

Abstract
Background and aims: Acute myeloid leukemia (AML) is a fatal hematological malignancy which is resistant to a variety of chemotherapy drugs. Phospho-ERK5 (p-ERK5) plays a novel role in chemoresistance in some cancer cells and this pathway is a central mediator of cell survival and apoptotic regulation. The aim of this study was to investigate the effect of a specific ERK5 small interference RNA (siRNA) on proliferation and the sensitivity of HL-60 acute myeloid leukemia (AML) cells to the chemotherapeutic drug cytarabine. Methods: The cells were transfected with siRNAs using Lipofectamine™ 2000 transfection reagent. Relative ERK5 mRNA and protein levels were measured by quantitative real-time PCR, immunocytochemical assay, and Western blotting, respectively. The cytotoxic effects of cytarabine and ERK5 siRNA, alone and in combination, on leukemic cells were determined using colony formation and MTT assay. Apoptosis was assessed by ELISA cell death assay. Results: ERK5 siRNA markedly reduced both mRNA and protein expression levels leading to distinct inhibition of cell proliferation and increased spontaneous apoptosis. Surprisingly, ERK5 siRNA synergistically increased the cell toxic effects of cytarabine. Conclusions: Our study suggests that down-regulation of ERK5 by siRNA can trigger apoptosis and overcome drug resistance of leukemia cells. Therefore, ERK5 siRNA may be an effective adjuvant in AML chemotherapy.
Keywords: Acute myeloid leukemia, chemotherapy, ERK5
Introduction
Acute myeloid leukemia (AML) accounts for one-fourth of acute leukemia in children, but is responsible for more than half of the leukemia deaths in this patient population [1]. Resistance to cytarabine (ara-C)-based chemotherapy is a major cause of treatment failure in this disease [2,3]. Therefore, new therapies for children with AML are urgently needed. Dysregulation of the apoptosis (Programmed cell death) machinery contributes to the formation of neoplasia and subsequent resistance to chemotherapy [4]. As most of the chemotherapy agents exert their anti-tumor effects by triggering apoptosis, new approaches for cancer treatment have focused on targeting mediators of this pathway [5].
The MEK5/ERK5 pathway is one of the lesser studied members of the mitogen-activated protein kinase (MAPK) family of protein kinases. This pathway has been implicated in cell survival, anti-apoptotic signaling, angiogenesis, cell motility, differentiation and cell proliferation [6-8].
The MEK5/ERK5 cascade mediates survival and proliferative signaling [9], particularly in transformed cells of hematopoietic origin. In myeloma cells, inhibition of ERK5 blocks proliferation and facilitates apoptosis induced by dexamethasone [10]. ERK5 expression is essential for survival of leukemic cells expressing Bcr/Abl [11]. Hodgkin lymphoma cells show constitutive activation of the ERK5 pathway [12]. Finally, expression of the microRNA miR-143 shows an inverse correlation with that of ERK5 [13], and the level of miR-143, along with miR-145, is decreased in most B cell malignancies, including chronic lymphocytic leukemias, B cell lymphomas, EBV-transformed B cell lines, and Burkitt lymphoma cell lines. These observations suggest that ERK5 plays an important, albeit poorly characterized, role in oncogenesis.
Some studies have examined the role of ERK5 in chemoresistance in MCF-7 breast cancer cells. Microarray analysis indicated a 22-fold increase in the levels of MEK5 in apoptically resistant cells (APO-cells). Transfection of these cells with dominant negative forms of ERK5 and subsequent treatment with and without several apoptotic inducing agents in addition to one cell survival compound revealed the antiapoptotic characteristics of MEK5/ERK5. This demonstrated that the MEK5/ERK5 pathway plays a novel role in chemoresistance and this pathway is a central mediator of cell survival and apoptotic regulation in MCF-7 cells [14].
Wang et al has found that ERK5 pathway plays a role in the control of monocytic differentiation, which is disturbed in myeloid leukemia [15]. A recent study has found stable expression of a small hairpin RNA for ERK5 (shERK5) leads to their elimination by NK cells in vivo. Coinjection of shERK5-expressing cells into the peritoneum diminishes survival of engrafted wild-type tumor cells. Moreover, s.c. injection of shERK5-expressing cells strongly diminishes tumor development by wild-type cells, suggesting that knockdown of ERK5 expression in leukemia cells effectively attenuates their tumor activity [16]. Garaude et al. shown ERK5 inhibition reduced cell viability, sensitized cells to death receptor-induced apoptosis, and blocked the palliative effects of phorbol ester in anti-Fas Ab-treated cells [17].
In this study, we investigated whether suppression of ERK5 could sensitize HL-60 human AML cell line to chemotherapeutic agent cytarabine. We therefore examined the effects of either ERK5 inhibition or cytarabine alone, versus their combination treatment in promoting tumor cells apoptosis.
Materials and methods
Cell culture
The HL-60 AML cell line was purchased from ATCC (Shanghai, China). It was grown in RPMI-1640 medium containing 15% fetal bovine serum (FBS), 100 IU/ml penicillin, 100 μg/ml streptomycin, 1% sodium pyruvate and 2 mM of glutamine, at 37°C in a humidified atmosphere containing 5% CO2. The cells were sub-cultured 24-48 h later with an initial concentration of 4 × 104 cells/ml and used in the logarithmic phase in all experiments.
Antibodies
ERK5 and phospho-ERK5 (Thr218/Tyr220) were purchased from Cell Signaling Technology (Danvers, MA). Anti-actin was from Sigma.
siRNA transfection
For ERK5 silencing, HL-60 cells were transfected with 100 μmol/L of specific human siRNAs against ERK5 by using Lipofectamine 2000 reagent (Invitrogen) following protocols provided by the manufacturer. Targeting sequences of siRNA is 5’-GGGCCTATATCCAGAGCUU-3’; Non-specific control siRNA was used as a negative control. Briefly, siRNAs and lipofectamine (4 μl/ml of transfection medium) were diluted in Opti-MEM I Reduced Serum Medium (Invitrogen) separately and incubated for 10 min at room temperature. The diluted solutions were then mixed and incubated for 20 min at room temperature. Subsequently, the mixtures were added to each well containing cells and medium. Moreover, the treated cells with only the transfection reagent were considered as a blank control. The cell culture plates were then incubated for 6 h at 37°C in a CO2 incubator. Following on, RPMI-1640 medium containing FBS (final FBS concentration of 15%) was added, with cells being incubated under the above mentioned conditions. To evaluate the effects of siRNA on gene silencing, transfections (5 × 105 cells/well) were performed in 6-well cell culture plates for 24-72 h, then the protein expression was determined by immunoblotting with antibodies as indicated above 3 and mRNA expression was determined by quantitative real-time RT-PCR (RT-qPCR). The siRNA showing the highest efficiency of ERK5 mRNA knocking-down in the cell lines was utilized for the experiments reported in the manuscript.
Stably expressed clones were selected by using medium containing G418 (400 ug/ml) for 28 days. The stable transfectants were named HL-60-siERK5 and HL-60-siControl respectively. To avoid colony-specific variation, we randomly selected about three to five cell clones and combined them for further analysis. Pooled stably transfected HL-60-siERK5 and HL-60-siControl cells were routinely maintained in selection media containing 200 ug/ml of G418-sulfate to avoid overgrowth of nontransfected cells.
Drug exposure
HL-60-siERK5 and HL-60-siControl were exposed to cytarabine for 2 h. Taking into account that the peak uptake level is 7.5 μg/ml for cytarabine in vitro [18], we tested 3.75 and 7.5 μg/ml concentrations for cytarabine. Evaluation of the cytotoxic effect was performed 48 h after the end of drug exposure.
Western blot
Cells were lysated and cellular proteins were denaturated, separated on 10% SDS-polyacrylamide gel and then electroblotted onto Hybond-C extra membrane. The membrane was stained with Ponceau S to verify equal amounts of sample loading and then incubated for 2 h at room temperature with T-PBS 5% non fat dry milk. The membrane was probed overnight at 4°C with the primary antibody and then with a horseradish peroxidase-conjugated secondary antibody diluted 1:1000 (Dako Corporation, Glostrup, Denmark). The bound antibodies were detected by enhanced chemiluminescence (ECL) using an ECL kit (Amersham Biosciences). The following primary antibodies were used: anti-ERK5, phospho-ERK5 (Thr218/Tyr220) and β-actin antibodies.
Immunocytochemical assay
Cells were grown on fibronectincoated coverslips, washed in PBS, and fixed for 15 min in 4% paraformaldehyde. Cell monola yers were permeabilized in 0.1% Triton X-100, washed, and blocked in 10% normal goat serum. Cells were incubated with the anti-ERK5 antibody overnight at 4°C. Immunolabelling was performed by a standard avidin-biotin (ABC) technique using a commercial kit (Maxim, China).Slides were examined under a microscope at a magnification of 200 ×. The results of immunohistochemical analyses were verified by three pathologists.
Quantitative real-time PCR
Total RNA was isolated with TRIzol reagent (Invitrogen) and first strand cDNA was synthesized from 1 μg total RNA using Oligo d (T) primer (Invitrogen) and ReveTra Ace (TOYOBO, Osaka, Japan). Quantitative real-time PCR was done using 2 μl of first-strand cDNA to quantitatively determine the relative amounts of ERK5 cDNAs. Primers used were for ERK5: F: 5’-CTGGCTGTCCAGATGTGAA-3’ and R: 5’-ATGGCACCATCTTTCTTTGG-3’. PCR was done with SYBR-green Real time PCR Master Mix (Toyobo, Osaka, Japan) and was run on a LightCycler instrument (Roche Molecular Biochemicals, Lewes, Sussex, UK). Thermal cycler parameters included one cycle at 94°C for 0.5 min, and 35 cycles involving denaturation at 94°C for 5 s annealing at 55°C for 5 s and extension at 72°C for 10 s. In each cycle, the amplified products were analyzed and the standard curve of ERK5 mRNA could be constructed and the copy numbers of samples were calculated by the Lightcycler software.
Colony formation assay
HL-60-siERK5 and HL-60-siControl cells (8 × 103/ml) or/and exposed to 3.75 μg/ml concentrations of cytarabine were maintained in 1 ml of 0.3% basal medium Eagle’s agar containing serum at 37°C in a humidified incubator for 14 days. Cell colonies were counted under a microscope using three different plates.
MTT assay
For quantitative viability assays, HL-60-siERK5 and HL-60-siControl cells were plated in 96-well plates (5 × 104 cells/well), or/and exposed to 3.75-and 7.5-μg/ml concentrations of cytarabine for 48 h. MTT assays were used to assess cell viability. 200 ul sterile MTT dye (5 mg/ml, sigma, USA) was added. After 4 h incubation at 37°C in 5% CO2, MTT medium mixture was removed and 200 ul of dimethyl sulfoxide (DMSO) was added to each well. Absorbance was measured at 490 nm using a multi-well spectrophotometer (Thermo Electron, Andover, USA). All experiments were carried out in triplicate.
ELISA assay for apoptosis
The experiment was divided into 2 parts: ① HL-60 leukemic cells were cultivated at a density of 5 × 104 cells/well in 24-well plates and then transciently transfected into siERK5 or siControl. After 72 h of transfection, cells were collected and apoptosis was detected using an ELISA cell death detection kit (Roche Diagnostics GmbH) according to the supplier’s recommendations; ② Stably transfected HL-60-siERK5 and HL-60-siControl cells at a density of 5 × 104 cells/well were exposed to 3.75- and 7.5-μg/ml concentrations of cytarabine for 48 h, apoptosis was detected using an ELISA cell death detection kit.
Statistical evaluation
Statistical analysis was performed using SPSS software (SPSS11.0; SPSS Inc). All results are expressed as the mean ± S.D. as indicated. For determination of the significance of differences the ANOVA and t test was performed.
Results
siRNA suppressed ERK5 mRNA and protein levels in HL-60 cells
Previous studies have reported on siRNA technique for successful silencing ERK5 expression in cancer cells [19]. In the present study, RNA interference technique was employed to knockdown ERK5 expression in HL-60 cells. siRNA was transciently transfected into the HL-60 cells for 24-72 h. As expected, expressions of ERK5 both mRNA and protein in siERK5 transfected HL-60 cells were significantly lower in a time-dependant way than those in siControl or nontransfected cells analyzed by immunocytochemical assay (Figure 1A), quantitative PCR (Figure 1B), Western blot (Figure 1C), respectively, suggesting that RNAi could effectively inhibit ERK5 expression in HL-60 cells.
Figure 1.

Effects of ERK5-siRNA on ERK5 mRNA and protein assessed by quantitative RT-PCR, western blot and immunocytochemical assays. siRNA was transciently transfected into the HL-60 cells for 24-72 h. ERK5 protein (A, B) and mRNA (C) and of siERK5 cells were significantly inhibited in a time-dependant way than those of nontransfection or siControl cells respectively (*P < 0.05, **P < 0.01). In the siERK5 stably transfected HL-60 cells, ERK5 protein (D, E) and Mrna (F) of siERK5 cells were also significantly inhibited (**P < 0.01).
In the siERK5 stably transfected HL-60 cells, ERK5 mRNA and protein was completed inhibited by immunocytochemical assay (Figure 1D), quantitative PCR (Figure 1E), and Western blot (Figure 1F), respectively.
Apoptosis assays
The HL-60 leukemic cells were cultivated at a density of 5 × 104 cells/well in 24-well plates and then transciently transfected into siERK5 or siControl. After 24-72 h of transfection, cells were collected and apoptosis was detected using an ELISA cell death detection kit. The results showed that suppression of ERK5 led to a time-dependant significant in apoptosis rate of HL-60 cells (Figure 2A).
Figure 2.

Effects of ERK5-siRNA on apoptosis, proliferation and colony formation. A. The effects of siERK5 at 24, 48, and 72 hs on apoptosis of HL-60 cell lines are shown. B. The effects of siERK5 at 24, 48, and 72 hs on viability of HL-60 cell lines are shown. C. The effects of siERK5 on colony formation of HL-60 cell lines are shown. *P < 0.05; **P < 0.01.
Proliferation assays
Next, we determined the proliferation of siERK5 or siControl on HL-60 cells. As shown in Figure 2B, compared with siControl and nontransfection HL-60 cells, siERK5 was significantly inhibited to 19.3% (P < 0.05), 26.4% (P < 0.05) and 39.2% (P < 0.01) at 24, 48, and 72 hs, respectively. There was no significant difference between siControl and nontransfection (P > 0.05).
Colony formation assay
HL-60-siERK5 and HL-60-siControl cells (8 × 103/ml) were maintained in 1 ml of 0.3% basal medium Eagle’s agar containing serum at 37°C in a humidified incubator for 14 days. Cell colonies were counted under a microscope using three different plates. As showed in Figure 3C, compared with siControl and nontransfection HL-60 cells, HL-60-siERK5 cells demonstrated a significant decrease in colony formation. After 14 days culture, the colonies that HL-60-siERK5 cells formed was 51.3% of siControl and 54.2% of nontransfection cells, respectively (Figure 2C, P < 0.05).
Figure 3.
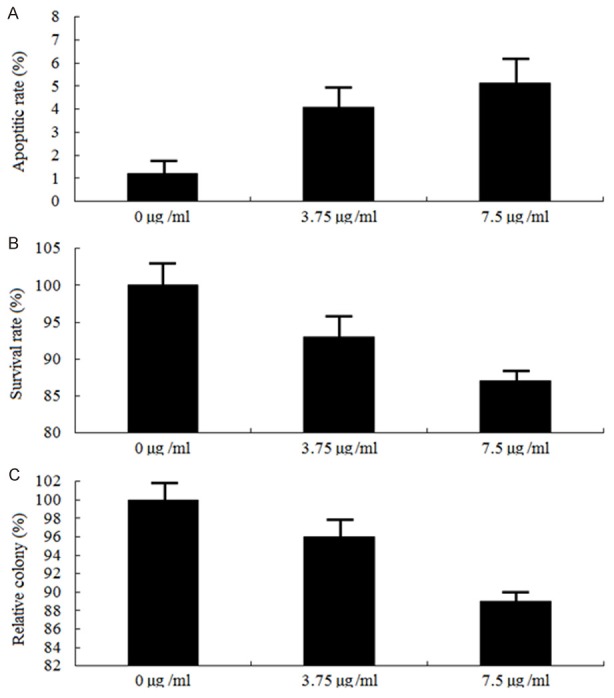
Effects of cytarabine on apoptosis, proliferation and colony formation. HL-60 cells were exposed to 3.75 and 7.5 μg/ml concentrations of cytarabine for 48 hs, ELISA assay (A) was used to detect apoptosis, MTT (B) was used to detect survival rate and colony formation assay was used to detect proliferation (P > 0.05).
Cytarabine treatment on apoptosis, proliferation and colony formation in HL-60 cells
HL-60 cells were exposed to 3.75- and 7.5 μg/ml concentrations of cytarabine for 48 hs, only low levels (5.7% and 7.4%) of apoptosis were detected (Figure 3A, P > 0.05). Proliferation and colony formation was also less inhibited (Figure 3B and 3C, P > 0.05, respectively). This might be due to the endogenous ERK5.
ERK5 depletion sensitized HL-60 cells to apoptosis induced by cytarabine
We treated the HL-60-siERK5 or HL-60-siControl cells with 3.75- and 7.5-μg/ml concentrations of cytarabine for 24 h and 3.75 μg/ml cytarabine for various time lengths (0, 12, 24, 36 and 48 h), respectively. The results showed that suppression of ERK5 led to a significant decrease in cell viability of HL-60-siERK5 cells in response to cytarabine in both time- (Figure 4A) and dose -dependent (Figure 4B) manners compared with those of HL-60-siControl or HL-60-nontransfection cells. Furthermore, suppression of ERK5 led to a significant increase in apoptotic rate in response to cytarabine in both time- (Figure 4C) and does-dependent (Figure 4D) manners compared with those of HL-60-siControl or HL-60-nontransfection cells. Colony formation assay showed that HL-60-siERK5 cells treated with 3.75 μg/ml cytarabine and at 37°C in a humidified incubator for 14 days resulted in fewer cell colonies compared with those of HL-60-siControl or HL-60-nontransfection cells treated with 3.75 μg/ml cytarabine (Figure 4E). The data suggest that ERK5 depletion may effectively enhance chemosensitivity of HL-60 cells to cytarabine.
Figure 4.

ERK5 modulation of cytarabine-induced cell death in HL-60 cells. HL-60-siERK5 or HL-60-siControl cells exposed to cytarabine (3.75- and 7.5-μg/ml) for 0, 12, 24, 36, and 48 h or cytarabine (3.75 μg/ml) for 24 h. A, B. MTT assay.*P < 0.05, **P < 0.01. C, D. ELISA assay (representative experiments); E. HL-60-siERK5 or HL-60-siControl cells were exposed to 3.75 μg/ml concentrations of cytarabine at 37°C in a humidified incubator for 14 days. Cell colonies were counted under a microscope using three different plates. *P < 0.05, **P < 0.01.
Discussion
Despite intensive efforts in the treatment of AML, it is unfortunately still deemed as an incurable disease with a high mortality rate. Owing to the occurrence of chemoresistance in leukemia cells, the majority of patients does not achieve CR or show relapse after first CR, following the standard therapy [20,21]. Therefore, development of new strategies for improved therapy is required. Aberrant mitogen/extracellular signal-regulated kinase 5 (MEK5)-extracellular signal-regulated protein kinase 5 (ERK5)-mediated signaling has been implicated in a number of tumor types, including AML, and the molecular basis of ERK5-driven carcinogenesis and its clinical relevance remain to be fully characterized. For example, ERK5-overexpressing PC3 cells have enhanced proliferative and invasive capabilities in vitro, and were dramatically more efficient in forming tumors, with a shorter mean time for tumors to reach a critical volume of 1000 mm3, in vivo, and vice versa [22,23].
ERK5 pathway also plays a role in the control of monocytic differentiation, which is disturbed in myeloid leukemia [24]. Inhibition of ERK5 auto-phosphorylation results in a particularly robust cell cycle arrest in G2 phase in AML cells [25]. Furthermore, inhibition of MEK5/ERK5 induces apoptosis of both FLT3-ITD transfected Ba/F3 cells as well as the FLT3-ITD carrying leukemic cell lines MV4-11 and MOLM-13, suggesting that MEK5/ERK5 is important for FLT3-ITD induced hematopoietic transformation and may thus represent an alternative therapeutic target in the treatment of FLT3-ITD positive leukemia [26]. Overexpression of s ERK5 is attributed to the chemoresistance of tumor cells [27,28]. On the contrary, different studies have shown that suppression of ERK5 expression can sensitize tumor cells to anti-cancer drugs and neurotrophin-3 induced apoptosis [29,30]. In the current study to investigate whether down-regulation of ERK5 affects chemosensitivity in AML cells, we examined the effect of ERK5 specific siRNA and cytarabine alone or in combination on HL-60 AML cells.
Immunocytochemical, (quantitative real time PCR) qRT-PCR and Western blot analysis revealed that transfection with pERK5 siRNA drastically reduced pERK5 mRNA and protein levels during the 72 h period. These findings suggest that pERK5 siRNA effectively had cleaved pERK5 mRNA and thereby blocked its translation to its corresponding protein. The results of the cell survival rate and colony formation assay showed that the down-regulation of pERK5 significantly inhibited the cell survival and number of colony of the HL-60 cells, demonstrating that ERK5 has an important role in the growth of leukemic cells. Most notably, the results of ELISA assay exhibited that down-regulation of pERK5 significantly promoted the apoptosis of HL-60 cells. This proposes that ERK5 could be a target for HL-60 cells treatment.
The study above showed that in addition to triggering apoptosis, recruiting surviving leukemia cells to a proliferative state, down-regulation of pERK5 may sensitize the leukemia cells to cytarabine. To further define the role of pERK5 in the drug resistance of leukemia cells, we tested the effect of pERK5 expression suppression on the apoptotic effect of cytarabine. Apoptosis assay findings indicated that monotherapy with cytarabine led to few apoptotic HL-60 cells. In addition, siRNA-mediated inhibition of pERK5 induced remarkable spontaneous apoptosis and enhanced sensitivity of the tumor cells to cytarabine -mediated apoptosis. In contrast, NC siRNA treatments did not alter the pERK5 gene expression, cell proliferation and cell toxicity of cytarabine, illustrating the specific impact of pERK5 siRNA. The above-mentioned subjects show that the presence of pERK5 is necessary for the survival and drug resistance of leukemic cells. Therefore, silencing of pERK5 expression could trigger spontaneous apoptosis and sensitize tumor cells to antileukemic drugs.
In conclusion, our data demonstrated that pERK5 has a critical role in the proliferation and drug resistance of HL-60 cells. Specific knockdown of pERK5 expression by siRNA induced apoptosis and synergistically enhanced sensitivity of leukemic cells to cytarabine. Our study emphasizes the ability of pERK5 siRNA for chemosensitization of leukemia cells to reduce the harmful side-effects of intensive chemotherapy. We suggest that the siRNA-silencing pERK5 may be considered as a novel treatment strategy in the future to overcome drug resistance of AML patients.
Disclosure of conflict of interest
None.
References
- 1.Meshinchi S, Arceci RJ. Prognostic factors and risk-based therapy in pediatric acute myeloid leukemia. Oncologist. 2014;12:341–355. doi: 10.1634/theoncologist.12-3-341. [DOI] [PubMed] [Google Scholar]
- 2.Kaspers GJ, Zwaan CM. Pediatric acute myeloid leukemia: towards high quality cure of all patients. Haematologica. 2007;92:1519–1532. doi: 10.3324/haematol.11203. [DOI] [PubMed] [Google Scholar]
- 3.Zwaan CM, Kaspers GJ. Possibilities for tailored and targeted therapy in paediatric acute myeloid leukaemia. Br J Haematol. 2014;127:264–279. doi: 10.1111/j.1365-2141.2004.05167.x. [DOI] [PubMed] [Google Scholar]
- 4.Li G, Chang H, Zhai YP, Xu W. Targeted silencing of inhibitors of apoptosis proteins with siRNAs: a potential anti-cancer strategy for hepatocellular carcinoma. Asian Pac J Cancer Prev. 2013;14:4943–52. doi: 10.7314/apjcp.2013.14.9.4943. [DOI] [PubMed] [Google Scholar]
- 5.Akagi H, Higuchi H, Sumimoto H. Suppression of myeloid cell leukemia-1 (Mcl-1) enhances chemotherapy-associated apoptosis in gastric cancer cells. Gastric Cancer. 2013;16:100–10. doi: 10.1007/s10120-012-0153-6. [DOI] [PubMed] [Google Scholar]
- 6.Hayashi M, Fearns C, Eliceiri B, Yang Y, Lee JD. Big mitogen-activated protein kinase 1/extracellular signal-regulated kinase 5 signaling pathway is essential for tumor-associated angiogenesis. Cancer Res. 2005;65:7699–706. doi: 10.1158/0008-5472.CAN-04-4540. [DOI] [PubMed] [Google Scholar]
- 7.Roberts OL, Holmes K, Muller J, Cross DA, Cross MJ. ERK5 and the regulation of endothelial cell function. Biochem Soc Trans. 2009;37:1254–9. doi: 10.1042/BST0371254. [DOI] [PubMed] [Google Scholar]
- 8.Wang X, Tournier C. Regulation of cellular functions by the ERK5 signalling pathway. Cell Signal. 2006;18:753–60. doi: 10.1016/j.cellsig.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 9.Wang X, Tournier C. Regulation of cellular functions by the ERK5 signalling pathway. Cell Signal. 2006;18:753–760. doi: 10.1016/j.cellsig.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Carvajal-Vergara X, Tabera S, Montero JC, Esparis-Ogando A, Lopez-Perez R, Mateo G, Gutierrez N, Parmo-Cabanas M, Teixido J, San Miguel JF, Pandiella A. Multifunctional role of Erk5 in multiple myeloma. Blood. 2005;105:4492–4499. doi: 10.1182/blood-2004-08-2985. [DOI] [PubMed] [Google Scholar]
- 11.Buschbeck M, Hofbauer S, Croce LD, Keri G, Ullrich A. Ablkinase-sensitive levels of ERK5 and its intrinsic basal activity contribute to leukaemia cell survival. EMBO Rep. 2014;6:63–69. doi: 10.1038/sj.embor.7400316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagel S, Burek C, Venturini L, Scherr M, Quentmeier H, Meyer C, Rosenwald A, Drexler HG, Macleod RA. Comprehensive analysis of homeobox genes in Hodgkin lymphoma cell lines identifies dysregulated expression of HOXB9 mediated via ERK5 signaling and BMI1. Blood. 2007;109:3015–3023. doi: 10.1182/blood-2006-08-044347. [DOI] [PubMed] [Google Scholar]
- 13.Akao Y, Nakagawa Y, Kitade Y, Kinoshita T, Naoe T. Downregulation of microRNAs-143 and -145 in B-cell malignancies. Cancer Sci. 2007;98:1914–1920. doi: 10.1111/j.1349-7006.2007.00618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weldon CB, Scandurro AB, Rolfe KW, Clayton JL, Elliott S, Butler NN, Melnik LI, Alam J, McLachlan JA, Jaffe BM, Beckman BS, Burow ME. Identification of mitogen-activated protein kinase as a chemoresistant pathway in MCF-7 cells by using gene expression microarray. Surgery. 2002;132:293–301. doi: 10.1067/msy.2002.125389. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Pesakhov S, Harrison JS, Danilenko M, Studzinski GP. ERK5 pathway regulates transcription factors important for monocytic differentiation of human myeloid leukemia cells. J Cell Physiol. 2014;229:856–67. doi: 10.1002/jcp.24513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Charni S, Aguilo JI, Garaude J, de Bettignies G, Jacquet C, Hipskind RA, Singer D, Anel A, Villalba M. ERK5 knockdown generates mouse leukemia cells with low MHC class I levels that activate NK cells and block tumorigenesis. J Immunol. 2009;182:3398–405. doi: 10.4049/jimmunol.0803006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garaude J, Cherni S, Kaminski S, Delepine E, Chable-Bessia C, Benkirane M, Borges J, Pandiella A, Iñiguez MA, Fresno M, Hipskind RA, Villalba M. ERK5 activates NF-kappaB in leukemic T cells and is essential for their growth in vivo. J Immunol. 2006;177:7607–17. doi: 10.4049/jimmunol.177.11.7607. [DOI] [PubMed] [Google Scholar]
- 18.Mulder JH, Harrap KR. Cytosine arabinoside uptake by tumour cells in vitro. Eur J Cancer. 1975;11:373–9. doi: 10.1016/0014-2964(75)90066-3. [DOI] [PubMed] [Google Scholar]
- 19.Yue B, Ren QX, Su T, Wang LN, Zhang L. ERK5 silencing inhibits invasion of human osteosarcoma cell via modulating the Slug/MMP-9 pathway. Eur Rev Med Pharmacol Sci. 2014;18:2640–2647. [PubMed] [Google Scholar]
- 20.Smits EL, Berneman ZN, Van Tendeloo VF. Immunotherapy of acute myeloid leukemia: current approaches. Oncologist. 2009;14:240–52. doi: 10.1634/theoncologist.2008-0165. [DOI] [PubMed] [Google Scholar]
- 21.Szer J. The prevalent predicament of relapsed acute myeloid leukemia. Hematol Am Soc Hematol Educ Program. 2012;2012:43–8. doi: 10.1182/asheducation-2012.1.43. [DOI] [PubMed] [Google Scholar]
- 22.McCracken SR, Ramsay A, Heer R, Mathers ME, Jenkins BL, Edwards J, Robson CN, Marquez R, Cohen P, Leung HY. Aberrant expression of extracellular signal-regulated kinase 5 in human prostate cancer. Oncogene. 2008;27:2978–8823. doi: 10.1038/sj.onc.1210963. [DOI] [PubMed] [Google Scholar]
- 23.Ramsay AK, McCracken SR, Soofi M, Fleming J, Yu AX, Ahmad I, Morland R, Machesky L, Nixon C, Edwards DR, Nuttall RK, Seywright M, Marquez R, Keller E, Leung HY. ERK5 signalling in prostate cancer promotes an invasive phenotype. Br J Cancer. 2011;104:664–72. doi: 10.1038/sj.bjc.6606062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang X, Pesakhov S, Harrison JS, Danilenko M, Studzinski GP. ERK5 pathway regulates transcription factors important for monocytic differentiation of human myeloid leukemia cells. J Cell Physiol. 2014;229:856–67. doi: 10.1002/jcp.24513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Pesakhov S, Weng A, Kafka M, Gocek E, Nguyen M, Harrison JS, Danilenko M, Studzinski GP. ERK 5/MAPK pathway has a major role in 1α, 25-(OH)2 vitamin D3-induced terminal differentiation of myeloid leukemia cells. J Steroid Biochem Mol Biol. 2014;144 Pt A:223–7. doi: 10.1016/j.jsbmb.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Razumovskaya E, Sun J, Rönnstrand L. Inhibition of MEK5 by BIX02188 induces apoptosis in cells expressing the oncogenic mutant FLT3-ITD. Biochem Biophys Res Commun. 2011;412:307–12. doi: 10.1016/j.bbrc.2011.07.089. [DOI] [PubMed] [Google Scholar]
- 27.Weldon CB, Scandurro AB, Rolfe KW, Clayton JL, Elliott S, Butler NN, Melnik LI, Alam J, McLachlan JA, Jaffe BM, Beckman BS, Burow ME. Identification of mitogen-activated protein kinase as a chemoresistant pathway in MCF-7 cells by using gene expression microarray. Surgery. 2002;132:293–301. doi: 10.1067/msy.2002.125389. [DOI] [PubMed] [Google Scholar]
- 28.Pi X, Yan C, Berk BC. Big mitogen-activated protein kinase (BMK1)/ERK5 protects endothelial cells from apoptosis. Circ Res. 2004;94:362–9. doi: 10.1161/01.RES.0000112406.27800.6F. [DOI] [PubMed] [Google Scholar]
- 29.Sayan M, Shukla A, MacPherson MB, Macura SL, Hillegass JM, Perkins TN, Thompson JK, Beuschel SL, Miller JM, Mossman BT. Extracellular signal-regulated kinase 5 and cyclic AMP response element binding protein are novel pathways inhibited by vandetanib (ZD6474) and doxorubicin in mesotheliomas. Am J Respir Cell Mol Biol. 2014;51:595–603. doi: 10.1165/rcmb.2013-0373TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sturla LM, Cowan CW, Guenther L, Castellino RC, Kim JY, Pomeroy SL. A novel role for extracellular signal-regulated kinase 5 and myocyte enhancer factor 2 in medulloblastoma cell death. Cancer Res. 2005;65:5683–9. doi: 10.1158/0008-5472.CAN-04-2283. [DOI] [PubMed] [Google Scholar]