

Abstract
Objectives: This study was conducted to investigate a molecular mechanism by which budesonide inhalation may mitigate pathological responses of cigarette smoke-induced COPD. Methods: Rats were exposed to air (control) and cigarette smoke (smoking) in the presence and absence of budesonide. Cell count in bronchoalveolar lavage fluid (BALF), lung function test, mean liner intercept in lung tissue, mean alveolar number, right ventricular hypertrophy index (RVHI) and morphological changes in lungs were assessed, respectively. Alpha-1 antitrypsin (A1AT) and neutrophil elastase (NE) mRNA expression in lung tissues and their protein productions in BALF were examined as well. Results: Smoking rats showed significant changes in the above assessments as compared to those of the control rats (all P < 0.01 or 0.05). Budesonide applied for the smoking rat significantly decreased differential cell counts in BALF and ameliorated lung function and RVHI (P < 0.01 or 0.05) with mitigated peribronchiolar inflammation and pulmonary bullae formation in the smoke-exposed lungs. Treatment with budesonide resulted in obvious decreases in NE mRNA and protein expression levels (both P < 0.05). Conclusion: Budesonide inhalation serves to improve lung function and right ventricular dysfunction through attenuating pulmonary inflammatory response and NE expression level in the diseased lungs.
Keywords: Budesonide, cigarette smoke, Alpha-1 antitrypsin, neutrophil elastase, pulmonary inflammation and COPD
Introduction
Chronic obstructive pulmonary disease (COPD) is a type of obstructive lung disease characterized by airflow limitation that is not fully reversible but usually progressive and associated with an inflammatory response of the lung [1]. The inflammatory cells involved mainly include neutrophil granulocytes and macrophages, two types of white blood cell [2,3]. The primary risk factor for COPD globally is tobacco smoking which subsequently leads to a series of pulmonary structure changes [4]. Moreover, advanced COPD can lead to pulmonary hypertension due to low oxygen levels in the alveoli and thereby harm heart [5]. Though there is no known cure for COPD that is an important cause of morbidity, mortality and hospital admissions, the symptoms are treatable and progression of the disease can be delayed, and most cases of COPD are potentially preventable.
Budesonide is a man-made glucocorticoid steroid related to the naturally-occurring hormone, cortisol or hydrocortisone which is produced in the adrenal glands and has a potent anti-inflammatory action [6]. When used as an inhaler, the agent goes directly to the inner lining of the inflamed airways to exert its effects. Although the role of many corticosteroids in COPD patients is controversial because of questionable benefit and potentially significant drug toxicity [7], regular treatment with budesonide inhalation has been shown to reduce the frequency of COPD exacerbation and improve health status in patients [8]. Budesonide therefore has widely been prescribed for COPD patients as the inhaled steroid may treat stable symptoms of COPD that are slowly getting worse. Since COPD is a multicomponent disease that includes airway inflammation, air flow limitation, airway structural changes and an imbalance between proteases and antiproteases found in the diseased lung tissues [9], it would be valuable to validate how this steroid may influence pathological changes in COPD with a pharmacological intervention applied for its clinical interest.
This study is to investigate the effect of inhaled budesonide on cigarette smoke-induced COPD with a possible mechanism by which this steroid may attenuate pulmonary inflammatory response, and functional and/or structural impairments of lungs and heart in its therapeutic effect.
Materials and methods
Animals and cigarette smoke exposure
Specific pathogen-free, male SD rats (6 wks) weighing about 180 g were purchased from the Experimental Centre of Animals at Hebei Medical University. The rats were housed in an environmentally controlled animal facility of our hospital for the duration of the experiments. All studies were conducted in accordance with the approval of Hospital Research Review Committee.
Rats were randomly divided into three groups of 10 each and the protocol for making animal model of cigarette smoke-induced COPD was modified with different treatments during a smoking period. The animal model of COPD was established with mainstream smoke exposure (20 cigarettes, twice a day) for 4 months in a cigarette smoke chamber. 2.0 mg budesonide (AstraZeneca, UK) dissolved in 2.0 ml phosphate buffered saline (PBS) was given by aerosol inhalation (once a day for a final month). The cigarette smoke-exposed rats inhaled an equivalent volume of PBS during the period of the last month. Rats in the control group were exposed to air and treated with aerosol of PBS at the same volume in the indicated time.
Cell counts and preparation of BALF and organ specimens
Experimental rats were sacrificed by blood loss from opening femoral artery and left lung of the rat lavaged 2 times with 3 ml of PBS via tracheal cannulation after the right main bronchus was ligated. An equal volume of BALF was collected and the BALF supernatants were obtained with centrifuge (1500 rpm × 15 min) at 4°C. The samples were stored at -70°C for the enzyme-linked immunosorbent assays (ELISA). The cell pellet collected from each sample was applied to a glass slide using a cytospin (1000 rpm × 10 min) and then the slide was stained with Hema 3 Stain Set (Fisher Scientific) for the differential count of cells. The relative proportion of different cells counted from a total of 300 cells/slide was factored to the number (×105/ml) of total BALF cells collected in each group.
Following the collection of BALF, the right lung and heart were harvested and blood accumulated in the right atrium was completely removed from the organ. Some of the lung tissues were immediately snap frozen in liquid nitrogen, then stored at -70°C until processed. Some of the lung tissues and heart were placed in a wide-mouth bottle filled up in 4% formaldehyde for histopathological examination. Briefly, the lung tissues from groups were embedded in paraffin and cut into a 4-µm section. The lung sections stained with hematoxylin and eosin (H&E) solution were applied to a glass slide and photographed by light microscope (Olympus, Japan) at a magnification of 400 for morphological analysis.
Measurement of lung function
Lung function was examined after rats were anaesthetized by an intraperitoneal injection of 10% Chloral hydrate (3 ml/kg) and maintained with an appropriate plane of the anesthesia. The trachea was opened with an inverted T-shaped incision in the position between the 2nd and the 3rd cartilage ring, rapidly intubated, and placed the animal into an apparatus for measuring the volume of air inspired and expired by the lungs. (Beijing Rambo Technology Co. Ltd. Beijing, China). The one of the exports of the T-typed cannula in the trachea was connected to a pressure transducer applied to a pulmonary mechanics analyzer and another one was used for administration of air to expand the lungs of rats.
A ratio of forced expiratory volume at 0.3 s and forced vital capacity (FEV0.3/FVC), dynamic lung compliance (Cdyn), and inspiratory (Ri) and expiratory (Re) resistance were measured with injecting 6.0 ml air into the T-typed cannula in the trachea of the anaesthetized rats and parameters of lung function were automatically recorded by the analyzer.
Morphometric study
Mean linear intercept (MLI) in lung tissue and mean alveolar number (MAN) were examined on two glass slides of each group in the same size of the field of view. Using light microscopy, MLI was determined for each region studied on an overlay consisting of horizontal and vertical lines. All intercepts with alveolar septal number (ASN) were counted at the intersection point of the two lines in the central field of the view under microscope. The total length (L) of all the lines together divided by the number of intercepts gives the mean linear intercept for the region studied. A formula is shown as MLI = L/ASN, which is used to estimate an average diameter of a single alveolus in size. MAN was determined according to alveolar number (AN) in each field of view and a square area (SA) of the field. A formula is shown as MAN = AN/SA (mm2), which is an indicator for density of alveoli.
The right ventricular free wall from its atrial, septal and valve ring attachments was dissected and carefully removed epicardial fat. The right ventricular free wall was then flattened out (endocardial surface down) and blotted on filter paper for mass determination by weight. The left ventricle, including the septum (S), was similarly weighed after removal of atrial and valvular attachments and epicardial fat. A right ventricular hypertrophy index (RVHI) is shown as RVHI = RV/(LV + S).
Determination of A1AT and NE mRNA and protein
A1AT and NE mRNA expression levels were assessed by using SYBR Premix Ex TaqTM (Takara, Japan). Briefly, the total RNA from lung tissue was extracted using a tissue homogenizer in Qiagen lysis buffer and purification of RNA was performed with Qiagen RNeasy minicolumns following the manufacturer’s protocol. RNA was quantified using the NanoDrop ND-1000 spectrophotometer and amplified and biotin-labeled with Nugen’s Ovation System, according to the manufacturer’s instructions (NuGEN Technologies, Inc., San Carlos, CA). The yield of total RNA per replicate varied from 0.6 g to 2.0 g. 50 ng of the RNA was added in a SYBR qPCR master mix for real-time RT-PCR (qPCR) which allows easy reaction assembly by simply adding primers, template and sterile distilled water, and therefore the intercalator-based qPCR can be performed. Contents of A1AT and NE mRNA were shown with a fold-change as compared to an expression level of the control sample.
The contents of A1AT and NE protein productions in the BALF were determined using antibodies against the murine A1AT and NE from Bio-Rad (Hercules, CAUS) according to the manufacturer’s directions. Briefly, 100 µl of the substrate solution was added to 100 µl of sample in microliter plates and incubated for 30 min at room temperature. The reaction was stopped by adding 50 µl of 4 M sulfuric acid, and the optical densities (OD) were read in a microtiter autoreader at 450 nm.
Statistical analysis
Values were expressed Mean ± Standard Deviation (SD). Statistical analysis was performed using Statistical Package for the Social Science (SPSS, version 13.0). Comparisons from groups were performed by one-way analysis of variance (ANOVA). Student’s paired t-test was used to compare measurements of individual groups. P values less than 0.05 were considered to be significant.
Results
Inflammatory cells in BALF
To clarify whether inhaled budesonide alone influenced the development of lung remodeling, we examined cellular composition of the BALF in smoking rats in absence and presence of the corticosteroid and the results are shown Figure 1A-C. A significant increase in the numbers of leukocytes such as neutrophils and macrophages was clearly observed in the smoking rats as compared to those of the control rats. Treatment with budesonide inhalation alone significantly decreased the numbers of these cells in the smoking rats (both P < 0.01). However, the budesonide-treated rats still showed high levels of the inflammatory cell infiltrates in the BALF as compared to the control animals (both P < 0.01).
Figure 1.
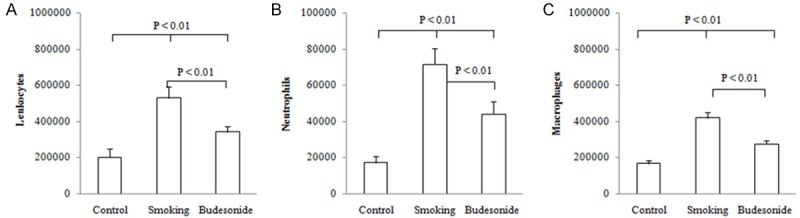
Differential cells in BALF. Rats were exposed to air (control) and cigarette smoke (smoking) in presence and absence of inhaled budesonide alone. The numbers of leukocytes (A) such as neutrophils (B) and macrophages (C) in the BALF from the experimental rats were assessed. Data were expressed as Mean ± SD (n = 10).
Change in lung function
FEV0.3/FVC and Cdyn were determined at the end of the challenge procedure and the results are shown in Figure 2A, 2B. The average values (%) for FVC/FVC0.3 and Cdyn (cmH2O.s.ml) were shown as 52.8 ± 8.1 and 0.0360 ± 0.0028 in control rats, 35.8 ± 6.7 and 0.0296 ± 0.0024 in smoking rats and 42.2 ± 5.1 and 0.0324 ± 0.0023 in the budesonide-treated rats, respectively. In contrast, the values of FVC/FVC0.3 and Cdyn in the smoking rats treated with and without budesonide were lower than those of the control animals (P < 0.01 or 0.05). Treatment with budesonide may partly reverse the lowering FVC/FVC0.3 and Cdyn with significant differences seen in both measurements between the smoking rats treated with and without budesonide (both P < 0.05).
Figure 2.
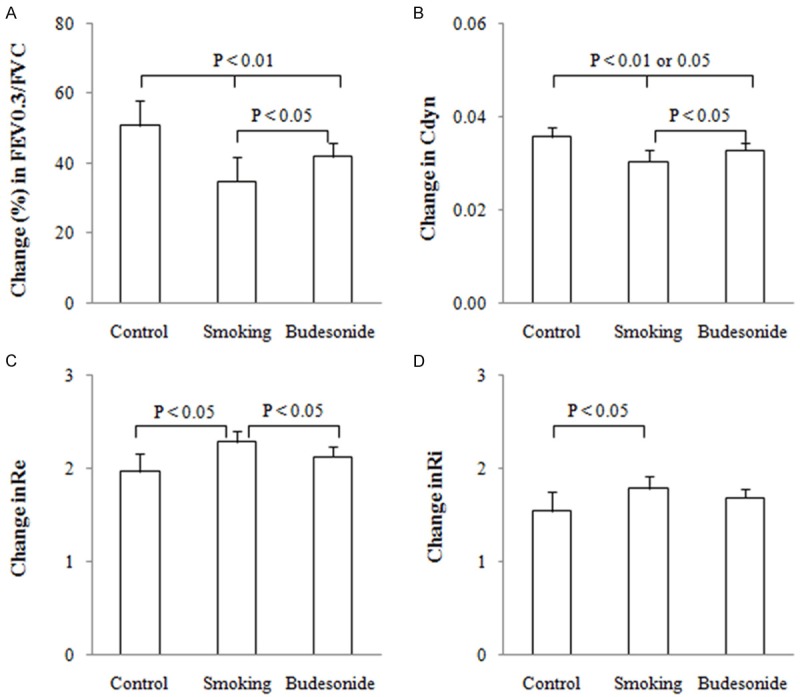
Lung function testing. A ratio of forced expiratory volume at 0.3 s and forced vital capacity (FEV0.3 /FVC) (A), dynamic lung compliance (Cdyn) (B) and respiratory resistance during expiratory (Re) (C) and inspiratory (Ri) (D) phases of tidal breathing were examined in experimental rats exposed to air (control) and cigarette smoke (smoking) in presence and absence of inhaled budesonide. Measurements were expressed as Mean ± SD (n = 10).
Ri and Re were synchronously determined in the rats and the results are shown in Figure 2C, 2D. The average values (cmH2O) for Re and Ri were shown as 2.01 ± 0.28 and 1.50 ± 0.14 in control rats, 2.56 ± 0.30 and 1.90 ± 0.17 in smoking rats and 2.29 ± 0.28 and 1.73 ± 0.18 in the budesonide-treated rats, respectively. In contrast, Re and Ri in the smoking rats were significantly increased over the control rats (both P < 0.05). Treatment with budesonide resulted in a decrease in Re but not Ri in the smoking rats (P < 0.05).
Change in cardiopulmonary structure
Lungs were surgically removed from the experimental rats and then applied to evaluate a change in size of air spaces and alveolar density in the lungs using MLI and MAN measurement technique. The results are shown in Figure 3A, 3B. The average values of MLI (m) and MAN (n/m2) were shown as 26.2 ± 4.3 and 20.3 ± 2.5 in control rats, 36.5 ± 3.1 and 12.5 ± 2.2 in smoking rats and 33.2 ± 2.5 and 14.5 ± 2.5 in the budesonide-treated rats, respectively. In contrast, enlargement of air spaces and a decrease in alveolar density was obviously observed in the lungs from smoking rats as compared to either control or the budesonide-treated rats (P < 0.01 or 0.05). Treatment with budesonide may slightly alter the values of MLI, MAN and BDW/D, but there were no significant differences seen in these measurements between the smoking rats treated with and without budesonide inhalation.
Figure 3.
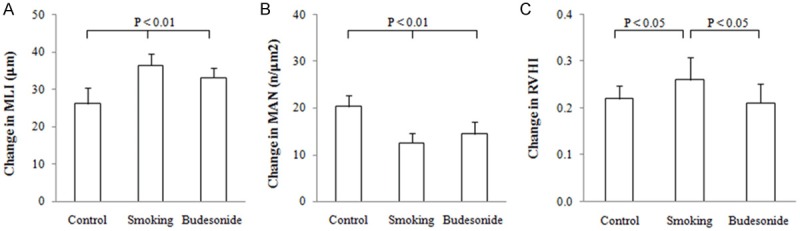
Morphometry of lung and right ventricular mass. Size of air spaces and alveolar density in lungs from rats exposed to air (control) and cigarette smoke (smoking) in presence and absence of inhaled budesonide was evaluated in MLI (A) and MAN (B) measurement technique in microscopic vision of lung tissue. Right ventricular hypertrophy index (RVHI) was examined by measuring mass of right and left ventricular wall and ventricle septal (C). All measurements were expressed as Mean ± SD (n = 10).
RVHI was determined in the rats and the results are shown in Figure 3C. The values for RVHI were shown as 0.221 ± 0.028, 0.261 ± 0.048 and 0.211 ± 0.042 in control, smoking and budesonide-treated rats, respectively. In contrast, the value for RVHI was significantly increased in smoking rats as compared to control ones (P < 0.05). Treatment with budesonide resulted in lowering the RVHI with a statistical difference in the measurement between the smoke- and the budesonide-exposed animals (P < 0.05).
Histopathologic analysis of lungs
The lower lobe of right lung was removed from experimental rats and sectional tissues of the lung were photographed in absence and presence of budesonide (Figure 4). Morphologically, rats in the control group showed a normal structure of lungs with an intact epithelium layer. Those rats were free of cellular infiltration and mucus observed in the lung sections. Examination by light microscope at ×400 magnification showed that an enlargement of alveolar airspaces and destruction of septal walls of alveoli with inflammatory cell infiltrates may be observed in the sectional tissue of the smoking rats. Treatment with budesonide may relieve accumulation of inflammatory cells around the airways, but focal emphysema still existed in the lungs of the treated rats.
Figure 4.
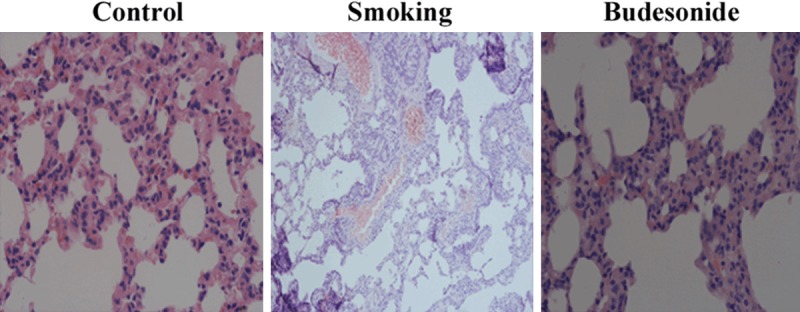
Morphological analysis in lungs. Histological structures of lungs, the status of airway epithelial layer and alveolar septa, and inflammatory cell infiltration were observed on frozen sections of lung specimens from rats exposed to air (A) and cigarette smoke (smoking) in absence (B) and presence (C) of inhaled budesonide. Histopathologic examination was performed under a light microscopy at a magnification of ×400.
A1AT and NE mRNA and protein expression levels
The A1AT and NE mRNA expression in right lung tissues were assayed by transcriptional profiling and represented as the fold change (Fc) of mRNA from the tissues of rats in reference to the mRNA expression level of the control animals. The results are shown in Figure 5A, 5B. A1AT and NE mRNA expression levels were shown as 1.00 and 1.00 in control rats, 0.55 ± 0.06 and 5.15 ± 1.15 in smoking rats, and 0.60 ± 0.08 and 2.75 ± 0.95 in the budesonide-treated rats, respectively. A1AT mRNA expression in the smoking rats treated with and without budesonide showed significant decreases as compared to the control rats (both P < 0.01). However, the NE mRNA expression in the smoking rats treated with and without the steroid showed 5.0- and 2.75-fold increases as compared to the controls (P < 0.01 or 0.05). Treatment with budesonide resulted in lowering the expression level of NE but not A1AT mRNA with a statistical difference observed between the smoking rats treated with and without the steroid (P < 0.05).
Figure 5.
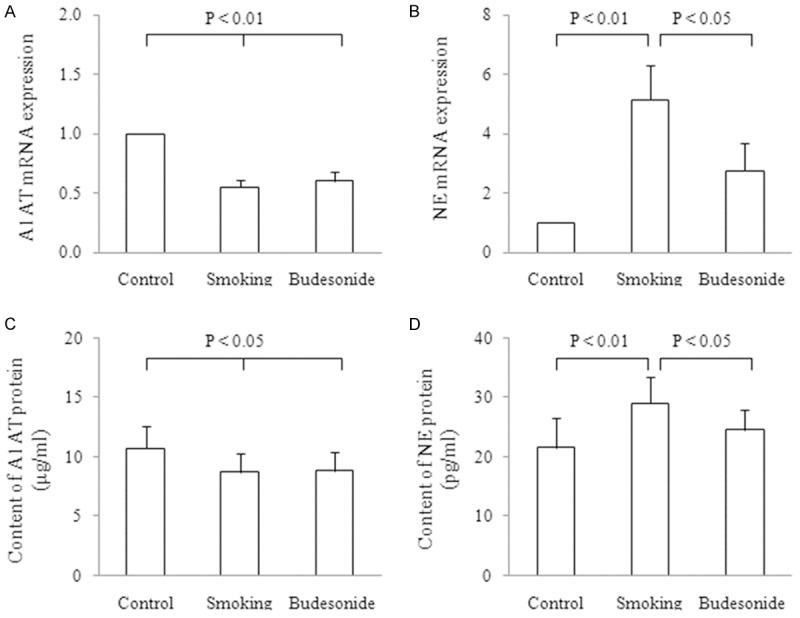
A1AT and NE mRNA and protein expression level. The A1AT and NE mRNA expression were assayed in lungs form rats exposed to air (control) and cigarette smoke (smoking) in absence and presence of budesonide inhalation. The data were represented as the fold change (Fc) of mRNA from the lungs of the treated rats. Expression levels (Fc) of A1AT (A) and NE (B) mRNA in the lungs were calculated in reference to the mRNA expression level of the control animals. A1AT (C) and NE (D) protein productions were determined in the BALF from the treated rats as well. Measurements were expressed as Mean ± SD (n = 10).
Levels of A1AT and NE protein productions in the BALF were assessed in the treated rats and the results are shown in Figure 5C, 5D. Contents of A1AT (g/ml) and NE (pg/ml) protein in the samples were 10.71 ± 1.91 and 21.52 ± 5.01 in control, 8.73 ± 1.62 and 29.04 ± 4.51 in smoking and 8.83 ± 1.61 and 24.51 ± 3.52 in the budesonide-treated rats, respectively. The level of A1AT production in BALF from the smoking rats in absence and presence of budesonide showed a significant decrease as compared to those of the control rats (both P < 0.05). However, the level of NE production in BALF from the smoking rats was obviously enhanced as compared to the control animals (P < 0.01). Budesonide inhalation significantly reduced NE but not A1AT protein content in the BALF from the challenged rats (P < 0.05).
Discussion
COPD is a deadly and costly disease with lots of studies emerged from the susceptibility to smoking-related lung functional damage [10-12]. In initial experiments, a rat COPD model was established based on the observation that a 4-month smoke challenge was able to model the abnormal changes of lung function. Our results revealed that this rat model was associated with a significant decrease in values of FEV0.3/FVC and Cdyn concomitantly with an obvious increase in Re as compared to the control animals. These findings demonstrated that lung function decline with certain degree of severity has been reached progressively with the challenge time in this model. In the measurements, the parameters of lung function tests were recorded by injecting 6.0 ml air into the lungs to expand chest of the animals. This offers an advantage over the agonist stimulation because the airflow response is more related to the condition of in vivo airway obstruction and/or narrowing. In terms of cell response, smoking rats showed a large inflammatory infiltrate, comprised mainly of leukocytes such as neutrophils and macrophages, into the BALF, clearly indicating that these cells were required for cigarette smoke-driven airway inflammation as the long-term remodeling effect associated with COPD. It has been found that the lungs of young smokers have an accumulation of macrophages in the bronchiolar region and bronchiolitis [13,14], which is considered as an important defense mechanism with which the cells may neutralize and clear the toxic particulate fraction [3]. The key contribution of neutrophils participated in the process of remodeling has been highlighted as a crucial role of mediators from the cells in animal models of COPD [15]. Furthermore, a change in an expression level of enzymes in the diseased lung tissues has led to understanding of neutrophils linked to development of the disease [2]. Treatment with inhaled budesonide not only partly reversed lung function decline but also markedly ameliorated inflammatory cell infiltrate, indicating efficacy of the anti-inflammatory steroid in treatment of COPD. It is more likely that budesonide protects lung function against the smoke-induced lung function damage through attenuating inflammatory response associated with neutrophil infiltrate.
Having demonstrated a significant role for budesonide in improving lung function damage and inflammatory cell infiltrate, we were curious as to the relationship of the steroid with a structural change in cardiopulmonary system. To investigate this interaction, MLI and MAN were measured in understanding distal air space size and alveolar density in the challenged lungs because the measurements of both morphometric techniques have been involved in the direct and unbiased estimation for the quantitative analysis of lung structure [16,17]. The results showed that enlargement of air spaces was obvious with a decreased alveolar number detected in the cigarette smoke-exposed lungs as compared to those of the control animals, whereas treatment with the steroid inhalation did not influence the pathological changes in the lungs. The findings led us to conclude that the glucocorticoid effects were weak in altering structural lesions of the lung tissues. Our data also showed a significant increase in RVHI of the rats exposed to cigarette smoke for a period of the months, indicating right ventricular dysfunction associated with the disease progresses. Budesonide successfully improved cardiac muscle dysfunction in the inhaled therapy, which is consistent with clinical observations that COPD patients treated with budesonide inhalation are less likely to experience cardiovascular events [18,19]. Since the steroid plays a protective role in the smoke-induced heart damage, it is conceivable that the corticosteroid therapy effect on RVHI is achieved probably due to relieving pulmonary cell infiltration which may promote compression of small pulmonary vessels [20] and induce pulmonary arterial hypertension as a common cause of right ventricular hypertrophy [21].
In association with the above-stated findings, photomicrograph of COPD-like changes in the challenged lung tissues was examined as well. The data showed peribronchiolar inflammation including a large amount of inflammatory cells infiltration around the small airways, an emphysema-like airspace enlargement, destruction of septal walls of alveoli and pulmonary bullae formed in the lungs [22]. These provided a direct evidence of lung injury, which was completely identical to the findings from the lung function tests. With regard to the observed histopathological changes, it indicated that the rat model of the cigarette smoke-induced COPD was successfully established. Treatment with budesonide slightly mitigated peribronchiolar inflammation and morphological impairments in the smoke-exposed lungs, suggesting that this steroid may help delay but not stop lung destruction in the progression of the disease.
In order to understand the tenuous balance between proteases and anti-proteases within the smoke-exposed lungs, A1AT and NE mRNA expression levels were assessed by transcriptional profiling. Our data revealed that A1AT and NE mRNA expression levels in the challenged lungs were down- and up-regulated in reference to those of the control animals, not only indicating that both gene products were involved in the event of COPD but also displayed highly disparate expression of the gene products. In contrast, the change in the expression level of NE mRNA was much more obvious than that of A1AT in this model, suggesting that the increase in the NE mRNA expression level referred to tissue destruction at the site of NE released since this may be an important factor in facilitating cell migration through the tight connective tissue matrix in the area of damage [23]. In support of the findings, A1AT and NE protein productions were determined in the BALF from the smoke-exposed rats and the results were completely identical to the findings at the mRNA levels of both gene products. These observations supported proteinase/antiproteinase theory of the pathogenesis of emphysema that is now accepted widely as a key mechanism of the disease development [9] and firmly established NE as the proteinase most responsible for tissue destruction in COPD. It has been known that NE is released from activated neutrophils which are increased in the airways of smokers and COPD patients [24], whereas experimental application of NE can reproduce many of the features of COPD patients [25]. Treatment with budesonide inhalation resulted in obvious influencing NE but not A1AT mRNA and protein expression levels, demonstrating that the glucocorticoid steroid acts predominantly on NE and regulates proteinase/antiproteinase imbalance through reducing the NE expression level within the diseased lungs. This finding provides a mechanism by which inhaled budesonide protects structure and function of heart and lungs against the NE-producing damages.
In conclusion, treatment with budesonide inhalation ameliorates lung function damage and right ventricular dysfunction through mitigating inflammatory cell infiltrates and reducing the NE expression level to mediate the proteinase/antiproteinase balance in the smoke-exposed lungs.
Disclosure of conflict of interest
None.
References
- 1.Rabe KF, Beghé B, Luppi F, Fabbri LM. Update in chronic obstructive pulmonary disease 2006. Am J Respir Crit Care Med. 2007;175:1222–32. doi: 10.1164/rccm.200704-586UP. [DOI] [PubMed] [Google Scholar]
- 2.Stockley RA. Neutrophils and the pathogenesis of COPD. Chest. 2002;121:151S–155S. doi: 10.1378/chest.121.5_suppl.151s. [DOI] [PubMed] [Google Scholar]
- 3.Tetley TD. Macrophages and the pathogenesis of COPD. Chest. 2002;121:156S–159S. doi: 10.1378/chest.121.5_suppl.156s. [DOI] [PubMed] [Google Scholar]
- 4.Bohadana A, Teculescu D, Martinet Y. Mechanisms of chronic airway obstruction in smokers. Respir Med. 2004;98:139–51. doi: 10.1016/j.rmed.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 5.Roberts DH, Lepore JJ, Maroo A, Semigran MJ, Ginns LC. Oxygen therapy improves cardiac index and pulmonary vascular resistance in patients with pulmonary hypertension. Chest. 2001;120:1547–55. doi: 10.1378/chest.120.5.1547. [DOI] [PubMed] [Google Scholar]
- 6.Mendes ES, Rebolledo P, Campos M, Wanner A. Immediate antiinflammatory effects of inhaled budesonide in patients with asthma. Ann Am Thorac Soc. 2014;11:706–11. doi: 10.1513/AnnalsATS.201307-220OC. [DOI] [PubMed] [Google Scholar]
- 7.Falk JA, Minai OA, Mosenifar Z. Inhaled and systemic corticosteroids in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2008;5:506–12. doi: 10.1513/pats.200707-096ET. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gunen H, Hacievliyagil SS, Yetkin O, Gulbas G, Mutlu LC, In E. The role of nebulised bude sonide in the treatment of exacerbations of COPD. Eur Respir J. 2007;29:660–7. doi: 10.1183/09031936.00073506. [DOI] [PubMed] [Google Scholar]
- 9.Greene CM, McElvaney NG. Proteases and antiproteases in chronic neutrophilic lung disease-relevance to drug discovery. Br J Phar macol. 2009;158:1048–58. doi: 10.1111/j.1476-5381.2009.00448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet. 2007;370:765–73. doi: 10.1016/S0140-6736(07)61380-4. [DOI] [PubMed] [Google Scholar]
- 11.Camiña-Tato M, Morcillo-Suárez C, Bustamante MF, Ortega I, Navarro A, Muntasell A, López-Botet M, Sánchez A, Carmona P, Julià E, Tortola MT, Audí L, Oksenberg JR, Martin R, Montalban X, Comabella M. Gender-associated differences of perforin polymorphisms in the susceptibility to multiple sclerosis. J Immunol. 2010;185:5392–404. doi: 10.4049/jimmunol.1000102. [DOI] [PubMed] [Google Scholar]
- 12.Celli BR, MacNee W ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23:932–46. doi: 10.1183/09031936.04.00014304. [DOI] [PubMed] [Google Scholar]
- 13.Jeffery PK. Structural and inflammatory changes in COPD: a comparison with asthma. Thorax. 1998;53:129–36. doi: 10.1136/thx.53.2.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finkelstein R, Ma HD, Ghezzo H, Whittaker K, Fraser RS, Cosio MG. Morphometry of small airways in smokers and its relationship to emphysema type and hyperresponsiveness. Am J Respir Crit Care Med. 1995;152:267–76. doi: 10.1164/ajrccm.152.1.7599834. [DOI] [PubMed] [Google Scholar]
- 15.Chung KF. Cytokines in chronic obstructive pulmonary disease. Eur Respir J Suppl. 2001;34:50s–59s. [PubMed] [Google Scholar]
- 16.Andersen MP, Parham AR, Waldrep JC, McKenzie WN, Dhand R. Alveolar fractal box dimension inversely correlates with mean linear intercept in mice with elastase-induced emphysema. Int J Chron Obstruct Pulmon Dis. 2012;7:235–43. doi: 10.2147/COPD.S26493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ochs M, Nyengaard JR, Jung A, Knudsen L, Voigt M, Wahlers T, Richter J, Gundersen HJ. The number of alveoli in the human lung. Am J Respir Crit Care Med. 2004;169:120–4. doi: 10.1164/rccm.200308-1107OC. [DOI] [PubMed] [Google Scholar]
- 18.Curkendall SM, Lanes S, de Luise C, Stang MR, Jones JK, She D, Goehring E Jr. Chronic obstructive pulmonary disease severity and cardiovascular outcomes. Eur J Epidemiol. 2006;21:803–13. doi: 10.1007/s10654-006-9066-1. [DOI] [PubMed] [Google Scholar]
- 19.Sin DD, Tu JV. Inhaled corticosteroids and the risk of mortality and readmission in elderly patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;164:580–4. doi: 10.1164/ajrccm.164.4.2009033. [DOI] [PubMed] [Google Scholar]
- 20.Peinado VI, Barberá JA, Abate P, Ramírez J, Roca J, Santos S, Rodriguez-Roisin R. Inflamm atory reaction in pulmonary muscular arteries of patients with mild chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;159:1605–11. doi: 10.1164/ajrccm.159.5.9807059. [DOI] [PubMed] [Google Scholar]
- 21.Chaouat A, Naeije R, Weitzenblum E. Pulmonary hypertension in COPD. Eur Respir J. 2008;32:1371–85. doi: 10.1183/09031936.00015608. [DOI] [PubMed] [Google Scholar]
- 22.H F. Animal models of pulmonary emphysema: a stereologist’s perspective. Eur Respir Rev. 2006;15:13. [Google Scholar]
- 23.Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A. Neutrophil Elastase Contributes to Cigarette Smoke-Induced Emphysema in Mice. Am J Pathol. 2003;163:7. doi: 10.1016/S0002-9440(10)63589-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev. 2007;87:1047–82. doi: 10.1152/physrev.00048.2006. [DOI] [PubMed] [Google Scholar]
- 25.Sommerhoff CP, Nadel JA, Basbaum CB, Caughey GH. Neutrophil elastase and cathepsin G stimulate secretion from cultured bovine airway gland serous cells. J Clin Invest. 1990;85:682–9. doi: 10.1172/JCI114492. [DOI] [PMC free article] [PubMed] [Google Scholar]