

Abstract
This study was to uncover the role of long non-coding RNA (lncRNA) in the process of endometrial cancer (EC) development using microarray technique to obtain the expression profiles of lncRNAs in EC and its adjacent normal tissues. A total of 45 pieces of pathologically-proven EC tissues were used in this study. All samples were frozen in liquid nitrogen immediately after resection and stored at -80°C for future use. The detection of lncRNA and transcripts was conducted using microarray analysis. To understand the biochemical function of lncRNA, bioinformatics analyses (gene ontology and pathway analyses) were performed. To further investigate the relationship between lncRNAs and EC, subgroup analysis was conducted. In order to validate the consistency of the lncRNAs with microarray data, qRT-PCR was performed. In this study, 30586 lncRNAs and 26109 transcripts (fold change ≥ 2.0) were found in the tested EC. In particular, compared with normal tissues, 4010 the lncRNA were up-regulated, and 3350 of them were down-regulated. Seven of the lncRNAs were in accordance with microarray data in qRT-PCR. Among these lncRNAs, 3 were up-regulated and 4 were down-regulated. Furthermore, pathway analysis revealed that 24 pathways were correlated to the up-regulated transcripts, while 27 pathways were associated with the down-regulated transcripts. Our study demonstrated that the expressions of a large amount of lncRNAs were altered in EC in comparison to normal tissues, suggesting that lncRNAs could potentially serve as a diagnostic biomarker that is beneficial for the diagnosis and therapy of EC.
Keywords: Long non-coding RNA, endometrial cancer, microarray analysis, gene ontology analysis, pathway analysis
Introduction
As the most common malignant tumor occurring in female reproductive system, endometrial cancer (EC) accounts for 7% of the total female malignant tumors. Obesity and diabetes are currently the most risky inducing factors [1,2]. The morbidity of EC has increased by 1 fold in the past twenty years [3]. According to the statistics from American Cancer Society, the number of new cases of EC in America was 49,560 in 2013, with the number of new deaths being 8,200. The average age of onset was 61, with most of the patients having 55-60 years of age [1,2], suggesting the occurrence tendency on young women in recent years [4]. Although the majority of EC cases are diagnosed at early stage, the disease seriously threats human health due to its relatively high mortality rate [5]. EC could be divided into estrogen-dependent type and estrogen-independent type. About 80% of EC patients belong to estrogen-dependent type, having better prognosis compared to estrogen-independent type [4]. Due to the high morbidity rate and recurrence rate of EC, the present diagnosis and therapy are not satisfactory. Therefore, understanding EC at molecular level and selecting remarkable biomarkers would play important roles in the diagnosis of EC as well as the improvement of survival rate and cure rate. Previous researches have confirmed the correlation between EC and certain genes, such as p15, p53, ki67, PTEN and k-ras [6-10]. However, the results were still unsatisfactory.
In the past few decades, researchers performed a great amount of studies on the biological function of non-coding RNA in mammal genomes, and recognized the significant regulating effects of non-coding RNA on various biological processes. Based on its length, non-coding RNA is classified into long non-coding RNA (lncRNA) and short non-coding RNA. Particularly, lncRNA is a kind of RNA with a length of transcription longer than 200 nucleotides [11], while the length of short non-coding RNA is in the range of 180 to 200 nucleotides. Large amounts of researches evidenced the abnormal expression of microRNA (miRNA or miR), one of the most frequently studied short non-coding RNA, in various tumors [12]. Several miRNAs, such as miR-205, miR-204 and miR-20a, were considered to be associated with EC morbidity [13,14]. Recent investigations on lncRNA showed that lncRNA exists in cytoplasm or nucleus, and exerts its regulatory effects on gene expression levels, including epigenetic regulation, transcriptional regulation and post-transcriptional regulation in the form of RNA [15]. LncRNA could be divided into different subgroups, such as lncRNAs with enhancer-like function (lncRNA-a), HOX lncRNA and large intergenic non-coding lncRNAs [16,17]. Recent studies revealed that these lncRNAs might be involved in the occurrence and development of multiple diseases, playing an important role in human cells [16,17]. Compared with other types of non-coding RNAs, lncRNA has much longer sequences and more complicated spatial construction. As a result, the regulation mechanisms of lncRNA in biological processes are more diverse and complicated [18]. It is well established that abnormal expression of lncRNA plays an important role in the development of many types of human tumors [19-22]. Despite progresses made in the studies on the role of lncRNA in various human diseases, the functions of lncRNA have rarely been confirmed [23].
In the present study, we used the third-generation microarray techniques to analyze the expression of a group of lncRNA and mRNA in EC and adjacent normal tissues, and validated 10 abnormally expressed lncRNA from 40 samples. The study also showed that large amounts of abnormally expressed genes are present in EC, playing a key role in the morbidity of EC. Our results will be beneficial for the early diagnosis and effective treatment of EC.
Materials and methods
Patients
Between September 2013 and December 2013, we obtained a total of 45 pieces of pathologically-proven EC tissues from patients at Shandong Provincial Hospital. We randomly chose one patient sample for the process of lncRNA expression profile. All samples were frozen in liquid nitrogen immediately after resection and stored at -80°C. This study was approved by the Research Ethics Committee of Shandong Provincial Hospital. Informed consent was obtained from all patients or their families.
Microarray analysis
Arraystar Human LncRNA Microarray V3.0 was designed for the global profiling of human lncRNAs and protein-coding transcripts. About 30,586 lncRNAs and 26,109 coding transcripts could be detected by the third-generation lncRNA microarray. The lncRNAs were carefully constructed using the most highly-respected public transcriptome databases (Refseq, UCSC knowngenes, Gencode, etc.), as well as landmark publications. Each transcript was represented by a specific exon or splice junction probe that could accurately identify individual transcripts. Positive probes for housekeeping genes and negative probes were also printed onto the array for hybridization quality control. Microarray analysis was performed by Kangchen Bio-tech, Shanghai, P. R. China.
RNA labeling and array hybridization
Sample labeling and array hybridization were performed according to Agilent One-Color Microarray-Based Gene Expression Analysis protocol (Agilent Technologies, Santa Clara, CA, USA) with minor modifications. Briefly, mRNA was purified from total RNA after removal of ribosomal RNA (mRNA-ONLY™ Eukaryotic mRNA Isolation Kit, Epicentre Technologies Corp., Chicago, IL, USA). Then, each sample was amplified and transcribed into fluorescent cRNA along the entire length of the transcripts without 3’ bias utilizing a random priming method. The labeled cRNAs were purified by RNeasy Mini Kit (Qiagen, Germany). The concentration and specific activity of the labeled cRNAs (pmol Cy3/μg cRNA) were measured by NanoDrop ND-1000 (Thermo Fisher Scientific Inc., Waltham, MA, USA). Each labeled cRNA (1 μg) was fragmented by adding 5 μl 10 × blocking agent and 1 μl of 25 × fragmentation buffer, before being heated at 60°C for 30 min, followed by addition of 25 μl 2 × GE Hybridization buffer. Hybridization solution (50 μl) was dispensed into the gasket slide and assembled to the lncRNA expression microarray slide. The slides were incubated for 17 hours at 65°C in an Agilent Hybridization Oven. The hybridized arrays were washed, fixed and scanned using the Agilent DNA Microarray Scanner (G2505C). The analysis was performed by Kangchen Bio-tech, Shanghai, P. R. China.
Agilent Feature Extraction software (version 11.0.1.1) was used to analyze the acquired array images. Quantile normalization and subsequent data processing were performed using the GeneSpring GX v11.5.1 software package (Agilent Technologies, Santa Clara, CA, USA). After quantile normalization of the raw data, lncRNAs and mRNAs with at least 1 out of 2 samples having flags in Present or Marginal (“All Targets Value”) were chosen for further data analysis. Differentially expressed lncRNAs and mRNAs were identified through fold change filtering. Heat map and hierarchical clustering were performed using the Agilent GeneSpring GX software (version 11.5.1).
Pathway and gene ontology (GO) analyses
Pathway analysis and GO analysis were performed using standard enrichment computation method, and were used to determine the roles of differentially expressed mRNAs that were playing important roles in these biological pathways or GO terms. Based on the latest Kyoto Encyclopedia of Genes and Genomes (http://www.genome.jp/kegg) database, we performed pathway analysis for differentially expressed mRNAs. Pathway analysis is an effective method to uncover the underlying biological function in response to abnormally expressed genes and proteins [24]. This analysis was used to determine the biological pathway that had significant enrichment of differentially expressed mRNAs. P values denoted the significance of the pathway. The smaller the P values were, the more significant the pathway was (P value cut-off was 0.05). GO analysis was a functional analysis associating differentially expressed mRNAs with GO categories. GO categories were derived from Gene Ontology (www.geneontology.org), which comprised three structured networks of defined terms that described gene product attributes. P values denoted the significance of GO term enrichment in the differentially expressed mRNA list. The smaller the P values were, the more significant the GO term was (P value ≤ 0.05 was recommended).
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated from frozen EC and normal tissue samples using TRIzol reagent (Life Technologies, Thermo Fisher Scientific Inc., Waltham, MA, USA), with its quantity and quality being examined by NanoDrop ND-1000 (Thermo Fisher Scientific Inc., Waltham, MA, USA). Then, total RNA was reversely transcribed according to the manufacturer’s recommendation. The expression of 6 up-regulated lncRNAs and 4 down-regulated lncRNAs in this study were tested by qRT-PCR using SYBR Green assays. qRT-PCR reaction conditions were as follows: a denaturation step of 10 min at 95°C, followed by 40 cycles of 10 s at 95°C and 60 s at 60°C, and a final step of 15 s at 90°C. All samples of this study were normalized to the internal control glyceraldehyde-3-phosphate dehydrogenase (GAPDH). For quantitative results, the 2-ΔΔCT method was used to calculate relative fold changes [25].
Statistical analysis
All data were analyzed using SPSS 17.0 software (IBM, USA). Different expressions of lncRNAs between EC and normal tissues were analyzed using Student’s t-test. P values < 0.05 were considered significant.
Results
Expression of lncRNA and mRNA in EC tissues is different from that in normal tissues
In order to compare the distributions of intensities from all samples, we used box plot to visualize the distributions of a dataset. In addition, scatter plot was used to assess lncRNA and mRNA expression variation or reproducibility between two samples or two groups of samples. Finally, hierarchical clustering was performed to show distinguishable lncRNA and mRNA expression patterns among samples. About 30,586 different lncRNAs can be detected between EC tissues and their paired adjacent noncancerous tissues using third-generation lncRNA microarray (fold change ≥ 2, P < 0.05). Among these lncRNAs, we found that a total number of 4,010 were up-regulated and 3,350 were down-regulated. The most up-regulated one was uc001tdk.2 (fold change: 85.810104) and the most down-regulated one was uc003zfx.3 (fold change: 117.568825). Similarly, a total of 26,109 dysregulated mRNA transcripts were detected, with 3,122 being up-regulated and 2,272 being down-regulated. Among them, NM_014420 was the most up-regulated one (fold change: 27.751808), whereas the most down-regulated one was NM_022580 (fold change: 2644.8286). Box plot showed the distributions of datasets. Scatter plot showed the lncRNA and mRNA expression variation or reproducibility between EC and normal tissues (Figure 1). Hierarchical clustering showed that lncRNA and mRNA expression patterns among samples were distinguishable (Figure 2). These data suggested that the expression of lncRNA and mRNA in EC tissues is different from that in normal tissues.
Figure 1.
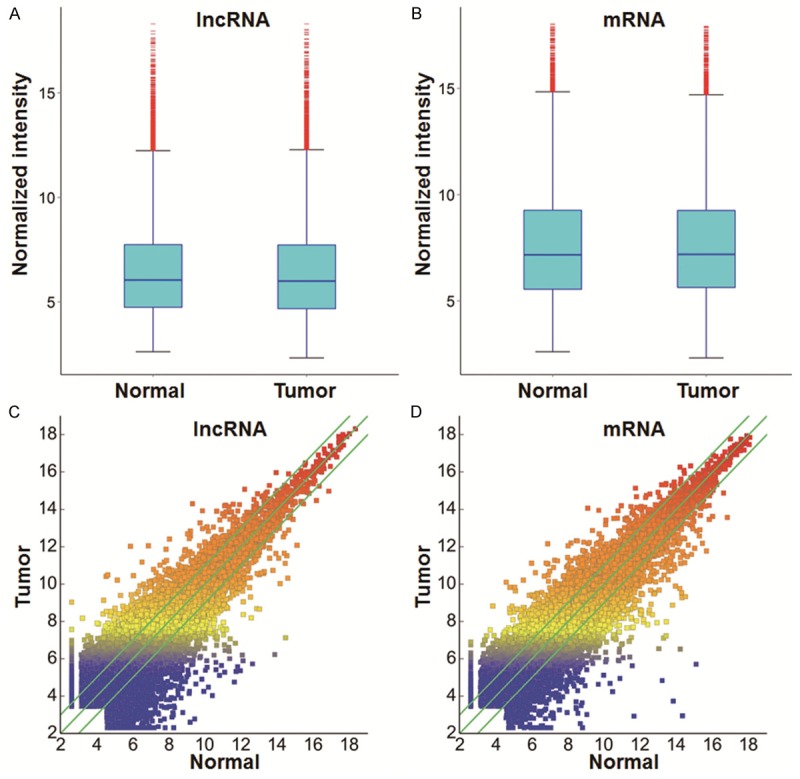
Expression profiles of lncRNAs and mRNAs in EC and adjacent normal tissues. Box plots of (A) lncRNAs and (B) mRNAs showed the distributions of intensities from all samples. Scatter plots showed (C) lncRNA and (D) mRNA expression variation between EC and adjacent normal tissues. Values of X and Y axes in the scatter plots are normalized signal values of the samples (log2 scaled). The green lines are fold-change lines, with the default fold change value being 2.0. LncRNAs above the top green line and below the bottom green line had more than 2.0-fold changes between EC and adjacent normal tissues.
Figure 2.
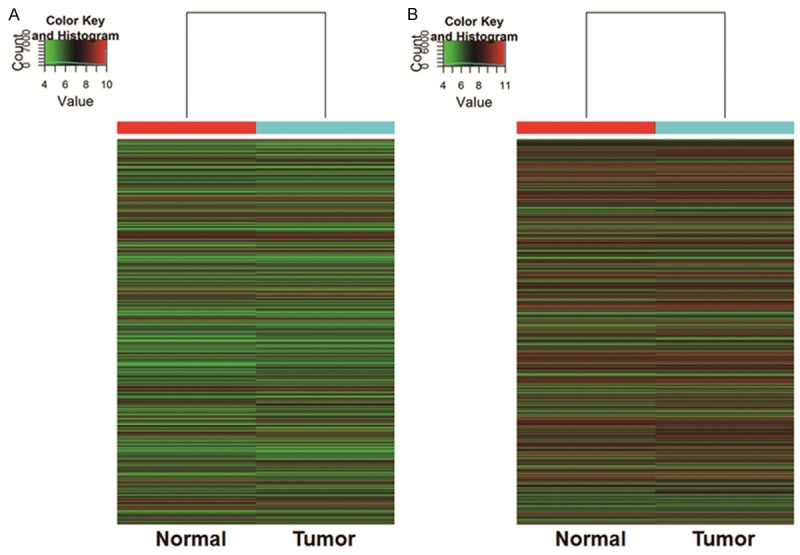
Heat map and hierarchical clustering showing (A) lncRNA and (B) mRNA expression profiling in EC and adjacent normal tissues. Cluster analysis arranges samples into groups based on their expression levels, which allows us to hypothesize the relationships among samples. “Red” indicates highly relative expression, and “green” indicates lowly relative expression.
Functional analysis for LncRNAs and mRNAs
To perform functional analysis for lncRNAs and mRNAs, GO and pathway analyses were used. In this study, we found that the most enrichment GO terms which were expressed in up-regulated transcripts were biological regulation (ontology: Biological Process), membrane part (ontology: Cellular Component) and binding (ontology: Molecular Function). Among the down-regulated transcripts, the most enrichment GO terms were biological regulation (ontology: Biological Process), membrane part (ontology: Cellular Component) and binding (ontology: Molecular Function). We also obtained the top 10 GO terms that were most associated with gene coding of up-regulated RNAs and down-regulated RNAs (Figure 3). Pathway analysis showed that a total of 24 pathways were related to up-regulated transcripts, and “Neuroactive ligand-receptor interaction-Homo sapiens (human)” that consisted of 62 targeted genes, was the most enrichment network. However, we found that 27 pathways were related to down-regulated transcripts and the most enrichment one was also “Neuroactive ligand-receptor interaction-Homo sapiens (human)”, which consisted of 48 targeted genes. Furthermore, out results also showed the top 5 pathways that were associated with gene coding of up-regulated RNAs and down-regulated RNAs (Figure 4).
Figure 3.

GO terms that are associated with coding gene functions of (A-C) up-regulated lncRNAs and (D-F) down-regulated lncRNAs. GO analysis is a functional analysis associating differentially expressed mRNAs with GO categories. GO categories were derived from Gene Ontology (www.geneontology.org), which comprised three structured networks of defined terms that described gene product attributes. P values denoted the significance of GO term enrichment in the differentially expressed mRNA list. The smaller the P values were, the more significant the GO term was (P value ≤ 0.05 was recommended).
Figure 4.

The top 10 pathways of (A) up-regulated and (B) down-regulated mRNAs in EC. Pathway analysis is an effective method to elaborate the underlying biological function. (C) Diagram showing focal adhesion that is closely related to EC pathogenesis.
LncRNA classification and subgroup analysis
To further investigate the relationship between lncRNAs and EC, subgroup analysis was conducted. GENCODE annotation of human genes was applied to the identification of lncRNA-a. The results showed that 2168 abnormally expressed enhancer lncRNAs were determined. The enhancer lncRNAs next to coding gene suggested that the enhancer lncRNA was able to regulate adjacent coding code. In addition, a total of 402 lncRNAs next to coding gene were detected, including 104 that were up-regulated and 83 that were down-regulated. In addition, 128 HOX clusters of four HOX foci were also found in the profiling of our study.
Seven lncRNAs are in accordance with microarray data according to qRT-PCR
In order to validate the consistency of the lncRNAs with microarray data, qRT-PCR was performed. Among the abnormally expressed lncRNAs, ten lncRNAs had fold changes > 5, including six up-regulated lncRNAs (ENST00000491264, NR_004053, uc001tdk.2, uc003xut.3, uc021re1.1, ENST00000445734) and four down-regulated lncRNAs (uc002nbr.3, ENST00000502941, ENST00000448093, ENST00000503710). The qRT-PCR results showed that seven of these lncRNAs were in accordance with microarray data, including three up-regulated lncRNAs (uc003xut., uc021re1.1, ENST00000445734) and four down-regulated ones (uc002nbr.3, ENST00000502941, ENST00000448093, ENST00000503710) (Figure 5). In particular, the alterations in uc003xut and ENST00000502941 were the most significant, representing the up-regulated lncRNAs and the down-regulated lncRNAs, respectively (Figure 6).
Figure 5.
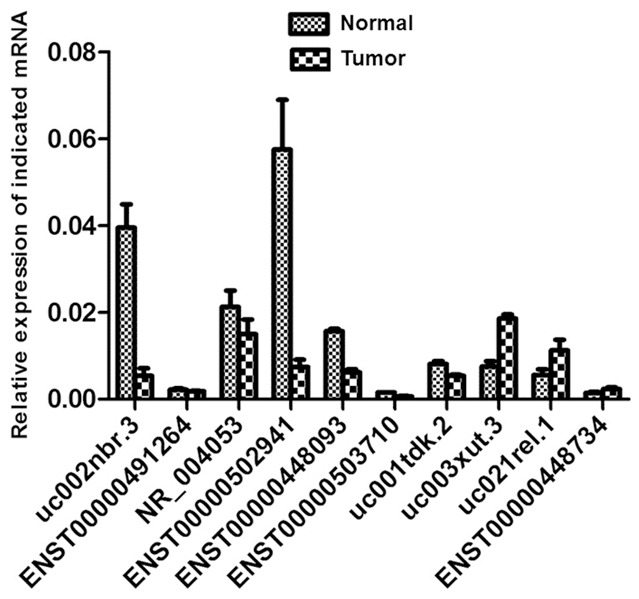
Comparison between microarray and quantitative real-time PCR results on 10 differentially expressed lncRNAs (ENST00000491264, NR_004053, uc001tdk.2, uc003xut.3, uc021re1.1, ENST00000445734, uc002nbr.3, ENST00000502941, ENST00000448093, ENST00000503710) in EC and normal tissues.
Figure 6.
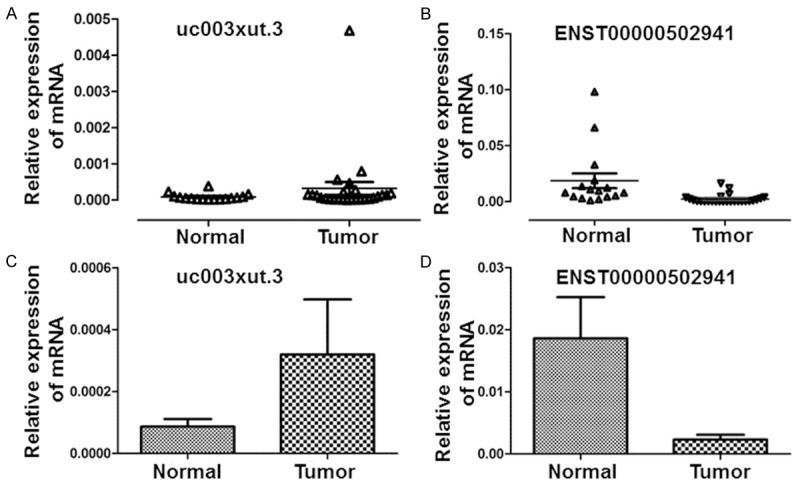
Distribution of (A) uc003xut.3 and (B) ENST00000502941 in EC and normal tissue samples. Triangle symbols show the distribution of mRNAs. Expression levels of (C) uc003xut.3 and (D) ENST00000502941 in EC and normal tissue samples. Histograms show the average expression levels of mRNAs and error bars indicate standard deviations.
Discussion
Recent studies on various tumors are focused on non-coding and coding genes, especially lncRNAs. Investigations on the relationship between EC and lncRNA are still at early stage with few articles reporting the correlation between the overexpression of lncRNA HOTAIR and EC [26,27]. Therefore, it is of great value to investigate EC pathogenesis and molecular markers with diagnostic significance.
Among mammalian genes, thousands of lncRNAs have been reported to play important roles in biological process through multiple mechanisms [18]. It is well established that lncRNA is critical to normal tissues and diseases [28]. Recently, studies have increasingly evidenced that several types of tumors are closely associated with the abnormal expression of lncRNA, which is involved in the occurrence, development and metastasis of tumor with a series of complicated mechanisms [19,29]. For instance, Wang et al. [30] found that the up-regulation of several lncRNAs was correlated with the occurrence of liver cancer. Through regulating the expression of p53, lncRNAMEG3 appeared to have a strong impact on osteosarcoma [31]. In addition, the correlation between the abnormal expression of lncRNA and several tumors, such as stomach cancer [32], renal cancer [33], pancreatic cancer [34], glioblastoma [35] and lung cancer [36,37] has been recognized. Therefore, lncRNA could be potentially employed to serve as indicators for tumor diagnosis and prognosis [29].
In the present study, a group of EC samples and adjacent normal tissue samples were analyzed through microarray technique, and validations of ten selected lncRNAs were carried out on 40 samples using qRT-PCR. The result of microarray expression profiles showed that compared with normal tissues, the expression of 30,586 lncRNAs and 26,109 mRNAs were detected to be abnormal in EC. In particular, 4008 lncRNAs were up-regulated, while 3350 lncRNAs were down-regulated. However, the functions of the majority of abnormally expressed genes have not been fully recognized.
In the process of biological development and differentiation, lncRNA generally acquires significant temporal and spatial specificity, and exerts its effect by means of a series of complicated mechanisms [16,38-42]. An important common function is realized by its impact on transcriptional process or by its direct enhancer-like effect that alters the expression of adjacent coding genes. In addition, the reduction of certain number of lncRNA would decrease the expression of adjacent coding genes [43-47]. To deepen the understanding of the correlation between lncRNA and EC, the classification and subgroup of lncRNA were further investigated, and enhancer lncRNAs and adjacent coding genes were determined. It is found that a total of 402 enhancer-like lncRNAs next to coding gene were abnormally expressed, and 104 of them were up-regulated while 38 of them were down-regulated. As mentioned previously, the abnormal expression of lncRNA in HOX loci emerged in the process of certain humor development [19]. In view of these considerations, we investigated HOX lncRNA clusters. Similar to previous report that confirmed the transcription of a large amount of lncRNAs in human HOX clusters [17], 128 HOX clusters of four HOX foci were presented in the profiling of our study.
According to microarray analysis, six up-regulated lncRNAs and four down-regulated lncRNAs were selected by verifying the expression consistency of twenty pairs of samples. In addition, the expression of the ten lncRNAs was further investigated in the present study. It was found that the result of seven tested lncRNAs was in line with microarray analysis. In particular, four of the lncRNAs were down-regulated with ENST0000502941 being significantly increased, while another three lncRNAs were up-regulated with uc003xut.3 having markedly increased expression. ENST00000502941, a product of gene ENSG00000237125, is a 358-bp bidirectional lncRNA with 3 exons transcribed from RP11-471J12.1 (chr4) gene located on chromosome 4. In addition, uc003xut.3 is an intergenic lncRNA with 2 exons transcribed from BC047540 (chr8) gene.
The result of qRT-PCR revealed that the expression of lncRNA was variable in different tissues. Furthermore, the consistency between the expression of the seven lncRNAs and the microarray data suggested a way to differentiate uterine tumor tissues from normal tissues. In comparison to the up-regulated genes, the extent of the down-regulated genes appeared to be much larger, indicating close relationship between certain down-regulated lncRNAs and the occurrence and development of EC. However, the biological functions of the above genes still remained unclear so far. Unluckily, the results in our study could not provide sufficient evidence to determine whether lncRNA qualifies as a biomarker. Further studies on lncRNAs, especially ENST0000502941 and uc003xut.3, are still necessary.
GO analysis could provide controlled vocabulary to illustrate related genes and gene products in EC. The result of GO analysis showed that three GO terms that were closely relevant to both up-regulated and down-regulated lncRNAs were biological regulation, membrane part and binding. In this study, fifty-one pathways were found to be relevant to the differentially expressed transcripts, in which twenty-four pathways were associated with down-regulated transcripts and twenty-seven with up-regulated transcripts. Previous researches proved the correlation between these pathways and multiple diseases including EC. Focal adhesion [48-50], MAPK [51], ECM-receptor [49] and PPAR [52-54] have been reported to be closely related to EC pathogenesis.
In conclusion, this study is the first work that uses microarray analysis to investigate abnormally expressed lncRNAs in EC and adjacent normal tissues. In addition, ten lncRNAs from 40 samples were validated using qRT-PCR. The result demonstrated that abnormal expression of lncRNAs was closely associated with EC. In the near future, we will select greater numbers of samples to deepen the research into the lncRNA molecular mechanism and biochemical function, in order to provide a novel accurate method for the early diagnosis and therapy of EC.
Acknowledgements
This work was supported by the Foundation for Outstanding Young Scientist in Shandong Province (BS2010YY057) and the Health Care and Family Planning Technology Development Plans in Shandong Province (2013WS0108).
Disclosure of conflict of interest
None.
References
- 1.Weiderpass E, Persson I, Adami HO, Magnusson C, Lindgren A, Baron JA. Body size in different periods of life, diabetes mellitus, hypertension, and risk of postmenopausal endometrial cancer (Sweden) Cancer Causes Control. 2000;11:185–92. doi: 10.1023/a:1008946825313. [DOI] [PubMed] [Google Scholar]
- 2.Parazzini F, La Vecchia C, Negri E, Riboldi GL, Surace M, Benzi G, Maina A, Chiaffarino F. Diabetes and endometrial cancer: an Italian case-control study. Int J Cancer. 1999;81:539–42. doi: 10.1002/(sici)1097-0215(19990517)81:4<539::aid-ijc6>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 3.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 4.Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol. 1983;15:10–7. doi: 10.1016/0090-8258(83)90111-7. [DOI] [PubMed] [Google Scholar]
- 5.Amant F, Moerman P, Neven P, Timmerman D, Van Limbergen E, Vergote I. Endometrial cancer. Lancet. 2005;366:491–505. doi: 10.1016/S0140-6736(05)67063-8. [DOI] [PubMed] [Google Scholar]
- 6.Lax SF. Molecular genetic pathways in various types of endometrial carcinoma: from a phenotypical to a molecular-based classification. Virchows Arch. 2004;444:213–23. doi: 10.1007/s00428-003-0947-3. [DOI] [PubMed] [Google Scholar]
- 7.Saal LH, Johansson P, Holm K, Gruvberger-Saal SK, She QB, Maurer M, Koujak S, Ferrando AA, Malmström P, Memeo L, Isola J, Bendahl PO, Rosen N, Hibshoosh H, Ringnér M, Borg A, Parsons R. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc Natl Acad Sci U S A. 2007;104:7564–9. doi: 10.1073/pnas.0702507104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park JY, Kim KR, Nam JH. Immunohistochemical analysis for therapeutic targets and prognostic markers in low-grade endometrial stromal sarcoma. Int J Gynecol Cancer. 2013;23:81–9. doi: 10.1097/IGC.0b013e3182738361. [DOI] [PubMed] [Google Scholar]
- 9.Yu W, Gius D, Onyango P, Muldoon-Jacobs K, Karp J, Feinberg AP, Cui H. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature. 2008;451:202–6. doi: 10.1038/nature06468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huarte M, Rinn JL. Large non-coding RNAs: missing links in cancer? Hum Mol Genet. 2010;19:R152–61. doi: 10.1093/hmg/ddq353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kowalczyk MS, Higgs DR, Gingeras TR. Molecular biology: RNA discrimination. Nature. 2012;482:310–1. doi: 10.1038/482310a. [DOI] [PubMed] [Google Scholar]
- 12.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–8. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu W, Lin Z, Zhuang Z, Liang X. Expression profile of mammalian microRNAs in endometrioid adenocarcinoma. Eur J Cancer Prev. 2009;18:50–5. doi: 10.1097/CEJ.0b013e328305a07a. [DOI] [PubMed] [Google Scholar]
- 14.Chung TK, Cheung TH, Huen NY, Wong KW, Lo KW, Yim SF, Siu NS, Wong YM, Tsang PT, Pang MW, Yu MY, To KF, Mok SC, Wang VW, Li C, Cheung AY, Doran G, Birrer MJ, Smith DI, Wong YF. Dysregulated microRNAs and their predicted targets associated with endometrioid endometrial adenocarcinoma in Hong Kong women. Int J Cancer. 2009;124:1358–65. doi: 10.1002/ijc.24071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caley DP, Pink RC, Trujillano D, Carter DR. Long noncoding RNAs, chromatin, and development. ScientificWorldJournal. 2010;10:90–102. doi: 10.1100/tsw.2010.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ørom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, Guigo R, Shiekhattar R. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–23. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904–14. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, Wang Y, Brzoska P, Kong B, Li R, West RB, van de Vijver MJ, Sukumar S, Chang HY. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–6. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hessels D, Schalken JA. The use of PCA3 in the diagnosis of prostate cancer. Nat Rev Urol. 2009;6:255–61. doi: 10.1038/nrurol.2009.40. [DOI] [PubMed] [Google Scholar]
- 21.Ifere GO, Ananaba GA. Prostate cancer gene expression marker 1 (PCGEM1): a patented prostate- specific non-coding gene and regulator of prostate cancer progression. Recent Pat DNA Gene Seq. 2009;3:151–63. doi: 10.2174/187221509789318360. [DOI] [PubMed] [Google Scholar]
- 22.Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, Fraser P. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science. 2008;322:1717–20. doi: 10.1126/science.1163802. [DOI] [PubMed] [Google Scholar]
- 23.Sun J, Zhou M, Mao ZT, Hao DP, Wang ZZ, Li CX. Systematic analysis of genomic organization and structure of long non-coding RNAs in the human genome. FEBS Lett. 2013;587:976–82. doi: 10.1016/j.febslet.2013.02.036. [DOI] [PubMed] [Google Scholar]
- 24.Khatri P, Sirota M, Butte AJ. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol. 2012;8:e1002375. doi: 10.1371/journal.pcbi.1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang J, Ke P, Guo L, Wang W, Tan H, Liang Y, Yao S. Lentivirus-mediated RNA interference targeting the long noncoding RNA HOTAIR inhibits proliferation and invasion of endometrial carcinoma cells in vitro and in vivo. Int J Gynecol Cancer. 2014;24:635–42. doi: 10.1097/IGC.0000000000000121. [DOI] [PubMed] [Google Scholar]
- 27.He X, Bao W, Li X, Chen Z, Che Q, Wang H, Wan XP. The long non-coding RNA HOTAIR is upregulated in endometrial carcinoma and correlates with poor prognosis. Int J Mol Med. 2014;33:325–32. doi: 10.3892/ijmm.2013.1570. [DOI] [PubMed] [Google Scholar]
- 28.Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–41. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 29.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–9. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Liu X, Wu H, Ni P, Gu Z, Qiao Y, Chen N, Sun F, Fan Q. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res. 2010;38:5366–83. doi: 10.1093/nar/gkq285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang LL. Biology of osteogenic sarcoma. Cancer J. 2005;11:294–305. doi: 10.1097/00130404-200507000-00005. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Feng X, Jia R, Liu G, Zhang M, Fan D, Gao S. Microarray expression profile analysis of long non-coding RNAs of advanced stage human gastric cardia adenocarcinoma. Mol Genet Genomics. 2014;289:291–302. doi: 10.1007/s00438-013-0810-4. [DOI] [PubMed] [Google Scholar]
- 33.Yu G, Yao W, Wang J, Ma X, Xiao W, Li H, Xia D, Yang Y, Deng K, Xiao H, Wang B, Guo X, Guan W, Hu Z, Bai Y, Xu H, Liu J, Zhang X, Ye Z. LncRNAs expression signatures of renal clear cell carcinoma revealed by microarray. PLoS One. 2012;7:e42377. doi: 10.1371/journal.pone.0042377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tahira AC, Kubrusly MS, Faria MF, Dazzani B, Fonseca RS, Maracaja-Coutinho V, Verjovski-Almeida S, Machado MC, Reis EM. Long noncoding intronic RNAs are differentially expressed in primary and metastatic pancreatic cancer. Mol Cancer. 2011;10:141. doi: 10.1186/1476-4598-10-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han L, Zhang K, Shi Z, Zhang J, Zhu J, Zhu S, Zhang A, Jia Z, Wang G, Yu S, Pu P, Dong L, Kang C. LncRNA profile of glioblastoma reveals the potential role of lncRNAs in contributing to glioblastoma pathogenesis. Int J Oncol. 2012;40:2004–12. doi: 10.3892/ijo.2012.1413. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y, Chen W, Chen J, Pan Q, Pan J. LncRNA expression profiles of EGFR exon 19 deletions in lung adenocarcinoma ascertained by using microarray analysis. Med Oncol. 2014;31:137. doi: 10.1007/s12032-014-0137-y. [DOI] [PubMed] [Google Scholar]
- 37.Xu G, Chen J, Pan Q, Huang K, Pan J, Zhang W, Chen J, Yu F, Zhou T, Wang Y. Long noncoding RNA expression profiles of lung adenocarcinoma ascertained by microarray analysis. PLoS One. 2014;9:e104044. doi: 10.1371/journal.pone.0104044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mattick JS, Gagen MJ. The evolution of controlled multitasked gene networks: the role of introns and other noncoding RNAs in the development of complex organisms. Mol Biol Evol. 2001;18:1611–30. doi: 10.1093/oxfordjournals.molbev.a003951. [DOI] [PubMed] [Google Scholar]
- 39.Mattick JS. Linc-ing Long noncoding RNAs and enhancer function. Dev Cell. 2010;19:485–6. doi: 10.1016/j.devcel.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 40.Popadin K, Gutierrez-Arcelus M, Dermitzakis ET, Antonarakis SE. Genetic and epigenetic regulation of human lincRNA gene expression. Am J Hum Genet. 2013;93:1015–26. doi: 10.1016/j.ajhg.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, Regev A, Lander ES, Rinn JL. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667–72. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang F, Zhang L, Huo XS, Yuan JH, Xu D, Yuan SX, Zhu N, Zhou WP, Yang GS, Wang YZ, Regev A, Lander ES, Rinn JL. Long noncoding RNA high expression in hepatocellular carcinoma facilitates tumor growth through enhancer of zeste homolog 2 in humans. Hepatology. 2011;54:1679–89. doi: 10.1002/hep.24563. [DOI] [PubMed] [Google Scholar]
- 43.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–7. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, Attardi LD, Regev A, Lander ES, Jacks T, Rinn JL. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–19. doi: 10.1016/j.cell.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ponjavic J, Oliver PL, Lunter G, Ponting CP. Genomic and transcriptional co-localization of protein-coding and long non-coding RNA pairs in the developing brain. PLoS Genet. 2009;5:e1000617. doi: 10.1371/journal.pgen.1000617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ebisuya M, Yamamoto T, Nakajima M, Nishida E. Ripples from neighbouring transcription. Nat Cell Biol. 2008;10:1106–13. doi: 10.1038/ncb1771. [DOI] [PubMed] [Google Scholar]
- 47.Sproul D, Gilbert N, Bickmore WA. The role of chromatin structure in regulating the expression of clustered genes. Nat Rev Genet. 2005;6:775–81. doi: 10.1038/nrg1688. [DOI] [PubMed] [Google Scholar]
- 48.Maurice-Duelli A, Ndoye A, Bouali S, Leroux A, Merlin JL. Enhanced cell growth inhibition following PTEN nonviral gene transfer using polyethylenimine and photochemical internalization in endometrial cancer cells. Technol Cancer Res Treat. 2004;3:459–65. doi: 10.1177/153303460400300507. [DOI] [PubMed] [Google Scholar]
- 49.Du XL, Jiang T, Zhao WB, Wang F, Wang GL, Cui M, Wen ZQ. Gene alterations in tumor-associated endothelial cells from endometrial cancer. Int J Mol Med. 2008;22:619–32. [PubMed] [Google Scholar]
- 50.Zhao Y, Yang Y, Trovik J, Sun K, Zhou L, Jiang P, Lau TS, Hoivik EA, Salvesen HB, Sun H, Wang H. A novel wnt regulatory axis in endometrioid endometrial cancer. Cancer Res. 2014;74:5103–17. doi: 10.1158/0008-5472.CAN-14-0427. [DOI] [PubMed] [Google Scholar]
- 51.Schointuch MN, Gilliam TP, Stine JE, Han X, Zhou C, Gehrig PA, Kim K, Bae-Jump VL. Simvastatin, an HMG-CoA reductase inhibitor, exhibits anti-metastatic and anti-tumorigenic effects in endometrial cancer. Gynecol Oncol. 2014;134:346–55. doi: 10.1016/j.ygyno.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ma JJ, Monsivais D, Dyson MT, Coon JS, Malpani S, Ono M, Zhao H, Xin H, Pavone ME, Kim JJ, Chakravarti D, Bulun SE. Ligand-activated peroxisome proliferator-activated receptor β/δ modulates human endometrial cancer cell survival. Horm Cancer. 2013;4:358–70. doi: 10.1007/s12672-013-0157-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nickkho-Amiry M, McVey R, Holland C. Peroxisome proliferator-activated receptors modulate proliferation and angiogenesis in human endometrial carcinoma. Mol Cancer Res. 2012;10:441–53. doi: 10.1158/1541-7786.MCR-11-0233. [DOI] [PubMed] [Google Scholar]
- 54.Holland CM, Saidi SA, Evans AL, Sharkey AM, Latimer JA, Crawford RA, Charnock-Jones DS, Print CG, Smith SK. Transcriptome analysis of endometrial cancer identifies peroxisome proliferator-activated receptors as potential therapeutic targets. Mol Cancer Ther. 2004;3:993–1001. [PubMed] [Google Scholar]