analyzed using a Biacore T100 system (GE Healthcare UK Ltd.). CM5 sensor chips were docked and GST-SPAK [T233E] was injected for 7 minutes at a flow rate of 10 μl/min over the surface after preactivation with a mixture of 0.2 M 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride and 0.05 M sulfo-N-hydroxysuccinimide. After the injection of GST-SPAK [T233E], the surface was deactivated with 150 μl of 1 M ethanolamine HCl (pH 8.5), giving an immobilization level for GST-SPAK [T233E] of approximately 10,000 resonance units. To reduce nonspecific binding, we used a reference surface immobilized with GST at 3000 resonance units. The TBS-based buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 10 mM MgCl2, 0.05% surfactant-P) with 5% DMSO was used as a running buffer to facilitate dissolution of the compounds, and the temperature was set to 25°C. Analysis of the interactions between the small-molecule compounds and GST-SPAK [T233E] was carried out using a one-shot kinetic approach. For each compound, a series of solutions of four different concentrations and a blank sample for double referencing were injected for 60 seconds at a flow rate of 90 μl/min. No regeneration was applied. The sensorgrams were processed and fitted to determine the dissociation constants using Biacore T100 evaluation software.
Kinase Profiling
Kinase profiling was conducted by PerkinElmer (Boston, MA) using a RapidKinase48 panel in duplicate wells. The compounds were tested at a final concentration of 1.0E-5 (M). Each test compound was incubated with each of the kinases and appropriate substrates according to PerkinElmer standard operating procedures. After the incubation period, the reaction products and remaining substrate were measured. According to PerkinElmer guidelines, compounds that show ≥50% inhibition are considered active.
Cell Culture and Inhibition Assay
The mpkDCT cell line kindly provided by A. Vandewalle was cultured in a defined medium as previously described.38 MOVAS cells were cultured in DMEM supplemented with 10% (v/v) FBS, 2 mM L-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin at 37°C in a humidified 5% CO2 incubator. mpkDCT and MOVAS cells were cultured on six-well dishes. Hypotonic stimulation was performed as previously reported.26 For the inhibition assay, cells were exposed to each compound for 30 minutes and then stimulated with a hypotonic low-chloride medium for 30 minutes.25 Cells were then lysed in 0.15 ml of ice-cold cell lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 50 mM sodium fluoride, 1 mM sodium orthovanadate, 1% Triton X-100, 0.27 M sucrose, 1 mM dithiothreitol) per well. After centrifugation at 12,000×g for 5 minutes at 4°C, the supernatants were denatured for 20 minutes at 60°C with SDS sample buffer and subjected to SDS-PAGE.
Animal Studies
Female C57BL/6 mice (10 weeks old) were obtained and received standard laboratory chow and water. The Animal Care and Use Committee of Tokyo Medical and Dental University approved the experimental protocol. In an acute treatment study, 1S-14279 and Closantel were both administered intraperitoneally at a dose of 20 mg/kg in 40 μl of DMSO; the control mice were administered 40 μl of DMSO alone. The mice were euthanized 30 minutes after injection. In the chronic treatment study, C57BL/6 mice were fed the mouse chow containing Closantel at a dosage of 300 mg/kg per day. The kidney was removed and rapidly homogenized in a 10-fold mass excess of ice-cold 1% (w/v) Triton X-100 lysis buffer. Whole homogenates without the nuclear fraction (600×g) were collected and denatured at 60°C for 20 minutes as previously reported.4 The aorta was rapidly isolated and frozen with liquid nitrogen. After being crushed by sonication, aortas were added to 150 μl of lysis buffer as previously reported,7 and centrifuged at 6000×g for 5 minutes at 4°C. A 120-μl aliquot of the supernatant was denatured at 60°C for 20 minutes. The total protein concentration was determined by the Bradford method using BSA as a standard, and lysates were stored at −80°C. Quantitative immunoblotting was performed as previously described.12 The relative intensities of immunoblot bands were determined by densitometry using ImageJ software.
The BP of the mice was measured using a radiotelemetric method in which a BP transducer (Data Sciences International, St. Paul, MN) was inserted into the left carotid artery. The mice were housed individually in a standard cage with a 12-hour/12-hour light/dark cycle and received standard laboratory chow and water as previously described.39 Seven days after transplantation, the systolic and diastolic arterial pressures were recorded 1 hour before and 2 hours after intraperitoneal injections of either 20 mg/kg Closantel dissolved in 40% DMSO or 40% DMSO alone as a vehicle control. All drugs were freshly prepared in DMSO each day, and the effect of each drug or vehicle was assessed on separate days. Blood was drawn from the retro-orbital sinus under light ether anesthesia. Serum data were determined using the i-STAT system (FUSO Pharmaceutical Industries, Osaka, Japan). Urine electrolytes were determined using a Fuji DRI-CHEM 4000 system (FujiFilm, Tokyo, Japan). Serum and urine creatinine concentrations were determined using a commercial kit (LabAssay Creatinine; Wako Chemical Inc., Osaka, Japan).
Statistical Analyses
Data are expressed as the mean±SEM. Comparisons between the two groups were performed with unpaired t tests. P values <0.05 were considered statistically significant.
Disclosures
None.
Supplementary Material
Acknowledgments
We thank Makiko Nawa (Laboratory of Cytometry and Proteome Research, Tokyo Medical and Dental University) for her help with Biacore analysis. We also thank Dr. K. Mutig and A. Vandewalle for the provision of pT2 antibody and mpkDCT cells, respectively.
This study was supported in part by Grants-in-Aid for Scientific Research (S) and (A) from the Japan Society for the Promotion of Science; a Grant-in-Aid for Young Scientists (B) from the Ministry of Education, Culture, Sports, Science and Technology of Japan; a Health and Labor Sciences Research Grant from the Ministry of Health, Labor and Welfare of Japan; and grants from the Salt Science Research Foundation (no. 1422), the Takeda Science Foundation, the Banyu Foundation, and the Vehicle Racing Commemorative Foundation.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2014060560/-/DCSupplemental.
References
- 1.Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, Gill JR, Jr, Ulick S, Milora RV, Findling JW, Canessa CM, Rossier BC, Lifton RP: Liddle’s syndrome: Heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell 79: 407–414, 1994 [DOI] [PubMed] [Google Scholar]
- 2.Achard JM, Disse-Nicodeme S, Fiquet-Kempf B, Jeunemaitre X: Phenotypic and genetic heterogeneity of familial hyperkalaemic hypertension (Gordon syndrome). Clin Exp Pharmacol Physiol 28: 1048–1052, 2001 [DOI] [PubMed] [Google Scholar]
- 3.Wilson FH, Disse-Nicodème S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP: Human hypertension caused by mutations in WNK kinases. Science 293: 1107–1112, 2001 [DOI] [PubMed] [Google Scholar]
- 4.Yang SS, Morimoto T, Rai T, Chiga M, Sohara E, Ohno M, Uchida K, Lin SH, Moriguchi T, Shibuya H, Kondo Y, Sasaki S, Uchida S: Molecular pathogenesis of pseudohypoaldosteronism type II: Generation and analysis of a Wnk4(D561A/+) knockin mouse model. Cell Metab 5: 331–344, 2007 [DOI] [PubMed] [Google Scholar]
- 5.Wakabayashi M, Mori T, Isobe K, Sohara E, Susa K, Araki Y, Chiga M, Kikuchi E, Nomura N, Mori Y, Matsuo H, Murata T, Nomura S, Asano T, Kawaguchi H, Nonoyama S, Rai T, Sasaki S, Uchida S: Impaired KLHL3-mediated ubiquitination of WNK4 causes human hypertension. Cell Reports 3: 858–868, 2013 [DOI] [PubMed] [Google Scholar]
- 6.Richardson C, Rafiqi FH, Karlsson HK, Moleleki N, Vandewalle A, Campbell DG, Morrice NA, Alessi DR: Activation of the thiazide-sensitive Na+-Cl- cotransporter by the WNK-regulated kinases SPAK and OSR1. J Cell Sci 121: 675–684, 2008 [DOI] [PubMed] [Google Scholar]
- 7.Zeniya M, Sohara E, Kita S, Iwamoto T, Susa K, Mori T, Oi K, Chiga M, Takahashi D, Yang SS, Lin SH, Rai T, Sasaki S, Uchida S: Dietary salt intake regulates WNK3-SPAK-NKCC1 phosphorylation cascade in mouse aorta through angiotensin II. Hypertension 62: 872–878, 2013 [DOI] [PubMed] [Google Scholar]
- 8.Meyer JW, Flagella M, Sutliff RL, Lorenz JN, Nieman ML, Weber CS, Paul RJ, Shull GE: Decreased blood pressure and vascular smooth muscle tone in mice lacking basolateral Na(+)-K(+)-2Cl(-) cotransporter. Am J Physiol Heart Circ Physiol 283: H1846–H1855, 2002 [DOI] [PubMed] [Google Scholar]
- 9.Akar F, Jiang G, Paul RJ, O’Neill WC: Contractile regulation of the Na(+)-K(+)-2Cl(-) cotransporter in vascular smooth muscle. Am J Physiol Cell Physiol 281: C579–C584, 2001 [DOI] [PubMed] [Google Scholar]
- 10.Garg P, Martin CF, Elms SC, Gordon FJ, Wall SM, Garland CJ, Sutliff RL, O’Neill WC: Effect of the Na-K-2Cl cotransporter NKCC1 on systemic blood pressure and smooth muscle tone. Am J Physiol Heart Circ Physiol 292: H2100–H2105, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiga M, Rai T, Yang SS, Ohta A, Takizawa T, Sasaki S, Uchida S: Dietary salt regulates the phosphorylation of OSR1/SPAK kinases and the sodium chloride cotransporter through aldosterone. Kidney Int 74: 1403–1409, 2008 [DOI] [PubMed] [Google Scholar]
- 12.Sohara E, Rai T, Yang SS, Ohta A, Naito S, Chiga M, Nomura N, Lin SH, Vandewalle A, Ohta E, Sasaki S, Uchida S: Acute insulin stimulation induces phosphorylation of the Na-Cl cotransporter in cultured distal mpkDCT cells and mouse kidney. PLoS ONE 6: e24277, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishida H, Sohara E, Nomura N, Chiga M, Alessi DR, Rai T, Sasaki S, Uchida S: Phosphatidylinositol 3-kinase/Akt signaling pathway activates the WNK-OSR1/SPAK-NCC phosphorylation cascade in hyperinsulinemic db/db mice. Hypertension 60: 981–990, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Talati G, Ohta A, Rai T, Sohara E, Naito S, Vandewalle A, Sasaki S, Uchida S: Effect of angiotensin II on the WNK-OSR1/SPAK-NCC phosphorylation cascade in cultured mpkDCT cells and in vivo mouse kidney. Biochem Biophys Res Commun 393: 844–848, 2010 [DOI] [PubMed] [Google Scholar]
- 15.Castañeda-Bueno M, Cervantes-Pérez LG, Vázquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, Gamba G: Activation of the renal Na+:Cl- cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci U S A 109: 7929–7934, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiga M, Rafiqi FH, Alessi DR, Sohara E, Ohta A, Rai T, Sasaki S, Uchida S: Phenotypes of pseudohypoaldosteronism type II caused by the WNK4 D561A missense mutation are dependent on the WNK-OSR1/SPAK kinase cascade. J Cell Sci 124: 1391–1395, 2011 [DOI] [PubMed] [Google Scholar]
- 17.Yang SS, Lo YF, Wu CC, Lin SW, Yeh CJ, Chu P, Sytwu HK, Uchida S, Sasaki S, Lin SH: SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J Am Soc Nephrol 21: 1868–1877, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Filippi BM, de los Heros P, Mehellou Y, Navratilova I, Gourlay R, Deak M, Plater L, Toth R, Zeqiraj E, Alessi DR: MO25 is a master regulator of SPAK/OSR1 and MST3/MST4/YSK1 protein kinases. EMBO J 30: 1730–1741, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vitari AC, Deak M, Morrice NA, Alessi DR: The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J 391: 17–24, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zagórska A, Pozo-Guisado E, Boudeau J, Vitari AC, Rafiqi FH, Thastrup J, Deak M, Campbell DG, Morrice NA, Prescott AR, Alessi DR: Regulation of activity and localization of the WNK1 protein kinase by hyperosmotic stress. J Cell Biol 176: 89–100, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP: A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 26: 127–132, 2008 [DOI] [PubMed] [Google Scholar]
- 22.Gagnon KB, England R, Delpire E: Characterization of SPAK and OSR1, regulatory kinases of the Na-K-2Cl cotransporter. Mol Cell Biol 26: 689–698, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Butini S, Gemma S, Brindisi M, Borrelli G, Lossani A, Ponte AM, Torti A, Maga G, Marinelli L, La Pietra V, Fiorini I, Lamponi S, Campiani G, Zisterer DM, Nathwani SM, Sartini S, La Motta C, Da Settimo F, Novellino E, Focher F: Non-nucleoside inhibitors of human adenosine kinase: Synthesis, molecular modeling, and biological studies. J Med Chem 54: 1401–1420, 2011 [DOI] [PubMed] [Google Scholar]
- 24.Koltsova SV, Kotelevtsev SV, Tremblay J, Hamet P, Orlov SN: Excitation-contraction coupling in resistance mesenteric arteries: Evidence for NKCC1-mediated pathway. Biochem Biophys Res Commun 379: 1080–1083, 2009 [DOI] [PubMed] [Google Scholar]
- 25.Naito S, Ohta A, Sohara E, Ohta E, Rai T, Sasaki S, Uchida S: Regulation of WNK1 kinase by extracellular potassium. Clin Exp Nephrol 15: 195–202, 2011 [DOI] [PubMed] [Google Scholar]
- 26.Mori T, Kikuchi E, Watanabe Y, Fujii S, Ishigami-Yuasa M, Kagechika H, Sohara E, Rai T, Sasaki S, Uchida S: Chemical library screening for WNK signalling inhibitors using fluorescence correlation spectroscopy. Biochem J 455: 339–345, 2013 [DOI] [PubMed] [Google Scholar]
- 27.Lin SH, Yu IS, Jiang ST, Lin SW, Chu P, Chen A, Sytwu HK, Sohara E, Uchida S, Sasaki S, Yang SS: Impaired phosphorylation of Na(+)-K(+)-2Cl(-) cotransporter by oxidative stress-responsive kinase-1 deficiency manifests hypotension and Bartter-like syndrome. Proc Natl Acad Sci U S A 108: 17538–17543, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uchida S: Pathophysiological roles of WNK kinases in the kidney. Pflugers Arch 460: 695–702, 2010 [DOI] [PubMed] [Google Scholar]
- 29.Kahle KT, Rinehart J, Giebisch G, Gamba G, Hebert SC, Lifton RP: A novel protein kinase signaling pathway essential for blood pressure regulation in humans. Trends Endocrinol Metab 19: 91–95, 2008 [DOI] [PubMed] [Google Scholar]
- 30.Richardson C, Alessi DR: The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signalling pathway. J Cell Sci 121: 3293–3304, 2008 [DOI] [PubMed] [Google Scholar]
- 31.Ashburn TT, Thor KB: Drug repositioning: Identifying and developing new uses for existing drugs. Nat Rev Drug Discov 3: 673–683, 2004 [DOI] [PubMed] [Google Scholar]
- 32.Garuti L, Roberti M, Bottegoni G: Non-ATP competitive protein kinase inhibitors. Curr Med Chem 17: 2804–2821, 2010 [DOI] [PubMed] [Google Scholar]
- 33.Pedersen SF, O’Donnell ME, Anderson SE, Cala PM: Physiology and pathophysiology of Na+/H+ exchange and Na+ -K+ -2Cl- cotransport in the heart, brain, and blood. Am J Physiol Regul Integr Comp Physiol 291: R1–R25, 2006 [DOI] [PubMed] [Google Scholar]
- 34.Bers DM, Barry WH, Despa S: Intracellular Na+ regulation in cardiac myocytes. Cardiovasc Res 57: 897–912, 2003 [DOI] [PubMed] [Google Scholar]
- 35.Ohno M, Uchida K, Ohashi T, Nitta K, Ohta A, Chiga M, Sasaki S, Uchida S: Immunolocalization of WNK4 in mouse kidney. Histochem Cell Biol 136: 25–35, 2011 [DOI] [PubMed] [Google Scholar]
- 36.Richardson C, Sakamoto K, de los Heros P, Deak M, Campbell DG, Prescott AR, Alessi DR: Regulation of the NKCC2 ion cotransporter by SPAK-OSR1-dependent and -independent pathways. J Cell Sci 124: 789–800, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mutig K, Paliege A, Kahl T, Jöns T, Müller-Esterl W, Bachmann S: Vasopressin V2 receptor expression along rat, mouse, and human renal epithelia with focus on TAL. Am J Physiol Renal Physiol 293: F1166–F1177, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Diepens RJ, den Dekker E, Bens M, Weidema AF, Vandewalle A, Bindels RJ, Hoenderop JG: Characterization of a murine renal distal convoluted tubule cell line for the study of transcellular calcium transport. Am J Physiol Renal Physiol 286: F483–F489, 2004 [DOI] [PubMed] [Google Scholar]
- 39.Susa K, Kita S, Iwamoto T, Yang SS, Lin SH, Ohta A, Sohara E, Rai T, Sasaki S, Alessi DR, Uchida S: Effect of heterozygous deletion of WNK1 on the WNK-OSR1/ SPAK-NCC/NKCC1/NKCC2 signal cascade in the kidney and blood vessels. Clin Exp Nephrol 16: 530–538, 2012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.