

Abstract
IL-6 can mediate proinflammatory effects, and IL-6 receptor (IL-6R) blockade as a treatment for inflammatory diseases has entered clinical practice. However, opposing effects of IL-6 have been observed in models of GN. Although IL-6 is proinflammatory in murine lupus nephritis, protective effects have been observed for IL-6 in the nephrotoxic nephritis (NTN) model of acute crescentic GN. In light of the potential dangers of IL-6–directed treatment, we studied the mechanisms underlying the contradictory findings in GN. IL-6 can signal through the membrane-bound IL-6R, which is expressed only on hepatocytes and certain leukocytes (classic), or through the soluble IL-6R, which binds the ubiquitously expressed gp130 (alternative). Preemptive treatment of mice with anti-IL-6R or anti-IL-6 worsened NTN, whereas selective blockade of alternative IL-6 signaling by the fusion protein sgp130Fc did not. FACS analysis of mouse spleen cells revealed proinflammatory macrophages express the highest levels of IL-6Rα, and in vitro treatment with IL-6 blocked macrophage proliferation. Furthermore, proinflammatory macrophages were expanded during inflammation in IL-6−/− mice. Late application of anti-IL-6 after establishment of adaptive nephritogenic immunity was sufficient to aggravate NTN within 2.5 days, a period when macrophages are active. Finally, NTN was aggravated in mice with macrophage-specific impairment of IL-6 classic signaling, coincident with enhanced macrophage proliferation and accumulation in the kidney. Our data thus reveal a novel mechanism in which IL-6–mediated dampening of macrophage activation protects tissues from overshooting immune responses. This finding has important implications for potential IL-6–directed therapies and supports the careful choice of recipient patients and timing.
Keywords: GN, immunology, pathology, cytokines
IL-6 is a pleiotropic cytokine that is involved in multiple pathways such as regulation of acute phase responses, cell growth and differentiation, as well as metabolic processes.1 Importantly, IL-6 has emerged as a potent regulator of immune responses and inflammation.2,3 Given these multiple functions, it is not surprising that signaling of IL-6 is complex and can occur via two different pathways. In the so-called classic IL-6 signaling pathway, IL-6 binds to the membrane-bound IL-6 receptor (IL-6R) which consists of two different subunits: the IL-6Rα chain to which IL-6 binds directly, and the signal-transducing membrane glycoprotein gp130. The IL-6Rα chain is exclusively expressed on the surface of hepatocytes and a number of distinct leukocyte subsets.4 Thus classic signaling can only occur on these defined cell types. However, in addition to this restricted signaling pathway, an alternative IL-6 signaling pathway exists which was termed IL-6 trans-signaling.5,6 Here, IL-6 forms a complex with a soluble IL-6 receptor α chain (sIL-6Rα) that binds to the ubiquitously expressed gp130. Therefore, in contrast with classic signaling, IL-6 trans-signaling can occur on all cells of the body. The complex role of IL-6 and the relative contribution of each signaling pathway in immunologically mediated disease has been the focus of many studies from the past few years.7 In this respect, IL-6 was initially identified as a B-cell differentiation factor.8 However, subsequent studies have also revealed effects on multiple other cells of the immune system such as neutrophils (PMNs), macrophages, dendritic cells (DCs), and T-helper (Th) cells. The discovery of IL-6 as a key cytokine required for development of the highly proinflammatory Th17 response has especially gained much attention.9,10 Together with TGF-β, IL-6 initiates cellular events leading to programming of Th17 differentiation.11,12 Many studies in Th17-mediated experimental models of immune-mediated diseases such as encephalitis, colitis, and arthritis have proven the proinflammatory effects of IL-6.13–15 As a consequence, IL-6–directed therapy has been introduced into clinical practice. The mAb tocilizumab, which is directed against the human IL-6R, is successfully used to treat rheumatoid arthritis and Still’s disease.16–18 However, some significant caveats remain. Besides its proinflammatory role, IL-6 signaling has also been shown to exert strong anti-inflammatory effects. Studies from the pre-Th17 era predominantly declared IL-6 to be an anti-inflammatory cytokine with IL-10–like properties.19–22 These effects include downregulation of macrophage proliferation and activation23–25 as well as inhibition of DC maturation.26 Most importantly, IL-6R blockade in humans has also been reported to cause inflammatory eye disease,27 psoriasis,28 as well as crescentic immune complex GN.29 Generally, the anti-inflammatory actions of IL-6 still remain very ill defined. This is also the case regarding inflammatory renal disease where conflicting data have been reported. Several studies have been performed in rodent models of SLE. Pioneering work by Kiberd showed an anti-inflammatory role of an IL-6R antibody in the Mrl/lpr model of lupus nephritis.30 Much later, a study in IL-6–deficient mice confirmed these findings in the same model.31 In a second model of SLE, three studies supported proinflammatory effects of IL-6. First, recombinant IL-6 aggravated pathogenic immune responses and the clinical course of SLE in NZW×NZB mice.32 Application of anti–IL-6R as well as anti–IL-6 antibodies were subsequently shown to ameliorate disease by two independent groups.33,34 Mechanistically, the deleterious effects of IL-6 were largely ascribed to production of pathogenic antibodies. Some authors also suspected IL-6 effects on T-cell activation; however, this was not addressed any further and effects on Th17 responses have not yet been studied.
Although these data are in line with and clearly indicate a proinflammatory role for IL-6 in lupus and lupus nephritis, results from another model of nephritis were contradictory. Two studies in the nephrotoxic nephritis (NTN) model of acute crescentic GN in the rat have documented anti-inflammatory effects of recombinant IL-6.35,36 Both the mechanisms underlying IL-6–mediated protection from nephritis and the responsible IL-6 signaling pathway (classic versus trans) remained unclear. Injury in the NTN model develops independently of antibodies37 but highly depends on Th1 and Th17 cell responses38–41 as well as macrophages.42 Therefore, it is very likely that the observed effects were mediated by these highly IL-6–responsive leukocyte subpopulations. In addition to the anti-inflammatory role of IL-6 in NTN, a more recent study found protective effects of IL-6 trans-signaling in AKI.43
Regarding the situation in humans, IL-6 secretion from PBMCs correlated with renal disease activity in lupus nephritis.44 Urinary IL-6 has also been suggested as a marker for renal inflammation in different forms of GN. Definite proof for a pathogenic role of IL-6 in human inflammatory renal disease is, however, still lacking. Few case reports from the literature describe successful treatment of renal disease by the IL-6R antibody tocilizumab,45 including one patient with pANCA-associated refractory GN.46 In addition, one pilot study assessed the effects of tocilizumab in patients with mild lupus nephritis.47 Although arthritis in these patients improved significantly, improvement of renal function was less evident. Nevertheless, given the positive results in animal studies of SLE and the paucity of effective therapeutic options for human lupus and ANCA-associated nephritis, it would be very tempting to initiate randomized clinical trials of IL-6R blockade in GN. However, for such studies to be launched, it is crucial to better understand which mechanisms underlie the strong anti-inflammatory and tissue-protective effects of IL-6. This study therefore aimed to (1) assess the effect of IL-6 pathway blockade on the clinical course of NTN, (2) identify the differential IL-6 signaling pathways that are involved (classic versus trans-signaling), and (3) identify which cell populations mediate the effects of IL-6 in NTN.
Results
IL-6R Blockade Aggravates NTN
NTN was induced in C57BL/6 mice and one group was treated with anti-IL-6R antibody,48 whereas controls were injected with control IgG. Analyses of kidneys at day 10 after NTN induction revealed significantly aggravated disease in anti-IL-6R–treated mice in terms of histologic and functional damage (Figure 1A). Renal leukocyte influx was significantly increased (Figure 1B) and renal Th1 and Th17 responses remained intact (Figure 1C). Analyses of systemic T- and B-cell immune responses showed multiple effects. Antigen-specific antibody production was impaired, confirming effective blockade of IL-6 signaling (Supplemental Figure 1A). Spleen cell numbers were significantly reduced but regulatory T-cell percentages remained unchanged (Supplemental Figure 1, B and C). By contrast, Th cell activation and proliferation were strongly downregulated by anti–IL-6R treatment (Supplemental Figure 1, D and E). In line, production of various T-cell cytokines was greatly diminished, including near absence of splenic IL-17 production in anti-IL-6R–treated mice (Supplemental Figure 1F).
Figure 1.

anti-IL-6R treatment aggravates GN. (A) Representative photographs of PAS-stained kidney sections (left) and quantification of renal histologic damage (middle) and function (right) in nephritic aIL-6R– or control IgG–treated mice at day 10 after induction of NTN. (B) Representative photographs (left) and quantification (right) of glomerular and interstitial T cells (CD3) and macrophages (MAC2, F4/80). (C) Representative FACS plots and quantification of renal Th17 and Th1 subsets. Circles represent individual animals, whereas horizontal lines indicate mean values. Error bars represent the SD. Numbers in FACS plots represent percentages of gated events. *P<0.05; **P<0.01; ***P<0.001. PAS, periodic acid–Schiff. Original magnification, ×400.
Blockade of IL-6 Classic Signaling but Not Trans-Signaling Aggravates NTN
In a next step, NTN was again induced and mice were treated with an antibody blocking IL-6 or the fusion protein sgp130Fc, which specifically blocks IL-6 trans-signaling or control IgG. Similar to anti-IL-6R–treated mice, nephritis was aggravated in the aIL-6 group in terms of histology and functional parameters (Figure 2A). By contrast, no significant proinflammatory effect was noted in sgp130Fc-treated mice. In line with histology, analyses of renal leukocytes showed enhanced infiltration in aIL-6–treated mice (Figure 2B). T-cell subtype characterization again revealed robust renal Th1 and Th17 responses in all experimental groups (Figure 2C). Systemic B- and T-cell immunity was much less affected than in the aIL-6R experiments. Antigen-specific IgG production was similar between the groups (data not shown). Spleen cell numbers in aIL-6–treated mice were slightly reduced (Supplemental Figure 2A), but production of most cytokines produced by spleen cells remained comparable (Supplemental Figure 2B).
Figure 2.
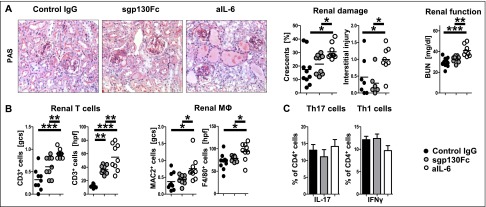
Blockade of the classic IL-6 pathway aggravates GN. (A) Representative photographs of PAS-stained kidney sections (left) and quantification of renal histologic damage (middle) and function (right) in nephritic control IgG–, sgp130Fc–, or aIL-6–treated mice at day 10 after induction of NTN. (B) Quantification of glomerular and interstitial T cells (CD3) and macrophages (MAC2, F4/80). (C) Representative FACS plots and quantification of renal Th17 and Th1 subsets. Circles represent individual animals, whereas horizontal lines indicate mean values. Error bars represent SD. *P<0.05; **P<0.01; ***P<0.001. PAS, periodic acid–Schiff. Original magnification, ×400.
IL-6 Reduces Macrophage Proliferation and Activation In Vitro and In Vivo
We next wanted to assess the effect of IL-6 on macrophages because anti-inflammatory effects have been postulated. FACS analyses of spleen cells indeed showed strong expression of IL-6Rα preferentially on inflammatory Ly6Chigh macrophages. Expression on all other myeloid cells measured, including Ly6Cint/lo macrophages, DCs, and neutrophils, was strikingly lower (Figure 3, A and B). This identified proinflammatory macrophages as primary targets for classic IL-6 signaling. We therefore isolated peritoneal macrophages and incubated them in the presence or absence of IL-6. Assessment of both numbers of colony forming units and cell numbers per colony, revealed strong inhibitory effects of IL-6 on macrophage proliferation (Figure 3C).
Figure 3.

Proinflammatory macrophages respond to IL-6. (A) Percentages of IL-6Rα–expressing cells of naïve splenic Ly6chigh and Ly6cint/low macrophages, DCs, and neutrophils (PMNs). (B) Quantification of IL-6Rα mean fluorescence intensity (MFI) in the respective populations. (C) Quantification of macrophage colony forming units (left) and cells per colony (right) with and without addition of IL-6. (D) FACS quantification of Ly6chigh macrophages in indicated compartments of IL-6−/− mice. (E) Quantification of MHCII expression on blood macrophages. Circles represent individual animals, whereas horizontal lines indicate mean values. Numbers in FACS plots represent percentages of gated events. ***P<0.001.
To assess in vivo effects of IL-6 on macrophages, IL-6−/− mice and age-matched wild-type controls were immunized with non-nephritogenic sheep IgG. Two days later, macrophages from bone marrow, spleen, and blood were analyzed by flow cytometry. Although analysis of bone marrow as the internal control did not reveal differences, percentages of highly activated Ly6Chigh macrophages were significantly enhanced in spleens and even more so in blood from IL6−/− mice (Figure 3D). These effects were specific for Ly6Chigh macrophages, because percentages of Ly6Cint/lo macrophages, DCs and neutrophils were slightly reduced in blood and spleens (not shown). In addition, blood monocytes from IL6−/− mice showed much higher MHCII expression (Figure 3E).
Blockade of IL-6 Classic Signaling in the Effector Phase Aggravates NTN
In order to exclude IL-6 effects on the development of adaptive immunity and to assess specific effects on the macrophage-dependent effector phase of nephritis, we switched to the model of accelerated NTN. Five days after preimmunization, NTN serum was injected. In order to minimize effects on neutrophils, which are very early mediators of NTN, anti-IL-6, sgp130Fc, or control IgG was administered at an interval of 36 hours after NTN induction. Treatment with aIL-6 resulted in significant aggravation at day 7 of nephritis in terms of histology, function, and leukocyte influx. Again, no deleterious effects were seen in sgp130Fc-treated mice (Figure 4, A and B). Next, we wanted to assess whether aggravation of nephritis could also be observed at earlier time points after anti–IL-6 application. Therefore, in a second set of experiments, accelerated NTN (aNTN) was again induced by the same protocol and organs were harvested as early as day 4. Again analyses showed significant aggravation of nephritis in the anti-IL-6–treated group only 2.5 days after antibody treatment, pointing to effects on rapidly acting cells like macrophages (Figure 4C).
Figure 4.
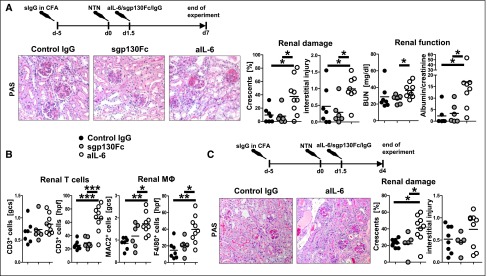
Blockade of classic IL-6 signaling in the effector phase aggravates GN. (A) Experimental outline (top), representative photographs of PAS-stained kidney sections (left) and quantification of renal histologic damage (middle) and function (right) in nephritic mice treated with control IgG, sgp130Fc or aIL-6 at day 7 after induction of aNTN. (B) Quantification of glomerular and interstitial T cells (CD3) and macrophages (MAC2, F4/80). (C) Experimental outline (top), representative photographs of PAS-stained kidney sections (left) and quantification of renal histologic damage (right) in nephritic control IgG–, sgp130Fc–, or aIL-6–treated mice at day 4 after induction of aNTN. Circles represent individual animals, whereas horizontal lines indicate mean values. *P<0.05; **P<0.01; ***P<0.001. PAS, periodic acid–Schiff. Original magnification, ×400.
Macrophage Restricted Abrogation of IL-6 Signaling Induces a Proinflammatory Phenotype and Aggravates NTN
Our previous experiments suggested a role for classic IL-6 signaling in macrophages as the underlying mechanism for the observed aggravation of nephritis. Therefore, mice lacking the IL-6R on macrophages but not on adaptive immune cells were generated by intercrossing LysMCre with IL-6Rαflox/flox mice. Effective knockout of IL-6Rα was confirmed by analysis of IL-6Rα expression of thioglycollate-elicited peritoneal myeloid cells49 (Supplemental Figure 3). NTN induction in LysMCre×IL-6Rαflox/flox mice again showed significant aggravation compared with IL-6R intact controls in terms of histology, functional impairment, and leukocyte infiltration (Figure 5, A and B). Importantly, FACS analyses of renal myeloid cells showed significant skewing in favor of proinflammatory Ly6Chigh macrophages, whereas percentages of infiltrating neutrophils remained unchanged (Figure 5C). In addition, proliferation rates of total macrophages as well as of the Ly6Chigh proinflammatory subset were enhanced in kidneys of LysMCre×IL-6Rαflox/flox mice (Figure 5D). By contrast, percentages of mannose receptor-positive anti-inflammatory M2 macrophages were greatly reduced (Figure 5E). DC percentages and proliferation remained unchanged (Supplemental Figure 4A). As expected, analyses of renal Th cells showed unaltered balances of Th1 and Th17 responses as well as regulatory T-cells (Supplemental Figure 4B). Systemic immunity including splenocyte composition, T-cell activation, and cytokine production was also similar between the groups (Supplemental Figure 4, C and D).
Figure 5.

Abrogation of IL-6 classic signaling in macrophages aggravates GN. (A) Representative photographs of PAS-stained kidney sections (left) and quantification of renal histologic damage (middle) and function (right) in nephritic LysMCre or LysMCre×IL-6Rαflox/flox mice at day 10 after induction of NTN. (B) Quantification of glomerular and interstitial T cells (CD3), macrophages (MAC2, F4/80), and neutrophils (GR-1). (C) Representative FACS plots (left) and quantification (right) of renal Ly6chigh and Ly6cint/low macrophage subsets as well as Ly6Ghi PMNs. (D) Quantification of renal macrophage subset proliferation. (E) Quantification of Mannose receptor/CD206 expression on renal macrophage subtypes. Circles represent individual animals, whereas horizontal lines indicate mean values. Error bars represent the SD. Numbers in FACS plots represent percentages of gated events. One representative of two experiments performed is shown. *P<0.05; **P<0.01; ***P<0.001. Original magnification, ×400.
Discussion
Our study aimed to evaluate the therapeutic potential of IL-6 blocking strategies in crescentic GN. Our initial hypothesis was that interference with IL-6 signaling would ameliorate disease by impairing generation of Th17 responses, which have been shown to be central mediators of GN.40,50 Antibody blockade of the IL-6R, however, surprisingly resulted in disease aggravation. As expected, antigen-specific antibody and systemic cytokine production were greatly reduced, indicating the potency of aIL-6R treatment. Th17 responses in the acutely inflamed kidney, however, remained intact. Various different mechanisms could account for this observation. First, IL-6 is needed for initiation of Th17 generation but not for their expansion so that preformed Th17 cells can proliferate in the absence of IL-6. In addition, some studies have identified so-called natural Th17 cells that are already generated in the thymus and do not depend on further IL-6.51,52 Second, antibody blockade is never complete and small remaining amounts of active IL-6 are probably sufficient to trigger Th17 differentiation, especially in a highly inflammatory environment like a nephritic kidney and the corresponding lymph node. Finally, it has been shown that other factors including IL-21 can substitute for IL-6 and can independently induce Th17 responses.10,53 Given the multiple effects of IL-6R blockade on a broad range of immune responses, we next wanted to investigate whether aggravation of nephritis was specifically attributable to the anti–IL-6R antibody we used or if they were more general in nature. We thus applied a different antibody that blocks IL-6 rather than IL-6R. Furthermore, we wanted to evaluate which IL-6 signaling pathway is involved in aggravation of disease and thus applied the designer protein sgp130Fc, which specifically blocks IL-6 trans-signaling.54 In line with our first experiment, application of anti–IL-6 aggravated nephritis. Trans-signaling blockade did not alter any of the examined end points, indicating that classic IL-6 signaling mediated the observed protective effects of IL-6. Interestingly, aggravation occurred although renal and systemic adaptive immune responses remained largely unchanged by anti–IL-6 treatment, which pointed toward a role for innate mediators. Because aggravation was mediated by classic signaling, the effects were unlikely to be mediated by resident nonhematopoietic renal cells as these do not express the IL-6R.43,55 Rather, aggravation of disease was more likely to be affected by leukocytes, which, except for hepatocytes, are the only cells known to express IL-6R on their surface.56 In this regard, we wanted to study the role of macrophages because macrophages are known to be potent effectors of tissue injury in NTN42 and IL-6 was postulated to downregulate macrophage activation and proliferation.23–25 In line, preferentially proinflammatory Ly6Chigh macrophages expressed the IL-6Rα. By contrast, expression levels on Ly6Cint/low macrophages, DCs, and especially neutrophils were much lower. Because high constitutive IL-6R expression was found on almost all Ly6chigh macrophages from naïve mice, inflammation-induced IL-6 could act as a rapid brake on these cells. Supporting this concept, immunization led to rapidly enhanced systemic numbers of proinflammatory macrophages in mice genetically lacking IL-6. Vice versa, macrophage proliferation could be greatly reduced by in vitro treatment with IL-6. In order to study the in vivo role of macrophages in nephritis, we used the model of accelerated NTN. In this model, mice are preimmunized with non-nephritogenic sheep IgG so that adaptive immune responses can develop during the following days. After establishment of adaptive immunity, NTN is induced by injection of nephritogenic sheep IgG, which binds to the glomerular basement membrane and acts as a planted glomerular antigen.41 The preformed adaptive immune responses then mediate tissue damage by recruitment of macrophages as effectors. Treatment with anti–IL-6 and sgp130Fc was started 36 hours after injection of the nephritogenic serum so that only the macrophage-dependent effector phase, but not the previous development of adaptive immunity, was affected. Importantly, by delaying treatment for 36 hours, this protocol also spares the very early neutrophil-dependent phase of NTN.57 Again we found significant aggravation of NTN by anti–IL-6 but not by sgp130Fc. Importantly, aggravation of nephritis was observed as early as 2.5 days after anti-IL-6 antibody application. This finding indicated IL-6 effects on rapid immune effectors such as macrophages. In order to provide compelling evidence that impaired classic IL-6 signaling in macrophages is causative for aggravation of nephritis, we generated mice specifically lacking the membrane-bound IL-6Rα subunit on the myeloid cell lineage including macrophages. In line with our hypothesis, induction of NTN indeed showed more severe disease in the knockout mice. Importantly, renal proinflammatory macrophage numbers and proliferative activity were significantly and selectively enhanced. Mannose receptor-positive M2 macrophages in contrast were reduced, indicating a switch toward a predominance of the M1 type.58 Further analyses showed that percentages of renal neutrophils were similar between the groups. Likewise, DC percentages and proliferative activity remained unchanged, as did T-cell activation and cytokine production. Although we cannot fully rule out contributions of neutrophils and DCs to the observed aggravation of nephritis, our data clearly support a key role for macrophages. It is likely that inflammation-induced IL-6 physiologically acts as a rapid downregulator of proinflammatory macrophage proliferation and an inductor of M2 polarization to protect the organism from overshooting immune reactions. Interestingly, a very recent independent study published during preparation of this article reported the same key finding of increased macrophage activation in the absence of IL-6Rα signaling that aggravated insulitis.59 This points toward a more general role of IL-6–mediated dampening of macrophage activation in inflammatory diseases. What implications do our observations have on IL-6 blocking strategies in renal inflammatory diseases? The consequences of IL-6 pathway blockade might be context dependent. Targeting IL-6 seems to be especially useful for diseases with certain characteristics, particularly those in which pathogenic IL-6–dependent autoantibodies are involved as SLE. Furthermore, in general, diseases with a more chronic course might be favorably influenced by continuous IL-6 pathway blockade. Conversely, however, during acute renal inflammation as in a disease flare, blockade might be deleterious. Finally, because the protective effects of IL-6 were mediated by classic signaling, specific blockade of IL-6 trans-signaling might be a promising alternative, especially because sgp130Fc is already being tested in clinical phase I studies.
In summary, our data reveal the novel finding that inflammation-induced IL-6 acts as a rapid brake on proinflammatory macrophage responses. This observation has great implications because it might constitute a common mechanism protecting the organism from overshooting immune responses during acute inflammatory diseases.
Concise Methods
Animals
IL6−/− and LysMCre (B6.129P2-Lyz2tm1(cre)Ifo/J) mice were provided by Gisa Tiegs (Hamburg, Germany). IL-6Rα floxed mice (B6;SJL-Il6ratm1.1Drew/J) were a kind gift from Juergen Scheller (Düsseldorf, Germany). All mice used were on a C57BL/6 background and initially derived from the The Jackson Laboratory and were bred at our animal facility.
Animal Experiments and Functional Studies
Nephrotoxic nephritis was induced in 8- to 10-week-old male mice of the indicated genotypes by intraperitoneal injection of 2.5 mg of nephrotoxic sheep serum per gram of body weight.60 Antibodies were applied in doses previously shown to be effective, including anti–IL-6R (750 μg, MR16-1; provided by Chugai Pharmaceutical Co. Ltd., Tokyo, Japan33), anti–IL-6 (500 μg, MP5-20F3), and sgp130Fc (250 μg),61 or control antibodies (750 μg rat IgG for MR16-1 or 500 μg hIgG for anti–IL-6 and sgp130Fc) were intraperitoneally injected 24 hours before and 5 days after NTN induction. For aNTN,41 mice were preimmunized with sheep IgG in complete Freund’s adjuvant at day 5. NTN was induced at day 0 and antibodies were administered intraperitoneally 36 hours later. Organs were harvested at day 10 after NTN induction and day 4 or 7 after aNTN induction as indicated. Animal experiments were performed according to national and institutional animal care and ethical guidelines and were approved by local committees (approval code G37/11). Urine samples were collected after housing the mice in metabolic cages. Albuminuria was determined by standard ELISA (Bethyl Laboratories). BUN and urinary creatinine were quantified using standard laboratory methods.
Morphologic Studies
Crescent formation and glomerular necrosis were determined in a minimum of 50 glomeruli per mouse in 2-µm–thick periodic acid–Schiff-stained kidney sections in a blinded manner. Semiquantitative analysis of tubulointerstitial damage was performed using 10 randomly selected cortical areas (×200) as previously described.41 Paraffin-embedded sections were stained with antibodies directed against CD3 (A0452; Dako, Hamburg, Germany), F4/80 (BM8; BMA Biomedicals, Hiddenhausen, Germany), MAC2 (M3/38; Cedarlane Laboratories, Burlington, ON, Canada), GR-1, or Foxp3 and developed with a polymer-based secondary antibody–alkaline phosphatase kit (POLAP; Zytomed, Berlin, Germany), as previously published.60 Fifty glomerular cross-sections and 30 tubulointerstitial high-power fields (×400 magnification) per kidney section were counted in a blinded fashion.
Isolation of Leukocytes from Various Tissues
Spleens were harvested in HBSS and passed through 70-µm nylon meshes. After lysis of erythrocytes with ammonium chloride, cells were washed and passed over 40-µm meshes. Cells were then washed again, counted, and resuspended in PBS for either culture or FACS analysis. Kidneys were minced and incubated in digestion-medium (RPMI 1640 medium containing 10% FCS, 1% HEPES, 1% penicillin/streptomycin, 8 µg/ml collagenase D, and 0.4 µg/ml DNase) at 37°C for 40 minutes. Tissues were then homogenized, passed over 70-µm nylon meshes, and centrifuged at 300g at 4°C for 5 minutes. After lysis of erythrocytes with ammonium chloride, cells were filtered over 40-µm meshes, washed again, and sedimented for 15 minutes at 4°C. The upper leukocyte fraction was aspirated and filtered through another 40-µm nylon mesh. Cells were washed, counted, and resuspended in PBS for staining and FACS analysis. Peripheral blood was drawn into EDTA-coated tubes and red blood cell lysis was performed after staining for surface markers.
Bone marrow cells were isolated from femoral and humeral bones, as previously described.62 In brief, bones were stored in ice-cold PBS, dissected, and flushed using a syringe filled with ice-cold PBS under sterile conditions. Afterward, cells were filtered through a 40-µm mesh and washed twice by centrifugation at 300g for 10 minutes at 4°C.
For analysis of peritoneal myeloid cells, mice were injected intraperitoneally with 1 ml thioglycollate. The peritoneal cavity was washed with 5 ml ice-cold PBS to harvest cells at day 3 (for real-time quantitative PCR) or day 8 (for flow cytometry).
Antigen-Specific Systemic Cellular and Humoral Immune Responses
Splenocytes (4×106 cells/ml) were cultured under standard conditions in the presence of normal sheep IgG (10 µg/ml; Sigma-Aldrich, Taufkirchen, Germany) and supernatants were harvested after 72 hours. Commercially available ELISAs were used for detection of IFN-γ, IL-4, IL-6, TNFα, IL-10, IL-17A (Biolegend, San Diego, CA) and TGF-β (R&D Systems, Minneapolis, MN). Circulating sheep-globulin–specific serum IgG titers were analyzed by ELISA (total IgG; Biozol, Eching, Germany).
Flow Cytometry
Cells were surface stained for 30 minutes at 4°C with fluorochrome-labeled antibodies against CD45, CD3, CD4, CD19, CD44, CD69, CD62L, γδ TCR, IL-6Rα, F4/80, Ly6C, Ly6G, CD206, and MHCII (all Ebioscience, San Diego, CA) and CD11b (BD Biosciences, Heidelberg, Germany) as previously described.60,62 After pregating on CD45+ live cells, macrophages were gated as CD11b+Ly6G−CD11c− and Ly6Chigh or Ly6Cint/low. DCs were gated as CD11b+CD11c+MHCII+, and neutrophils were gated as CD11b+Ly6G+. For intracellular and intranuclear staining, samples were processed using a commercial intranuclear staining kit (Foxp3 Staining Kit; Ebioscience). Fluorochrome-labeled antibodies against IL-17, IFN-γ, Foxp3, and Ki67 (Ebioscience) were utilized as recently published.60 For intracellular cytokine staining, cells were activated with PMA (50 ng/ml; Sigma-Aldrich) and ionomycin (1 µg/ml; Calbiochem-Merck) for 5 hours. After 30 minutes of incubation, brefeldin A (10 µg/ml; Sigma-Aldrich) was added. LIVE/DEAD staining (Invitrogen Molecular Probes, Eugene, OR) was used to exclude dead cells during flow cytometry and to ensure viability of the cells after the stimulation procedure. Experiments were performed on a BD LSRII Cytometer (Becton Dickinson, Germany).
In Vitro Macrophage Proliferation
Mice were injected intraperitoneally with 1 ml thioglycollate. To isolate macrophages, the peritoneal cavity was washed with 5 ml ice-cold PBS at day 4. Peritoneal macrophages were washed and resuspended in RPMI 1640 with L-glutamine containing 1% penicillin-streptomycin, 1% HEPES, 20% FCS, and 20% L-cell conditioned medium. Three milliliters of macrophage culture was plated at a concentration of 3.3×102 cells/ml on a six-well plate. Cells were cultivated in a humidified incubator with 5% CO2 at 37°C. Colony formation in presence or absence of 100 U/ml IL-6 was monitored for 14 days. Analysis was performed by counting colony forming units and cells per colony forming unit. Colony forming units were determined as a group of cells with cell–cell contacts or at least a distance of <1×cell width and a total number of cells of n>3.
Real-Time Quantitative PCR Analyses
IL-6Rα mRNA expression was determined from thioglycollate-elicited peritoneal myeloid cells of LysMCre× IL-6Rαflox/flox mice or controls by standard real-time PCR using the TaqMan probe Mm00439653_m1 (Life Technologies, Carlsbad, CA). Expression levels were normalized to 18s mRNA.
Statistical Analyses
Results are expressed as the mean±SD. Groups were compared by the t test and a P value<0.05 was considered statistically significant. In the case of multiple comparisons, one-way ANOVA was applied using Tukey post hoc testing.
Disclosures
S.-R.J. is a shareholder of the CONARIS Research Institute AG (Kiel, Germany), which is commercially developing sgp130Fc as a therapy for inflammatory diseases, and is an inventor on gp130 patents owned by CONARIS.
Supplementary Material
Acknowledgments
We thank Professor Gisa Tiegs for providing IL-6−/− and LysMCre mice. MR16-1 antibody was kindly provided by Tadamitsu Kishimoto and Chugai Pharma (Osaka, Japan). We thank M. Schaper and M. Reszka for their excellent technical help.
This work was supported by grants from the German Research Foundation (STE 1822/2-1 and KFO 228 STE 1822/3-1 to O.M.S., KFO 228 to H.-W.M., and SFB841 [project C1] and SFB877 [project A1] to S.-R.J.).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “The Janus Faces of IL-6 in GN,” on pages 1480–1482.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2014060620/-/DCSupplemental.
References
- 1.Neurath MF, Finotto S: IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev 22: 83–89, 2011 [DOI] [PubMed] [Google Scholar]
- 2.Kishimoto T: Interleukin-6: From basic science to medicine—40 years in immunology. Annu Rev Immunol 23: 1–21, 2005 [DOI] [PubMed] [Google Scholar]
- 3.Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S: The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta 1813: 878–888, 2011 [DOI] [PubMed] [Google Scholar]
- 4.Taga T, Kishimoto T: Gp130 and the interleukin-6 family of cytokines. Annu Rev Immunol 15: 797–819, 1997 [DOI] [PubMed] [Google Scholar]
- 5.Rose-John S, Neurath MF: IL-6 trans-signaling: The heat is on. Immunity 20: 2–4, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Rose-John S, Scheller J, Elson G, Jones SA: Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: Role in inflammation and cancer. J Leukoc Biol 80: 227–236, 2006 [DOI] [PubMed] [Google Scholar]
- 7.Jones SA, Scheller J, Rose-John S: Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest 121: 3375–3383, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirano T, Yasukawa K, Harada H, Taga T, Watanabe Y, Matsuda T, Kashiwamura S, Nakajima K, Koyama K, Iwamatsu A, Tsunasawa S, Sakiyama F, Matsui H, Takahara Y, Taniguchi T, Kishimoto T: Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature 324: 73–76, 1986 [DOI] [PubMed] [Google Scholar]
- 9.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK: Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441: 235–238, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR: IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol 8: 967–974, 2007 [DOI] [PubMed] [Google Scholar]
- 11.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT: Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441: 231–234, 2006 [DOI] [PubMed] [Google Scholar]
- 12.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B: TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24: 179–189, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Serada S, Fujimoto M, Mihara M, Koike N, Ohsugi Y, Nomura S, Yoshida H, Nishikawa T, Terabe F, Ohkawara T, Takahashi T, Ripley B, Kimura A, Kishimoto T, Naka T: IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 105: 9041–9046, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamamoto M, Yoshizaki K, Kishimoto T, Ito H: IL-6 is required for the development of Th1 cell-mediated murine colitis. J Immunol 164: 4878–4882, 2000 [DOI] [PubMed] [Google Scholar]
- 15.Fujimoto M, Serada S, Mihara M, Uchiyama Y, Yoshida H, Koike N, Ohsugi Y, Nishikawa T, Ripley B, Kimura A, Kishimoto T, Naka T: Interleukin-6 blockade suppresses autoimmune arthritis in mice by the inhibition of inflammatory Th17 responses. Arthritis Rheum 58: 3710–3719, 2008 [DOI] [PubMed] [Google Scholar]
- 16.Tanaka T, Narazaki M, Kishimoto T: Anti-interleukin-6 receptor antibody, tocilizumab, for the treatment of autoimmune diseases. FEBS Lett 585: 3699–3709, 2011 [DOI] [PubMed] [Google Scholar]
- 17.Schoels MM, van der Heijde D, Breedveld FC, Burmester GR, Dougados M, Emery P, Ferraccioli G, Gabay C, Gibofsky A, Gomez-Reino JJ, Jones G, Kvien TK, Murakami M, Nishimoto N, Smolen JS: Blocking the effects of interleukin-6 in rheumatoid arthritis and other inflammatory rheumatic diseases: Systematic literature review and meta-analysis informing a consensus statement. Ann Rheum Dis 72: 583–589, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanaka T, Narazaki M, Ogata A, Kishimoto T: A new era for the treatment of inflammatory autoimmune diseases by interleukin-6 blockade strategy. Semin Immunol 26: 88–96, 2014 [DOI] [PubMed] [Google Scholar]
- 19.Aderka D, Le JM, Vilcek J: IL-6 inhibits lipopolysaccharide-induced tumor necrosis factor production in cultured human monocytes, U937 cells, and in mice. J Immunol 143: 3517–3523, 1989 [PubMed] [Google Scholar]
- 20.Ulich TR, Yin S, Guo K, Yi ES, Remick D, del Castillo J: Intratracheal injection of endotoxin and cytokines. II. Interleukin-6 and transforming growth factor beta inhibit acute inflammation. Am J Pathol 138: 1097–1101, 1991 [PMC free article] [PubMed] [Google Scholar]
- 21.Ulich TR, Guo KZ, Remick D, del Castillo J, Yin SM: Endotoxin-induced cytokine gene expression in vivo. III. IL-6 mRNA and serum protein expression and the in vivo hematologic effects of IL-6. J Immunol 146: 2316–2323, 1991 [PubMed] [Google Scholar]
- 22.Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei XF, Achong MK: IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest 101: 311–320, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riedy MC, Stewart CC: Inhibitory role of interleukin-6 in macrophage proliferation. J Leukoc Biol 52: 125–127, 1992 [DOI] [PubMed] [Google Scholar]
- 24.Tilg H, Trehu E, Atkins MB, Dinarello CA, Mier JW: Interleukin-6 (IL-6) as an anti-inflammatory cytokine: Induction of circulating IL-1 receptor antagonist and soluble tumor necrosis factor receptor p55. Blood 83: 113–118, 1994 [PubMed] [Google Scholar]
- 25.Clutterbuck R, Powles R, Millar J, Catovsky D: Interleukin-6 and other gp130-dependent cytokines selectively inhibit proliferation of macrophage-lineage hemopoietic progenitor cells. Exp Hematol 28: 1120–1128, 2000 [DOI] [PubMed] [Google Scholar]
- 26.Park SJ, Nakagawa T, Kitamura H, Atsumi T, Kamon H, Sawa S, Kamimura D, Ueda N, Iwakura Y, Ishihara K, Murakami M, Hirano T: IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J Immunol 173: 3844–3854, 2004 [DOI] [PubMed] [Google Scholar]
- 27.Wendling D, Paccou J, Berthelot JM, Flipo RM, Guillaume-Czitrom S, Prati C, Dernis E, Direz G, Ferrazzi V, Ristori JM, CRI : New onset of uveitis during anti-tumor necrosis factor treatment for rheumatic diseases. Semin Arthritis Rheum 41: 503–510, 2011 [DOI] [PubMed] [Google Scholar]
- 28.Wendling D, Letho-Gyselinck H, Guillot X, Prati C: Psoriasis onset with tocilizumab treatment for rheumatoid arthritis. J Rheumatol 39: 657, 2012 [DOI] [PubMed] [Google Scholar]
- 29.Matsuo Y, Mizoguchi F, Kohsaka H, Ito E, Eishi Y, Miyasaka N: Tocilizumab-induced immune complex glomerulonephritis in a patient with rheumatoid arthritis. Rheumatology (Oxford) 52: 1341–1343, 2013 [DOI] [PubMed] [Google Scholar]
- 30.Kiberd BA: Interleukin-6 receptor blockage ameliorates murine lupus nephritis. J Am Soc Nephrol 4: 58–61, 1993 [DOI] [PubMed] [Google Scholar]
- 31.Cash H, Relle M, Menke J, Brochhausen C, Jones SA, Topley N, Galle PR, Schwarting A: Interleukin 6 (IL-6) deficiency delays lupus nephritis in MRL-Faslpr mice: The IL-6 pathway as a new therapeutic target in treatment of autoimmune kidney disease in systemic lupus erythematosus. J Rheumatol 37: 60–70, 2010 [DOI] [PubMed] [Google Scholar]
- 32.Ryffel B, Car BD, Gunn H, Roman D, Hiestand P, Mihatsch MJ: Interleukin-6 exacerbates glomerulonephritis in (NZB x NZW)F1 mice. Am J Pathol 144: 927–937, 1994 [PMC free article] [PubMed] [Google Scholar]
- 33.Mihara M, Takagi N, Takeda Y, Ohsugi Y: IL-6 receptor blockage inhibits the onset of autoimmune kidney disease in NZB/W F1 mice. Clin Exp Immunol 112: 397–402, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang B, Gardner DB, Griswold DE, Bugelski PJ, Song XY: Anti-interleukin-6 monoclonal antibody inhibits autoimmune responses in a murine model of systemic lupus erythematosus. Immunology 119: 296–305, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karkar AM, Smith J, Tam FW, Pusey CD, Rees AJ: Abrogation of glomerular injury in nephrotoxic nephritis by continuous infusion of interleukin-6. Kidney Int 52: 1313–1320, 1997 [DOI] [PubMed] [Google Scholar]
- 36.Karkar AM, Tam FW, Proudfoot AE, Meager A, Rees AJ: Modulation of antibody-mediated glomerular injury in vivo by interleukin-6. Kidney Int 44: 967–973, 1993 [DOI] [PubMed] [Google Scholar]
- 37.Li S, Holdsworth SR, Tipping PG: Antibody independent crescentic glomerulonephritis in mu chain deficient mice. Kidney Int 51: 672–678, 1997 [DOI] [PubMed] [Google Scholar]
- 38.Tipping PG, Huang XR, Qi M, Van GY, Tang WW: Crescentic glomerulonephritis in CD4- and CD8-deficient mice. Requirement for CD4 but not CD8 cells. Am J Pathol 152: 1541–1548, 1998 [PMC free article] [PubMed] [Google Scholar]
- 39.Tipping PG, Holdsworth SR: T cells in glomerulonephritis. Springer Semin Immunopathol 24: 377–393, 2003 [DOI] [PubMed] [Google Scholar]
- 40.Turner JE, Paust HJ, Steinmetz OM, Panzer U: The Th17 immune response in renal inflammation. Kidney Int 77: 1070–1075, 2010 [DOI] [PubMed] [Google Scholar]
- 41.Steinmetz OM, Summers SA, Gan PY, Semple T, Holdsworth SR, Kitching AR: The Th17-defining transcription factor RORγt promotes glomerulonephritis. J Am Soc Nephrol 22: 472–483, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holdsworth SR, Tipping PG: Leukocytes in glomerular injury. Semin Immunopathol 29: 355–374, 2007 [DOI] [PubMed] [Google Scholar]
- 43.Nechemia-Arbely Y, Barkan D, Pizov G, Shriki A, Rose-John S, Galun E, Axelrod JH: IL-6/IL-6R axis plays a critical role in acute kidney injury. J Am Soc Nephrol 19: 1106–1115, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Esposito P, Balletta MM, Procino A, Postiglione L, Memoli B: Interleukin-6 release from peripheral mononuclear cells is associated to disease activity and treatment response in patients with lupus nephritis. Lupus 18: 1329–1330, 2009 [DOI] [PubMed] [Google Scholar]
- 45.Iijima T, Suwabe T, Sumida K, Hayami N, Hiramatsu R, Hasegawa E, Yamanouchi M, Hoshino J, Sawa N, Takaichi K, Oohashi K, Fujii T, Ubara Y: Tocilizumab improves systemic rheumatoid vasculitis with necrotizing crescentic glomerulonephritis [published online ahead of print February 18, 2014]. Mod Rheumatol 10.3109/14397595.2013.874748 [DOI] [PubMed] [Google Scholar]
- 46.Sumida K, Ubara Y, Suwabe T, Hayami N, Hiramatsu R, Hasegawa E, Yamanouchi M, Hoshino J, Sawa N, Takemoto F, Takaichi K, Ohashi K: Complete remission of myeloperoxidase-anti-neutrophil cytoplasmic antibody-associated crescentic glomerulonephritis complicated with rheumatoid arthritis using a humanized anti-interleukin 6 receptor antibody. Rheumatology (Oxford) 50: 1928–1930, 2011 [DOI] [PubMed] [Google Scholar]
- 47.Illei GG, Shirota Y, Yarboro CH, Daruwalla J, Tackey E, Takada K, Fleisher T, Balow JE, Lipsky PE: Tocilizumab in systemic lupus erythematosus: Data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheum 62: 542–552, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okazaki M, Yamada Y, Nishimoto N, Yoshizaki K, Mihara M: Characterization of anti-mouse interleukin-6 receptor antibody. Immunol Lett 84: 231–240, 2002 [DOI] [PubMed] [Google Scholar]
- 49.McFarland-Mancini MM, Funk HM, Paluch AM, Zhou M, Giridhar PV, Mercer CA, Kozma SC, Drew AF: Differences in wound healing in mice with deficiency of IL-6 versus IL-6 receptor. J Immunol 184: 7219–7228, 2010 [DOI] [PubMed] [Google Scholar]
- 50.Kitching AR, Holdsworth SR: The emergence of TH17 cells as effectors of renal injury. J Am Soc Nephrol 22: 235–238, 2011 [DOI] [PubMed] [Google Scholar]
- 51.Marks BR, Nowyhed HN, Choi JY, Poholek AC, Odegard JM, Flavell RA, Craft J: Thymic self-reactivity selects natural interleukin 17-producing T cells that can regulate peripheral inflammation. Nat Immunol 10: 1125–1132, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanaka S, Yoshimoto T, Naka T, Nakae S, Iwakura Y, Cua D, Kubo M: Natural occurring IL-17 producing T cells regulate the initial phase of neutrophil mediated airway responses. J Immunol 183: 7523–7530, 2009 [DOI] [PubMed] [Google Scholar]
- 53.Kimura A, Naka T, Kishimoto T: IL-6-dependent and -independent pathways in the development of interleukin 17-producing T helper cells. Proc Natl Acad Sci U S A 104: 12099–12104, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Waetzig GH, Rose-John S: Hitting a complex target: An update on interleukin-6 trans-signalling. Expert Opin Ther Targets 16: 225–236, 2012 [DOI] [PubMed] [Google Scholar]
- 55.Waiser J, Budde K, Katalinic A, Kuerzdörfer M, Riess R, Neumayer HH: Interleukin-6 expression after renal transplantation. Nephrol Dial Transplant 12: 753–759, 1997 [DOI] [PubMed] [Google Scholar]
- 56.Taga T: The signal transducer gp130 is shared by interleukin-6 family of haematopoietic and neurotrophic cytokines. Ann Med 29: 63–72, 1997 [DOI] [PubMed] [Google Scholar]
- 57.Cochrane CG, Unanue ER, Dixon FJ: A role of polymorphonuclear leukocytes and complement in nephrotoxic nephritis. J Exp Med 122: 99–116, 1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anders HJ, Ryu M: Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int 80: 915–925, 2011 [DOI] [PubMed] [Google Scholar]
- 59.Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, Theurich S, Hausen AC, Schmitz J, Brönneke HS, Estevez E, Allen TL, Mesaros A, Partridge L, Febbraio MA, Chawla A, Wunderlich FT, Brüning JC: Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol 15: 423–430, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kluger MA, Luig M, Wegscheid C, Goerke B, Paust HJ, Brix SR, Yan I, Mittrücker HW, Hagl B, Renner ED, Tiegs G, Wiech T, Stahl RA, Panzer U, Steinmetz OM: Stat3 programs Th17-specific regulatory T cells to control GN. J Am Soc Nephrol 25: 1291–1302, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoge J, Yan I, Jänner N, Schumacher V, Chalaris A, Steinmetz OM, Engel DR, Scheller J, Rose-John S, Mittrücker HW: IL-6 controls the innate immune response against Listeria monocytogenes via classical IL-6 signaling. J Immunol 190: 703–711, 2013 [DOI] [PubMed] [Google Scholar]
- 62.Kluger MA, Zahner G, Paust HJ, Schaper M, Magnus T, Panzer U, Stahl RA: Leukocyte-derived MMP9 is crucial for the recruitment of proinflammatory macrophages in experimental glomerulonephritis. Kidney Int 83: 865–877, 2013 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.