

Abstract
Saikosaponin-d (Ssd) is one of the major pharmacologically active molecules present in Bupleurum falcatum L, a medical herb against inflammatory diseases in the traditional Chinese medicine. In the current study, we investigated the protective activity of Ssd on diabetic nephropathy along with the underlying mechanisms using renal tubular epithelial cell line (NRK-52E). Our study showed that high glucose stimulation significantly increased NRK-52E cell proliferation. Ssd administration dramatically inhibited high glucose-induced proliferation and DNA synthesis in NRK-52E cell. In addition, high glucose treatment resulted in oxidative stress as shown by increased production of ROS, higher concentration of MDA, and decreased activity of SOD. However, incubation with Ssd reversed such changes in NRK-52E cells. On the molecular level, Ssd also increased the mRNA levels of IDH2 and MnSOD. Moreover, Ssd-treated NRK-52E cells displayed a dramatic enhancement in SIRT3 expression both at mRNA and protein levels. Down-regulation of SIRT3 abolished the protective effects of Ssd on NRK-52E cells. These findings demonstrated that Ssd protected renal tubular epithelial cell against high glucose induced injury via upregulation of SIRT3.
Keywords: Diabetic nephropathy, saikosaponin-d, oxidative stress, SIRT3
Introduction
Diabetic nephropathy (DN), one of the most severe microvascular complications of diabetes mellitus, is the leading cause of end-stage renal disease [1,2]. DN is considered to be a progressive kidney disease caused by angiopathy of capillaries in the kidney glomeruli and contributes to morbidity and mortality of diabetic patients in the world [3]. Pathological manifestations of DN include thickening of basement membrane and excessive accumulation of extracellular matrix, which ultimately progress to glomerulosclerosis [4]. Nowadays, the pathophysiology of DN has not been fully elucidated. Accumulating knowledge indicate that hyperglycemia-induced metabolic and hemodynamic alterations, such as abnormal cell proliferation, increased reactive oxygen species (ROS) production, enhanced generation of advanced glycation end products (AGEs), and elevated levels of inflammatory cytokines, play critical roles in the development and progression of DN [5-8].
Saikosaponin-d (Ssd) is one of the major triterpenoid saponins derived from Bupleurum falcatum L, which is a commonly prescribed agent against inflammatory and infectious diseases in China, Japan and other Asian countries [9]. Recent studies have shown that Ssd has anti-inflammatory, immune-modulatory, and antiviral activities, making it a potential chemotherapeutic drug for clinical application [10,11]. It has been reported that Ssd inhibits the proliferation and migratory activity of rat hepatic stellate cells and attenuates the development of liver fibrosis by preventing hepatocyte injury [12,13]. Furthermore, Ssd administration attenuates oxidative damage in pig kidney proximal tubular cells by increasing the expression of anti-oxidant enzymes and heat shock protein 72 (HSP72) at both the mRNA and protein levels [14]. In the present study, we aimed to test the hypothesis if Ssd has a protective effects against high glucose-induced tubular epithelial cells damage.
Materials and methods
Cell culture
NRK-52E cells were purchased from Chinese academy of sciences and cultured in DMEM (Dulbecco’s modified Eagle’s medium) supplemented with 10% fetal calf serum, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in an atmosphere containing 5% CO2.
CCK-8 assay
The cell viability was assessed using a colorimetric cell counting kit (CCK-8; Dojindo, Japan). After incubation in DMEM supplemented with 0.5% fetal calf serum for 24 h, NRK-52E cells were treated with normal glucose (5.6 mM glucose), high glucose (30 mM glucose), and high glucose plus Ssd for indicated time points. Then 10 μL/well CCK-8 solution (Dojindo, Kumamoto, Japan) was added, the plates incubated for 3 h, and absorbance was measured at 450 nm using an MRX II microplate reader (Dynex, Chantilly , VA, USA).
EdU incorporation assay
The DNA synthesis was determined by using a commercially Click-iT EdU Imaging Kit (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instruction. Briefly, NRK-52E cell were exposed to high glucose alone or high glucose combined with Ssd for 24 h and 48 h. Cells were exposed to EdU for 2 h and fixed for 20 min with 4% paraformaldehyde. The number of EdU-positive and total cells was counted using fluorescence microscopy (Leica, Germany) in four non-overlapping fields per coverslip.
Measurement of ROS production
Cells were incubated with 2,7-dichlorofluorescein diacetate (DCF-DA) (Sigma, St. Louis, MO, USA) for 1 h at 37°C in the dark, and then resuspended in PBS. Intracellular ROS production was assessed using a fluorescence microscope (OLYMPUS, Germany) with ex/em λ= 488 nm/515 nm.
Measurement of enzymes activities
The enzymatic activity of MDA and SOD was measured by use of a commercially available assay kits according to the manufacturer’s instruction (Cayman Chemical, Ann Arbor, MI, USA).
Real time PCR
RNA was extracted using the Trizol reagent, and reversely transcribed with an RT kit using oligo (dT) 18 primer according to manufacturer’s protocol. Real-time PCR was performed on a ABI 7500 using SYBR Green PCR Master Mix (Takara, Japan). The primers used were: IDH2 forward: 5’-AATTTTAGGACCCCCGTCTG-3’, reverse: 5’-GGGGTGAAGACCATTTTGAA-3’; Mn-SOD forward: 5’-AAGGAGCAAGGTCGCTTACAGA-3’, reverse: 5’-CAAATGGCTTTCAGATAGTCAGGTC-3’. The relative amounts of mRNA were determined by the 2-ΔΔCt calculations.
siRNA transfection
NRK-52E cells were transfected with SIRT3 siRNA or control siRNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. The transfection medium (Opti-MEM; Gibco) was replaced with complete medium 12 h after transfection, and the cells were incubated for the indicated times.
Western blot
Cells were lysed in 50 μl cell lysis buffer containing protease inhibitors (Sigma, USA). The protein concentration was quantified using the BCA Protein Kit (Thermo, Rockford, IL, USA). Cell lysates were separated by 10% SDS-PAGE and proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The membranes were then incubated with anti-SIRT3 and anti-GAPDH primary antibodies (Abcam, Cambridge, USA) at 4°C overnight. The membranes were washed three times with TBS/T and then incubated with the appropriate HRP-conjugated secondary antibodies for 1 h at room temperature. Protein expression was detected by chemiluminescence (GE Healthcare, Piscataway, NJ, USA).
Statistical analysis
Each experiment was performed in triplicate, and repeated at least three times. All the data were presented as means ± SD and treated for statistics analysis by SPSS program. Comparison between groups was made using ANOVA and statistically significant difference was defined as P < 0.05.
Results
Inhibitory effects of Ssd on high glucose-induced cell proliferation
NRK-52E cell proliferation was evaluated using CCK-8 analysis. Compared with the control group, we found that glucose alone (30 mM) significantly promoted NRK-52E cell proliferation at 24 and 48 h time points. We then examined the effect of different doses of Ssd (15, 30, 45, 60 μmol/L) on cell growth. Results showed that Ssd had little effect on cell proliferation between 15~30 μmol/L (Figure 1A and 1B). However, Ssd at 45 and 60 μmol/L dramatically inhibited high glucose-induced NRK-52E cell proliferation (Figure 1C and 1D). Consistent with data from CCK-8 analysis, EdU incorporation assay also showed that Ssd suppressed high glucose-induced DNA synthesis both at 45 and 60 μmol/L doses (Figure 1E and 1F). Furthermore, we treated cells with different concentrations of Ssd alone for 24 or 48 h to determine the cytotoxic effects of drugs. Results showed that low concentrations of Ssd (15, 30, 45, 60 μmol/L) had little inhibitory effects on cell growth, whereas Ssd at 90 μmol/L suppressed NRK-52E cell proliferation (Figure 2A and 2B). Thus, Ssd at 45 and 60 μmol/L was used for further experimental analysis.
Figure 1.
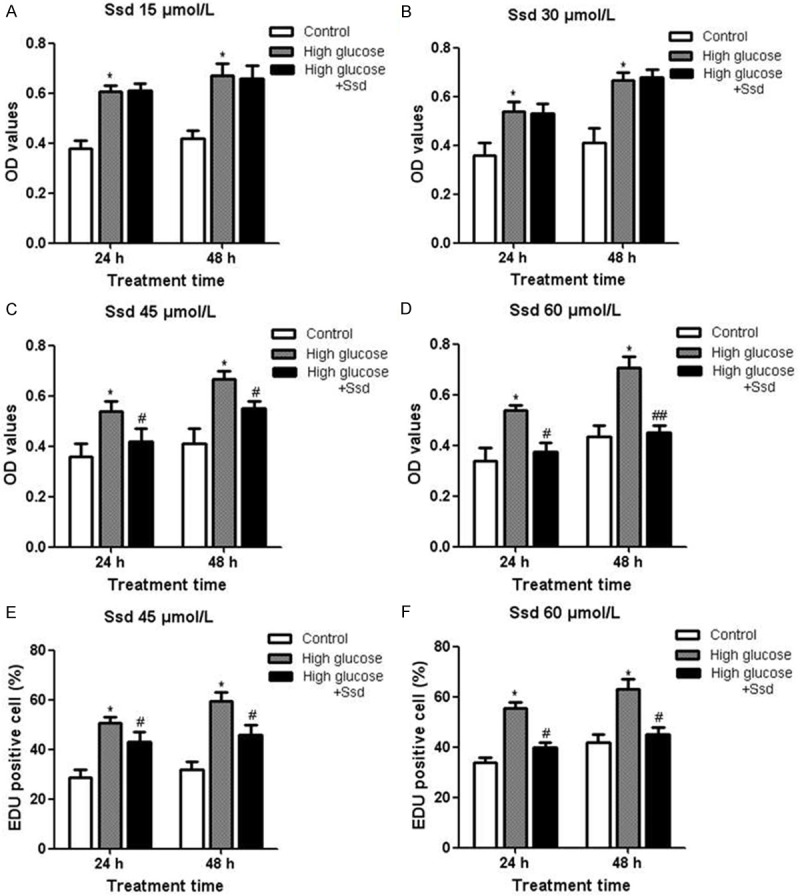
Inhibitory effects of Ssd on high glucose-induced cell proliferation NRK-52E cells were treated with high glucose with or without Ssd for 24 or 48 h. CCK-8 analysis was used to measure cell proliferation under different concentrations (15, 30, 45, 60 μmol/L) of Ssd treatment (A-D). EdU incorporation assay was performed to detect the DNA synthesis in NRK-52E cells treated with Ssd at 45 and 60 μmol/L for 48 h (E and F). *P < 0.05 vs. control; #P < 0.05 vs. High glucose; ##P < 0.01 vs. High glucose.
Figure 2.
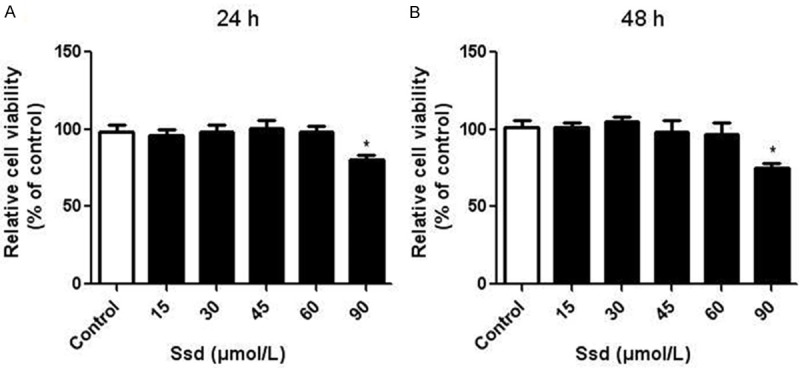
Low concentrations of Ssd had little cytotoxic effects on NRK-52E cells NRK-52E cells were treated with different concentrations (15, 30, 45, 60, 90 μmol/L) of Ssd for 24 (A) or 48 h (B). Cell viability in different groups was measured by CCK-8 analysis. *P < 0.05 vs. control.
Ssd alleviated high glucose-induced oxidative stress in NRK-52E cells
Oxidative stress is a well-established event in the pathology of many diseases, including DN. We found that the high glucose treatment significantly increased the ROS generation, whereas Ssd attenuated the intracellular ROS levels (Figure 3A). In addition, the concentration of MDA was higher, whereas the activity of SOD was lower in the high glucose group than in the control group. Treatment with Ssd significantly reduced the concentrations of MDA (Figure 3B) and enhanced SOD activity (Figure 3C). We also measured the mRNA expression of IDH2 and MnSOD by real-time PCR and found that the mRNA levels of IDH2 (Figure 4A) and MnSOD (Figure 4B) were both obviously increased after Ssd incubation. These results suggested that Ssd alleviated oxidative stress in high glucose treated NRK-52E cells.
Figure 3.
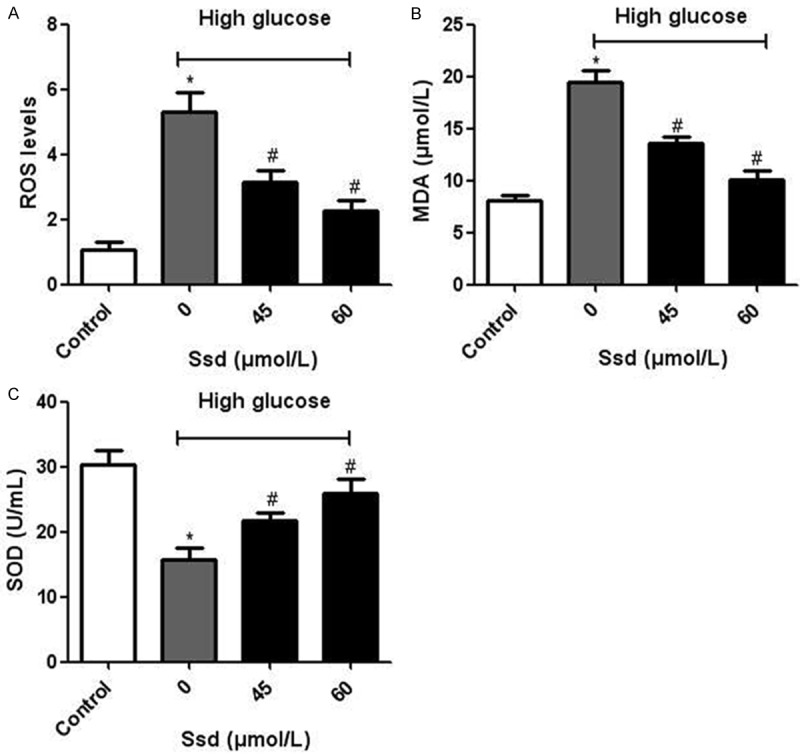
Effects of Ssd on ROS production and the enzymatic activities of MDA and SOD. NRK-52E cells were treated with high glucose with or without Ssd (45, 60 μmol/L) for 48 h. A. Determination of ROS production. B and C. Enzymatic activities of MDA and SOD. *P < 0.05 vs. control; #P < 0.05 vs. Ssd (0 μmol/L).
Figure 4.
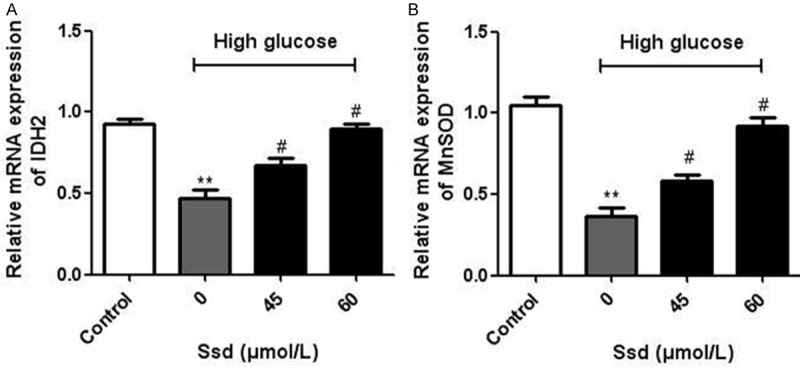
Effects of Ssd on the mRNA expression of IDH2 and MnSOD NRK-52E cells were treated with high glucose with or without Ssd (45, 60 μmol/L) for 48 h. The mRNA levels of IDH2 (A) and MnSOD (B) were measured by real-time PCR. *P < 0.05 vs. control; #P < 0.05 vs. Ssd (0 μmol/L).
Up-regulation of SIRT3 by Ssd administration in NRK-52E cells
We further investigated the molecular mechanism underlying the protective effects of Ssd on NRK-52E cells. Since SIRT3 has been reported to be involved in cell proliferation and oxidative stress, we hypothesized that SIRT3 may play an important role in NRK-52E cells. Real time PCR showed that the mRNA expression of SIRT3 was significantly increased after Ssd treatment for 48 h (Figure 5A). Similarly, the protein level of SIRT3 was also enhanced more than three folds in Ssd-treated cells, even in the presence of high glucose (Figure 5B and 5C), suggesting that Ssd could promote SIRT3 expression in NRK-52E cells.
Figure 5.

Ssd treatment promoted the expression of SIRT3. NRK-52E cells were treated with Ssd (60 μmol/L) with or without high glucose for 48 h. Real time PCR was used to detect the mRNA expression of SIRT3 (A). Protein levels of SIRT3 were measured by western blot (B) and the band intensity was quantified, normalized to GAPDH (C). *P < 0.05 vs. control.
Down-regulation of SIRT3 diminished the protective effects of Ssd on NRK-52E cells
In order to further confirm that SIRT3 is involved in the protective effects by Ssd, we silenced SIRT3 with a specific small interfering RNA (siRNA) in NRK-52E cells. The expression of SIRT3 was significantly reduced both at mRNA and protein levels (Figure 6A and 6B). As a result, we found that Ssd failed to inhibit high glucose-induced cell proliferation (Figure 6C). Moreover, there were little changes in ROS production (Figure 6D), SOD activities (Figure 6E) and MDA content (Figure 6F) in the Ssd group compared with that in the high glucose group. These results demonstrated that SIRT3 knockdown reversed the protective effects of Ssd on NRK-52E cells.
Figure 6.
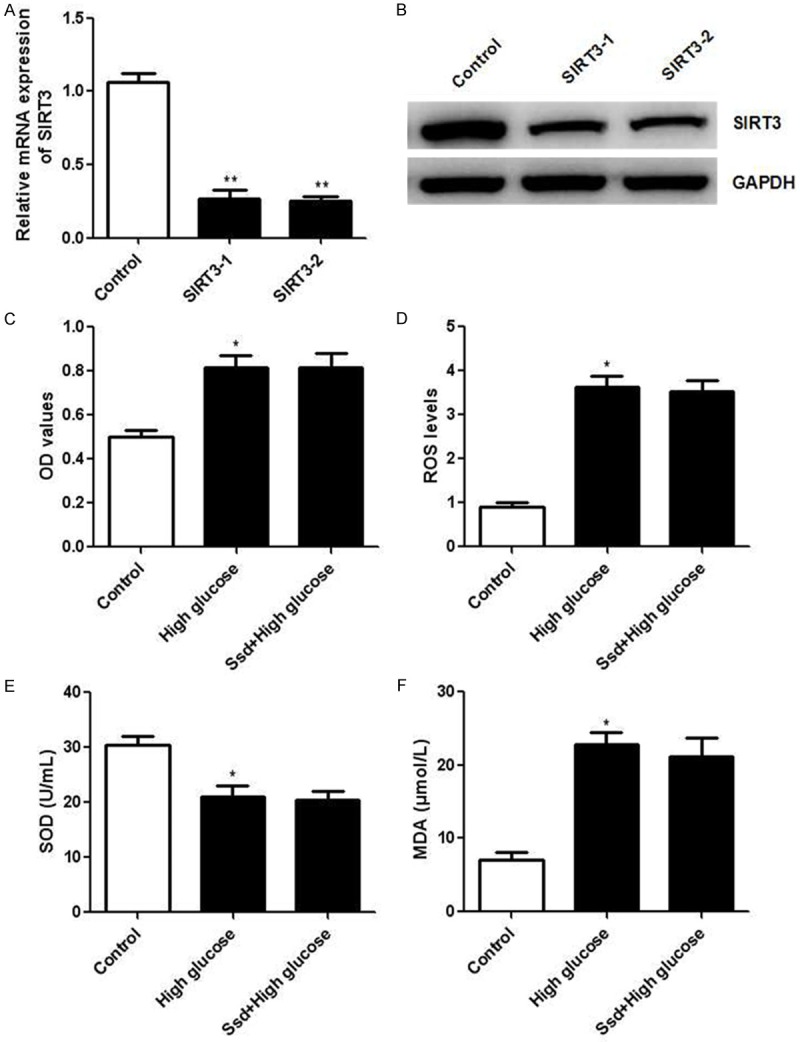
SIRT3 knockdown diminished the protective effects of Ssd on NRK-52E cells. NRK-52E cells were transfected with SIRT3 siRNA or control siRNA for 24 h. Real time PCR and western blot were applied to detect the mRNA (A) and protein (B) levels of SIRT3. NRK-52E cells transfected with SIRT3 siRNA were treated with Ssd (60 μmol/L) with or without high glucose for 48 h. followed by the determination of cell proliferation (C), ROS production (D) and enzymatic activities of SOD (E) and MDA (F). *P < 0.05 vs. control; **P < 0.01 vs. control.
Discussion
Diabetes mellitus causes macrovascular and microvascular complications including heart disease, stroke and kidney failure, which affects the function of many organs [15]. One of them is the injury to the kidney tissue that results in renal dysfunction, and currently there are no effective medicines [16]. It has been suggested that high glucose contributes to the proliferation of renal tubular epithelial cells, vascular smooth muscle cells and mesangial cells under diabetic condition [17]. Tubular epithelial cell is the main component of the total kidney volume and the hypertrophy of tubular epithelial cell plays an important role in causing kidney hypertrophy [18,19]. Thus, inhibition of tubular epithelial cells growth could help to control the DN progression. In the present study, we found that high glucose promoted NRK-52E cell proliferation after stimulation for 24 and 48 h. However, co-treatment with Ssd at 45 and 60 μmol/L suppressed high glucose-induced NRK-52E cell proliferation. In addition, the DNA synthesis in NRK-52E cells was significantly augmented after high glucose incubation, which was reduced by Ssd administration. These results showed that Ssd inhibited high glucose induced proliferation and DNA synthesis in NRK-52E cells.
There is growing evidence that oxidative stress is involved in the pathogenesis of chronic diseases such as diabetes and its complications including nephropathy [20-22]. A number of ROS-generating pathways including xanthine oxidase, glycolysis, uncoupling of nitric oxide synthase, specific defects in the polyol pathway, advanced glycation, and NAD(P)H oxidase have been identified as potential contributors to the development and progression of DN [23]. Furthermore, it is suggested that mitochondrial production of ROS due to the chronic hyperglycemia is a key initiator for these pathogenic pathways [24]. In our study, high glucose stimulation significantly enhanced ROS production, whereas Ssd attenuated the intracellular ROS levels. Persistent oxidative stress results in lipid peroxidation, formation of harmful products, and decreased activity of antioxidant defense enzymes [25,26]. We found that the concentration of MDA was increased, whereas the SOD activity was decreased after high glucose intervention. However, Ssd treatment markedly reduced MDA content and enhanced SOD activity. In addition, the mRNA levels of IDH2 and MnSOD, two important endogenous anti-oxidative enzymes [27] were significantly increased in Ssd treated NRK-52E cells. Taken together, these results demonstrated that Ssd could protect NRK-52E cells against oxidative stress induced by high glucose.
The sirtuins are a conserved family of class III histone deacetylases, which are involved in genetic control of aging, transcriptional regulation, and longevity of organisms ranging from yeasts to humans [28-30]. Among the known sirtuin members, SIRT3 is emerging as a pivotal regulator of cell proliferation and oxidative stress [31]. Studies on SIRT3 deficient mice have reported that SIRT3 protects cells from oxidative stress by activating superoxide dismutase 2 through deacetylation of lysine residues [32]. A recent study has demonstrated that SIRT3 modulates the deacetylation of FOXO3 and attenuates mitochondrial dysfunction induced by oxidative stress [33]. In our study, we hypothesized that SIRT3 may play a key role in NRK-52E cells. Interestingly, results showed that Ssd could promote the expression of SIRT3 in NRK-52E cells. In addition, we knockdown SIRT3 with specific SIRT3 siRNA and observed no changes in cell proliferation, ROS production, and activities of ROS and MDA in the Ssd group compared with that in the high glucose group. These results demonstrated that Ssd protected NRK-52E cells against high glucose induced injury through regulation of SIRT3.
In summary, our study for the first time demonstrates that Ssd treatment protects renal tubular epithelial cell against high glucose induced injury. SIRT3 upregulation may represent a novel mechanism by which Ssd exerts the therapeutic effects on hyperglycemia-associated diseases.
Disclosure of conflict of interest
None.
References
- 1.Giunti S, Barit D, Cooper ME. Diabetic nephropathy: from mechanisms to rational therapies. Minerva Med. 2006;97:241–262. [PubMed] [Google Scholar]
- 2.Waanders F, Visser FW, Gans RO. Current concepts in the management of diabetic nephropathy. Neth J Med. 2013;71:448–458. [PubMed] [Google Scholar]
- 3.Zhou X, Feng Y, Zhan Z, Chen J. Hydrogen sulfide alleviates diabetic nephropathy in a streptozotocin-induced diabetic rat model. J Biol Chem. 2014;289:28827–28834. doi: 10.1074/jbc.M114.596593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanwar YS, Wada J, Sun L, Xie P, Wallner EI, Chen S, Chugh S, Danesh FR. Diabetic nephropathy: mechanisms of renal disease progression. Exp Biol Med (Maywood) 2008;233:4–11. doi: 10.3181/0705-MR-134. [DOI] [PubMed] [Google Scholar]
- 5.Pal PB, Sinha K, Sil PC. Mangiferin attenuates diabetic nephropathy by inhibiting oxidative stress mediated signaling cascade, TNFalpha related and mitochondrial dependent apoptotic pathways in streptozotocin-induced diabetic rats. PLoS One. 2014;9:e107220. doi: 10.1371/journal.pone.0107220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kitada M, Kume S, Imaizumi N, Koya D. Resveratrol improves oxidative stress and protects against diabetic nephropathy through normalization of Mn-SOD dysfunction in AMPK/SIRT1-independent pathway. Diabetes. 2011;60:634–643. doi: 10.2337/db10-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang SC, Lee SF, Wang CJ, Lee CH, Lee WC, Lee HJ. Aqueous Extract from Hibiscus sabdariffa Linnaeus Ameliorate Diabetic Nephropathy via Regulating Oxidative Status and Akt/Bad/14-3-3 gamma in an Experimental Animal Model. Evid Based Complement Alternat Med. 2011;2011:938126. doi: 10.1093/ecam/nep181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sourris KC, Forbes JM. Interactions between advanced glycation end-products (AGE) and their receptors in the development and progression of diabetic nephropathy-are these receptors valid therapeutic targets. Curr Drug Targets. 2009;10:42–50. doi: 10.2174/138945009787122905. [DOI] [PubMed] [Google Scholar]
- 9.Lu CN, Yuan ZG, Zhang XL, Yan R, Zhao YQ, Liao M, Chen JX. Saikosaponin a and its epimer saikosaponin d exhibit anti-inflammatory activity by suppressing activation of NF-kappaB signaling pathway. Int Immunopharmacol. 2012;14:121–126. doi: 10.1016/j.intimp.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 10.Wong VK, Zhou H, Cheung SS, Li T, Liu L. Mechanistic study of saikosaponin-d (Ssd) on suppression of murine T lymphocyte activation. J Cell Biochem. 2009;107:303–315. doi: 10.1002/jcb.22126. [DOI] [PubMed] [Google Scholar]
- 11.Ying ZL, Li XJ, Dang H, Wang F, Xu XY. Saikosaponin-d affects the differentiation, maturation and function of monocyte-derived dendritic cells. Exp Ther Med. 2014;7:1354–1358. doi: 10.3892/etm.2014.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen MF, Huang CC, Liu PS, Chen CH, Shiu LY. Saikosaponin a and saikosaponin d inhibit proliferation and migratory activity of rat HSC-T6 cells. J Med Food. 2013;16:793–800. doi: 10.1089/jmf.2013.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan J, Li X, Li P, Li N, Wang T, Shen H, Siow Y, Choy P, Gong Y. Saikosaponin-d attenuates the development of liver fibrosis by preventing hepatocyte injury. Biochem Cell Biol. 2007;85:189–195. doi: 10.1139/O07-010. [DOI] [PubMed] [Google Scholar]
- 14.Zhang BZ, Guo XT, Chen JW, Zhao Y, Cong X, Jiang ZL, Cao RF, Cui K, Gao SS, Tian WR. Saikosaponin-D attenuates heat stress-induced oxidative damage in LLC-PK1 cells by increasing the expression of anti-oxidant enzymes and HSP72. Am J Chin Med. 2014;42:1261–1277. doi: 10.1142/S0192415X14500797. [DOI] [PubMed] [Google Scholar]
- 15.Gross JL, de Azevedo MJ, Silveiro SP, Canani LH, Caramori ML, Zelmanovitz T. Diabetic nephropathy: diagnosis, prevention, and treatment. Diabetes Care. 2005;28:164–176. doi: 10.2337/diacare.28.1.164. [DOI] [PubMed] [Google Scholar]
- 16.Schena FP, Gesualdo L. Pathogenetic mechanisms of diabetic nephropathy. J Am Soc Nephrol. 2005;16(Suppl 1):S30–S33. doi: 10.1681/asn.2004110970. [DOI] [PubMed] [Google Scholar]
- 17.Sodhi CP, Phadke SA, Batlle D, Sahai A. Hypoxia and high glucose cause exaggerated mesangial cell growth and collagen synthesis: role of osteopontin. Am J Physiol Renal Physiol. 2001;280:F667–F674. doi: 10.1152/ajprenal.2001.280.4.F667. [DOI] [PubMed] [Google Scholar]
- 18.Habib SL. Alterations in tubular epithelial cells in diabetic nephropathy. J Nephrol. 2013;26:865–869. doi: 10.5301/jn.5000287. [DOI] [PubMed] [Google Scholar]
- 19.Tang SC, Lai KN. The pathogenic role of the renal proximal tubular cell in diabetic nephropathy. Nephrol Dial Transplant. 2012;27:3049–3056. doi: 10.1093/ndt/gfs260. [DOI] [PubMed] [Google Scholar]
- 20.Kashihara N, Haruna Y, Kondeti VK, Kanwar YS. Oxidative stress in diabetic nephropathy. Curr Med Chem. 2010;17:4256–4269. doi: 10.2174/092986710793348581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamagishi S, Matsui T. Advanced glycation end products, oxidative stress and diabetic nephropathy. Oxid Med Cell Longev. 2010;3:101–108. doi: 10.4161/oxim.3.2.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wada J, Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci (Lond) 2013;124:139–152. doi: 10.1042/CS20120198. [DOI] [PubMed] [Google Scholar]
- 23.Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes. 2008;57:1446–1454. doi: 10.2337/db08-0057. [DOI] [PubMed] [Google Scholar]
- 24.Zhou X, Feng Y, Zhan Z, Chen J. Hydrogen sulfide alleviates diabetic nephropathy in a streptozotocin-induced diabetic rat model. J Biol Chem. 2014;289:28827–28834. doi: 10.1074/jbc.M114.596593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dentelli P, Rosso A, Zeoli A, Gambino R, Pegoraro L, Pagano G, Falcioni R, Brizzi MF. Oxidative stress-mediated mesangial cell proliferation requires RAC-1/reactive oxygen species production and beta4 integrin expression. J Biol Chem. 2007;282:26101–26110. doi: 10.1074/jbc.M703132200. [DOI] [PubMed] [Google Scholar]
- 26.Karadeniz A, Simsek N, Karakus E, Yildirim S, Kara A, Can I, Kisa F, Emre H, Turkeli M. Royal jelly modulates oxidative stress and apoptosis in liver and kidneys of rats treated with cisplatin. Oxid Med Cell Longev. 2011;2011:981793. doi: 10.1155/2011/981793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai SH, Chen T, Wang YH, Zhu J, Luo P, Rao W, Yang YF, Fei Z, Jiang XF. Sirt3 protects cortical neurons against oxidative stress via regulating mitochondrial Ca2+ and mitochondrial biogenesis. Int J Mol Sci. 2014;15:14591–14609. doi: 10.3390/ijms150814591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michan S, Sinclair D. Sirtuins in mammals: Insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haigis MC, Guarente LP. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 30.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, Samant S, Ravindra PV, Isbatan A, Gupta MP. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J Biol Chem. 2010;285:3133–3144. doi: 10.1074/jbc.M109.077271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Y, Zhang J, Lin Y, Lei Q, Guan KL, Zhao S, Xiong Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011;12:534–541. doi: 10.1038/embor.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tseng AH, Shieh SS, Wang DL. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic Biol Med. 2013;63:222–234. doi: 10.1016/j.freeradbiomed.2013.05.002. [DOI] [PubMed] [Google Scholar]