

Abstract
The histone deacetylase (HDACs) family contains a family of enzymes, which are involved in modulating a wide range of cellular processes, such as proliferation, differentiation, apoptosis, and cell cycle progression. However, the biological function of HDAC5 in colorectal cancer has not been well established. In the current research, our data showed that the mRNA and protein levels of HDAC5 were up-regulated in human colorectal cancer cell lines. CCK-8 assay showed that overexpression of HDAC5 significantly promoted the proliferation of colorectal cancer cell lines including SW480 and HCT116. On the contrary, HDAC5 knockdown using small interfering RNA suppressed cell growth in colorectal tumor cells. At the molecular level, we demonstrated that HDAC5 promoted the expression of DLL4. In addition, down-regulation of DLL4 diminished the proliferative effects of HDAC5 in human colorectal cancer cells. Taken together, these results suggest that HDAC5 elevates the proliferation of colorectal cancer cells through up-regulation of DLL4. The current study might provide novel potential therapeutic targets in the treatment of colorectal cancer.
Keywords: Colorectal cancer, HDAC5, proliferation, DLL4
Introduction
Colorectal cancer (CRC) is the second leading cause of cancer death in the United States [1]. Despite new treatment options developed in the last decade, the prognosis for patients with advanced or recurrent CRC remains poor [2]. It is estimated that over 1 million new cases are diagnosed each year worldwide, and approximately 50% of these patients die of colorectal tumor [3]. Currently, surgical resection is the optimal treatment for colorectal cancer, and chemotherapy serves as one of the important adjuvant therapies for its treatment [4]. However, increasing studies have shown that the resistance of CRC cells to conventional drugs is becoming a challenging problem. Such limitation highlights the imperative need for identifying novel and effective therapeutic methods for the treatment of CRC.
The histone deacetylase (HDACs) regulate the acetylation level of histones and non-histone proteins and thus regulate the genes expression and chromatin structure [5]. HDACs family is composed of 18 proteins and these proteins are classified into classes I-IV based on their homology and structure [6]. Accumulating evidences suggested that HDAC family functions as an important regulator in tumor progression and metastasis [7,8]. Recently, several HDAC inhibitors have been shown to exhibit anti-tumor activity in cancer cells and animal models [9,10]. HDAC5, a member of the class II histone deacetylase family, has been shown to be aberrantly expressed in several types of tumors and play critical role in cellular behaviors including cell proliferation, cell cycle progression and apoptosis [11-13]. However, the biological function of HDAC5 in human CRC has not been fully elucidated and, thus the present study aimed to investigate the role of HDAC5 in human colorectal cancer using an in vitro cell model.
Materials and methods
Cell culture
The human CRC cell line SW480, HCT116, SW620 and a non-malignant colonic epithelial cell lines NCM460 were purchased from the American Type Culture Collection (Rockville, MD) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) in an atmosphere containing 5% CO2 at 37°C .
Cell viability assay
CRC cells including SW480 and HCT116 were seeded onto 96-well plates at 3 × 103 cells/well. The medium was replaced with the corresponding serum-free medium for 24 h, then serum-free medium was replaced with complete medium. Then 10 μL/well CCK-8 solution (Dojindo, Kumamoto, Japan) was added and incubated with the plates for 3 h, and the absorbance was determined at 450 nm using an MRX II microplate reader (Dynex, Chantilly, VA, USA).
Plasmid construction, siRNA and transfection
The cDNA fragment encoding HDAC5 was isolated with Takara RNA PCR kit (Takara, Japan) using total RNAs from SW480 cells. PCR products were cloned into pcDNA3.1 (+) (Invitrogen, Carlsbad, CA). HDAC5 siRNA, DLL4 siRNA and negative controls were purchased from Invitrogen (Carlsbad, CA, USA). Cells were transfected with lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the instruction.
Quantitative real-time PCR (qRT-PCR)
Total RNAs were isolated from cancer cells by TRIzol reagent, and reverse transcriptions were performed by Takara RNA PCR kit (Takara, Japan) according to the manufacturer’s instructions. In order to quantify the transcripts of the interest genes, quantitative Real-time PCR (qRT-PCR) was performed using a SYBR Green Premix Ex Taq ((Takara, Tokyo, Japan) on ABI 7500 system (Applied Biosystems, Foster, CA, USA). The transcript levels of genes of interest were normalized to GAPDH and were calculated using the 2-ΔΔCt method.
Western blotting analysis
Total cell extracts were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose filter membrane. Then the membranes were incubated with primary antibodies against HDAC5 (dilution, 1:1000), DLL4 (dilution, 1:1000) and GAPDH (dilution, 1:2000) (Santa Cruz, CA, USA). GAPDH was used as a loading control. The membranes were then incubated with the appropriate HRP-conjugated secondary antibodies for 1 h at room temperature. The proteins were visualized by the enhanced chemiluminescence method and intensity of protein bands was quantified by densitometry.
Statistical analysis
All data were presented as mean ± SD and treated for statistics analysis by SPSS program. Comparison between groups were determined by ANOVA and statistical significance was indicated as *P<0.05.
Results
Up-regulation of HDAC5 in CRC cell lines
Firstly, we detected the expression of HDAC5 in CRC cell lines and normal colonic epithelial cells by qRT-PCR. Compared with normal colonic epithelial cell lines NCM460, we found that the mRNA expression of HDAC5 was dramatically elevated in several CRC cell lines including SW480, HCT116, SW620 (Figure 1A). Furthermore, HDAC5 protein expression in CRC cells were analyzed by western blotting. Data also showed that the protein levels of HDAC5 was obviously up-regulated (Figure 1B) in the four CRC cell lines compared with that in control cells. These data suggested that the expression of HDAC5 was significantly increased in CRC cell lines.
Figure 1.
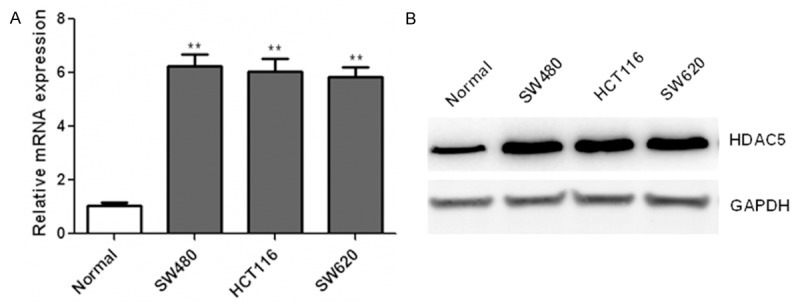
Up-regulation of HDAC5 in CRC cell lines. The mRNA and protein levels of HDAC5 were determined by qRT-PCR (A) and western blotting (B) in human colorectal cancer cell lines SW480, HCT116, SW620 and a non-malignant colonic epithelial cell lines NCM460. **P<0.01.
Overexpression of HDAC5 promoted the proliferation of CRC cells
To further elucidate the biological role of HDAC5 in CRC cells, SW480 and HCT116 cells were transfected with plasmids encoding HDAC5. Then CCK-8 assay was performed and data showed that HDAC5 overexpression obviously enhanced the proliferation of SW480 and HCT116 cells (Figure 2A and 2B). By contrast, down-regulation of HDAC5 with small interfering RNA (siRNA) in SW480 cells was confirmed by qRT-PCR (Figure 3A) and western blotting (Figure 3B and 3C). Consequently, cell growth was obviously suppressed after HDAC5 knockdown in SW480 cells (Figure 3D). Similar results were also observed in HCT116 cells (data not shown). These results indicated that HDAC5 up-regulation could promote the proliferation of CRC cells.
Figure 2.
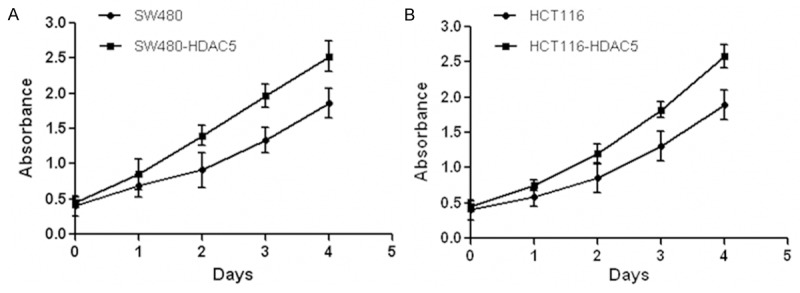
Enforced expression of HDAC5 promoted CRC cells growth. Human colon cancer cell lines were transfected with plasmids encoding HDAC5 and control plasmids. Then CCK-8 assay was performed to evaluate the time course changes of SW480 (A) and HCT116 (B) cells proliferation.
Figure 3.
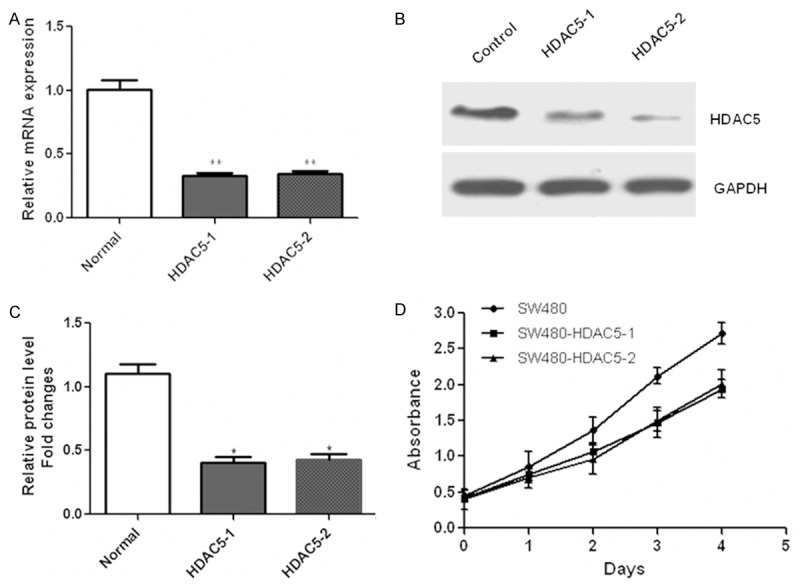
Down-regulation of HDAC5 inhibited the proliferation of CRC cells. Down-regulation of HDAC5 was detected with qRT-PCR (A) and western blotting (B and C) in SW480 cells transfected with siRNA oligos targeting HDAC5. Then, cells proliferation was determined using the CCK-8 (D) in SW480 cells transfected with HDAC5 siRNA oligos. *P<0.05; **P<0.01.
HDAC5 increased the expression of DLL4 in CRC cells
We further investigated the molecular mechanism underlying the proliferative effects of HDAC5 on CRC cells. Western blotting analysis showed that protein expression of DLL4 was increased after over-expression of HDAC5 in SW480 cells (Figure 4A and 4B). On the contrary, down-regulation of HDAC5 by siRNA significantly reduced the DLL4 expression (Figure 4C and 4D). These results showed that HDAC5 served as a positive regulator of DLL4 expression in CRC cells.
Figure 4.
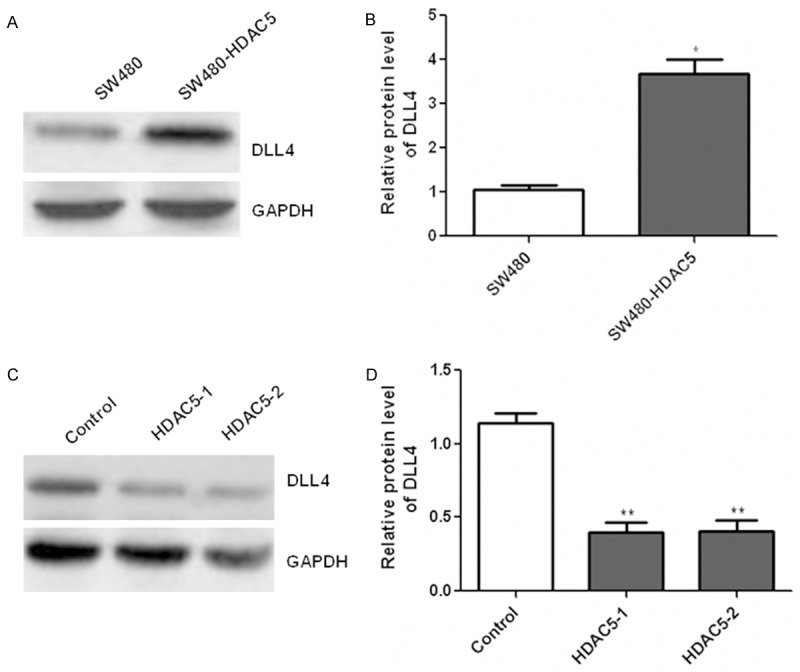
HDAC5 enhanced the expression of DLL4 in CRC cells. SW480 cells were transfected with plasmids encoding HDAC5 and then the expression of DLL4 was detected with western blotting (A and B). After transfection with HDAC5 siRNA, western blotting was used to determine the DLL4 expression (C and D). Relative band intensities of each protein were quantified by densitometry. *P<0.05; **P<0.01.
HDAC5 enhanced tumor cells proliferation by up-regulation of DLL4
In order to investigate how HDAC5 exhibited the proliferative effects on CRC cells, we silenced the expression of DLL4 via transfection with DLL4 siRNA. Western blotting was applied to confirm the down-regulation of DLL4 (Figure 5A). As a result, the proliferative effects of HDAC5 on tumor cells were diminished after down-regulation of DLL4 in SW480 cells (Figure 5B). Taken together, these results demonstrated that HDAC5 could promote CRC cell proliferation by up-regulation of DLL4.
Figure 5.
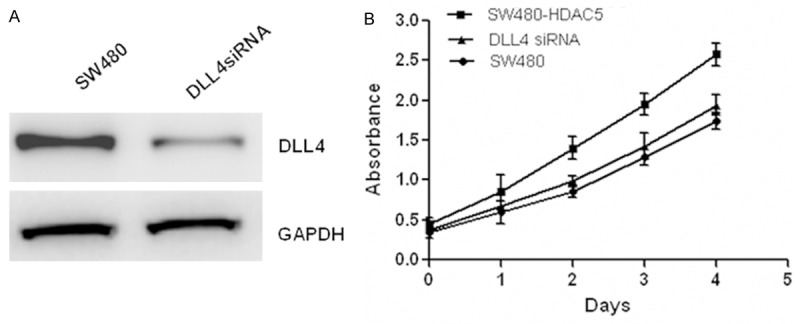
HDAC5 promotes cells proliferation via DLL4 in CRC cells. The expression of DLL4 was decreased after transfection with specific DLL4 siRNA (A). And CCK-8 assay (B) was performed to measure cell proliferation in SW480 cells overexpressing HDAC5 with or without DLL4 siRNA.
Discussion
A large body of literature indicates that HDAC family members function as critical regulators of cell growth, differentiation, and apoptotic programs. Their aberrant expression is closely related with tumor development and progression [14]. Based on such studies, several HDAC inhibitors, such as SAHA, depsipeptide, valproic acid and MS-275, are in clinical trials for the treatment of solid and hemopoetic tumors [15]. HDAC5 belongs to the class II histone deacetylase family and is a critical regulator in cell proliferation in many cancer cell lines [16]. It has been reported that HDAC5 was significantly over-expressed in high-risk medulloblastoma in comparison with low-risk medulloblastoma, and its expression was associated with poor survival, suggesting that HDAC5 may be an important marker for risk stratification [13]. Recently, another study demonstrated that HDAC5 promoted the twist 1 expression and highlighted a potential link between HDAC5 and osteosarcoma progression [17]. In glioma cells, HDAC5 was shown to promote glioma cells proliferation via up-regulation of Notch 1 expression, which might provide novel therapeutic targets in the treatment of gliomas [18]. The above results support the crucial roles of HDAC5 in the carcinogenesis. However, the relationship between HDAC5 and colorectal cancer has not been reported. Thus, the present study examined the expression of HDAC5 in colorectal cancer cells and found that its expression was significantly increased both at the mRNA and protein levels. In addition, enforced expression of HDAC5 in colon tumor cells significantly promoted the proliferation rate, whereas HDAC5 knockdown suppressed cancer cell growth, suggesting that HDAC5 acts as a positive regulator of growth in colorectal cancer cells.
Notch signaling is a conserved pathway affecting numerous cell fate/lineage decisions in multicellular organisms during embryogenesis, postnatal development, and in the adult [19]. Delta-like 4 ligand (DLL4), the latest identified Notch ligand, is predominantly expressed in arterial endothelial cells during embryonic development, which is a critical component of Notch-mediated stem cell self-renewal and vascular development [20,21]. It has been shown that haploinsufficiency of DLL4 results in severe vascular defects and embryonic lethality [22]. Moreover, various studies demonstrate that inhibition of DLL4 leads to broad spectrum of antitumor activity, which makes DLL4 a potential therapeutic target [23-25]. Our study found that up-regulation of HDAC5 resulted in elevated expression of DLL4; conversely, down-regulation of HDAC5 reduced DLL4 levels in colorectal cancer cells. Furthermore, our data showed that the proliferative effects of HDAC5 on tumor cells was suppressed due to DLL4 knockdown in colon tumor cells, indicating that the proliferative activity of HDAC5 was mediated by DLL4.
In conclusion, our results demonstrated that HDAC5 enhanced the proliferation of colorectal cancer cells through up-regulation of DLL4 expression. Our study provides further insight into the pathogenic mechanisms of colorectal cancer, and indicates that HDAC5 may be a potential therapeutic target for colorectal cancer treatment.
Disclosure of conflict of interest
None.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Rehman MU, Buttar QM, Khawaja MI, Khawaja MR. An impending cancer crisis in developing countries: are we ready for the challenge? Asian Pac J Cancer Prev. 2009;10:719–20. [PubMed] [Google Scholar]
- 3.DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD. Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin. 2014;64:252–71. doi: 10.3322/caac.21235. [DOI] [PubMed] [Google Scholar]
- 4.Chibaudel B, Tournigand C, Andre T, de Gramont A. Therapeutic strategy in unresectable metastatic colorectal cancer. Ther Adv Med Oncol. 2012;4:75–89. doi: 10.1177/1758834011431592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kahali S, Sarcar B, Chinnaiyan P. The emerging role of histone deacetylases (HDACs) in UPR regulation. Methods Enzymol. 2011;490:159–74. doi: 10.1016/B978-0-12-385114-7.00010-6. [DOI] [PubMed] [Google Scholar]
- 6.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heideman MR, Wilting RH, Yanover E, Velds A, de Jong J, Kerkhoven RM, Jacobs H, Wessels LF, Dannenberg JH. Dosage-dependent tumor suppression by histone deacetylases 1 and 2 through regulation of c-Myc collaborating genes and p53 function. Blood. 2013;121:2038–50. doi: 10.1182/blood-2012-08-450916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mottet D, Castronovo V. [Histone deacetylases: a new class of efficient anti-tumor drugs] . Med Sci (Paris) 2008;24:742–6. doi: 10.1051/medsci/20082489742. [DOI] [PubMed] [Google Scholar]
- 9.Insinga A, Monestiroli S, Ronzoni S, Gelmetti V, Marchesi F, Viale A, Altucci L, Nervi C, Minucci S, Pelicci PG. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat Med. 2005;11:71–6. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- 10.Wagner JM, Hackanson B, Lubbert M, Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics. 2010;1:117–136. doi: 10.1007/s13148-010-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lai F, Jin L, Gallagher S, Mijatov B, Zhang XD, Hersey P. Histone deacetylases (HDACs) as mediators of resistance to apoptosis in melanoma and as targets for combination therapy with selective BRAF inhibitors. Adv Pharmacol. 2012;65:27–43. doi: 10.1016/B978-0-12-397927-8.00002-6. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Matkovich SJ, Duan X, Diwan A, Kang MY, Dorn GW 2nd. Receptor-independent protein kinase C alpha (PKCalpha) signaling by calpain-generated free catalytic domains induces HDAC5 nuclear export and regulates cardiac transcription. J Biol Chem. 2011;286:26943–51. doi: 10.1074/jbc.M111.234757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Milde T, Oehme I, Korshunov A, Kopp-Schneider A, Remke M, Northcott P, Deubzer HE, Lodrini M, Taylor MD, von Deimling A, Pfister S, Witt O. HDAC5 and HDAC9 in medulloblastoma: novel markers for risk stratification and role in tumor cell growth. Clin Cancer Res. 2010;16:3240–52. doi: 10.1158/1078-0432.CCR-10-0395. [DOI] [PubMed] [Google Scholar]
- 14.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 15.Li Z, Zhu WG. Targeting histone deacetylases for cancer therapy: from molecular mechanisms to clinical implications. Int J Biol Sci. 2014;10:757–70. doi: 10.7150/ijbs.9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marek L, Hamacher A, Hansen FK, Kuna K, Gohlke H, Kassack MU, Kurz T. Histone deacetylase (HDAC) inhibitors with a novel connecting unit linker region reveal a selectivity profile for HDAC4 and HDAC5 with improved activity against chemoresistant cancer cells. J Med Chem. 2013;56:427–36. doi: 10.1021/jm301254q. [DOI] [PubMed] [Google Scholar]
- 17.Chen J, Xia J, Yu YL, Wang SQ, Wei YB, Chen FY, Huang GY, Shi JS. HDAC5 promotes osteosarcoma progression by upregulation of Twist 1 expression. Tumour Biol. 2014;35:1383–7. doi: 10.1007/s13277-013-1189-x. [DOI] [PubMed] [Google Scholar]
- 18.Liu Q, Zheng JM, Chen JK, Yan XL, Chen HM, Nong WX, Huang HQ. Histone deacetylase 5 promotes the proliferation of glioma cells by upregulation of Notch 1. Mol Med Rep. 2014;10:2045–50. doi: 10.3892/mmr.2014.2395. [DOI] [PubMed] [Google Scholar]
- 19.Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–89. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 20.Oon CE, Harris AL. New pathways and mechanisms regulating and responding to Delta-like ligand 4-Notch signalling in tumour angiogenesis. Biochem Soc Trans. 2011;39:1612–18. doi: 10.1042/BST20110721. [DOI] [PubMed] [Google Scholar]
- 21.Huang F, Zhu X, Hu XQ, Fang ZF, Tang L, Lu XL, Zhou SH. Mesenchymal stem cells modified with miR-126 release angiogenic factors and activate Notch ligand Delta-like-4, enhancing ischemic angiogenesis and cell survival. Int J Mol Med. 2013;31:484–92. doi: 10.3892/ijmm.2012.1200. [DOI] [PubMed] [Google Scholar]
- 22.Gale NW, Dominguez MG, Noguera I, Pan L, Hughes V, Valenzuela DM, Murphy AJ, Adams NC, Lin HC, Holash J, Thurston G, Yancopoulos GD. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci U S A. 2004;101:15949–54. doi: 10.1073/pnas.0407290101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding XY, Ding J, Wu K, Wen W, Liu C, Yan HX, Chen C, Wang S, Tang H, Gao CK, Guo LN, Cao D, Li Z, Feng GS, Wang HY, Xu ZF. Cross-talk between endothelial cells and tumor via delta-like ligand 4/Notch/PTEN signaling inhibits lung cancer growth. Oncogene. 2012;31:2899–906. doi: 10.1038/onc.2011.467. [DOI] [PubMed] [Google Scholar]
- 24.Hu W, Lu C, Dong HH, Huang J, Shen DY, Stone RL, Nick AM, Shahzad MM, Mora E, Jennings NB, Lee SJ, Roh JW, Matsuo K, Nishimura M, Goodman BW, Jaffe RB, Langley RR, Deavers MT, Lopez-Berestein G, Coleman RL, Sood AK. Biological roles of the Delta family Notch ligand Dll4 in tumor and endothelial cells in ovarian cancer. Cancer Res. 2011;71:6030–9. doi: 10.1158/0008-5472.CAN-10-2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu SK, Bham SA, Fokas E, Beech J, Im J, Cho S, Harris AL, Muschel RJ. Delta-like ligand 4-notch blockade and tumor radiation response. J Natl Cancer Inst. 2011;103:1778–98. doi: 10.1093/jnci/djr419. [DOI] [PubMed] [Google Scholar]