

Abstract
In the present study, the genotype of two Canine distemper virus (CDV) strains, namely, ZJJ-SD and ZJJ-LN, were investigated, based on the whole hemagglutinin (HA) gene. The CDV strains were obtained from two foxes in Shandong Province and Liaoning Province in 2011. Phylogenetic analyses were carried out for 260 CDV strains worldwide, and a statistical analysis was performed in the amino acid substitutions at positions 530 and 549 of the HA protein. Phylogenetic analyses revealed that the two strains, ZJJ-SD and ZJJ-LN, belonged to the CDV Asia I lineage. Site 530 of HA protein was found to be relatively conserved within CDV lineages in different host species by combining the genetic sequence data with the published data from 260 CDV strains worldwide. The data analysis showed a bias toward the predicted substitution Y549H for the non-dog strains in Asia I and Europe lineages. The ratio of site 549 genetic drift in the HA gene were significantly different between dogs and non-dogs in the two lineages. The strain ZJJ-SD, from wild canid, has an Y549H substitution. It is one of three Y549H substitution for wild canids in Asia I lineages. Site 530 of HA protein was not immediately relative to CDV genetic drift from dogs to non-dogs. Statistical analysis indicated that non-dog strains have a high probability to contain Y549H than dog strains in Asia I and Europe lineages. Thus, site 549 is considered important in genetic drift from dogs to non-dogs, at least in Asia I and Europe lineages.
Keywords: Canine distemper virus (CDV), hemagglutinin (HA), phylogenetic analysis, genetic drift
Introduction
Canine distemper virus (CDV) is an enveloped, single-stranded, negative RNA virus from the Morbillivirus genus and family Paramyxoviridae. CDV is prevalent among the dog population and causes disease in several types of carnivores worldwide [1-3]. The CDV hemagglutinin (HA) gene encodes the HA protein, which has a significant role in virus invasion. This protein is an important factor that binds the cell receptor-signaling lymphocytic activation molecule SLAM (CD150) in all morbilliviruses [3-5].
Experiments have shown that the CDV HA protein is the most variable protein in any member of the genus Morbillivirus. This finding could explain why CDV has a broader host range compared with other morbilliviruses [2]. The SLAM binding region for CDV is important when CDV enters into the host cells [5]. Phylogenetic analyses of the sequence data of strains from domestic dogs and non-dog species indicate that positive selection drives amino acid substitutions at positions 530 and 549 within the SLAM binding region of the HA gene [6]. Although six different amino acids [aspartic acid (D), glutamic acid (E), glycine (G), asparagine (N), arginine (R), and serine (S)] were observed at site 530, majority of CDV strains retrieved from domestic dogs had either 530G or 530E. However, strains from terrestrial non-dog carnivores and one strain from an aquatic carnivore (Baikal seal, Pusa sibirica) had R, D, and N residues [3,6]. In different hosts, CDV strains retrieved from domestic dogs typically showed tyrosine (Y) at site 549. However, 7 out of 12 strains from non-dog hosts encoded histidine (H) at site 549 [3,6]. The wild-type domestic dog CDV strain in ferrets (Mustela putorius) showed a genetic drift in Y549H, which is also 106 expected in non-dog hosts [7]. Analysis of amino acid substitutions at the known functional positions 530 and 549 within the SLAM binding region of the HA gene indicated that the significant differences between the prevalence of 549Y and 549H in wild canid and non-canid strains suggested a degree of virus adaptation to the categories of the host [3].
Most phylogenetic analyses that focused on the HA gene from CDV strains from different parts of the world have shown that CDV strains usually clustered within different geographical areas (America I, America II, Asia I, Asia II, Europe, European wildlife, Arctic, South America, and Southern Africa) [8-10]. These results indicate that the HA gene has undergone genetic drift in different geographical regions [8].
In the current study, the genotype of two CDV strains, ZJJ-SD and ZJJ-LN, were analyzed. Phylogenetic analyses revealed that the strains examined belonged to the CDV Asia I lineage. The combination of the genetic sequence data with the published data from 262 CDV strains worldwide confirmed the importance of amino acid substitutions in the HA protein at position 549 (Y to H/L) to the spread of domestic-dog-adapted CDV strains to other animals and vaccine strains, especially in Asia I and Europe lineages. Strains from non-dog hosts were biased toward the predicted substitution Y549H in Asia I lineages Europe and vaccine strain lineages. Site 530 of the HA protein was found to be conserved within CDV lineages from different host species, but has undergone genetic drift from G/E to A in the Asia I lineage (GenBank No. HM623893.1 and HM623891.1), V in the European wildlife lineage (GenBank No. JN153021.1 and JN153022.1), and K in the Asia II lineage (GenBank No. AY297453.1).
Materials and methods
Clinical specimens
The viscera of the foxes, which were suspected to have CDV infection, were collected from the provinces of China. CDV-positive samples were identified by RT-PCR, which amplifies a 335 bp long fragment of the nucleoprotein gene [11].
RT-PCR
The total RNA was extracted from 100 mg of homogenized tissue samples (lung, spleen, and liver) by using the TRIzol reagent (Invitrogen). The extracted RNA was eluted using 30 μL of diethyl pyrocarbonate-treated water. Reverse transcription was performed in a final volume of 20 μL containing 12 μL of RNA solutions, 4 μL of 5×RT buffer, 1 μL of dNTP (10 mM each), 1 μL (50 pmol) of random hexamers, 1 μL of reverse transcriptase (AMV) (TaKaRa), and 1 μL of RNase inhibitor, and was incubated at 42°C for 1 h and at 70°C for 15 min. PCR amplification was performed with primers targeting the HA gene, H-F (5’-TTAGGGCTCAGGTAGTCCA-3’), and H-R (5’-CTAAGKCCAATTGARA TGTGT-3’; K = G/T, R = A/G). PCR amplicons of the expected size (1879 bp) were obtained. After pre-denaturation at 95°C for 5 min, the PCR amplification cycle was optimized at 94°C for 45 s; 52°C for 45 s; and 72°C for 2 min, for 35 cycles with a final extension step at 72°C for 10 min. Approximately 10 μl of the amplified product was visualized using gel electrophoresis in 1.0% agarose gel in the presence of ethidium bromide. Bands were visualized by ultraviolet light transillumination.
Data set collection and phylogenetic analysis
Sequences were extracted from the GenBank database. Datasets for the analysis included CDV HA sequences acquired from dog and non-dog hosts. Datasets were constructed for 262 HA genes with accession numbers shown in Table S1 and Figure 1. Sequences were aligned using Clustal X [12]. A neighbor-joining tree was generated with MEGA 5 [13]: (i) using 2 complete HA gene sequences obtained in the current study; and (ii) 260 published HA sequences from GenBank. Bootstrap consensus trees were inferred from 1000 replicates using the close-neighbor-interchange algorithm. The two complete CDV HA gene sequences used in the current study were provided by Jianjun Zhao. These sequences were recovered from two domesticated foxes caught in Shandong Province and Liaoning Province, China, in 2011.
Figure 1.
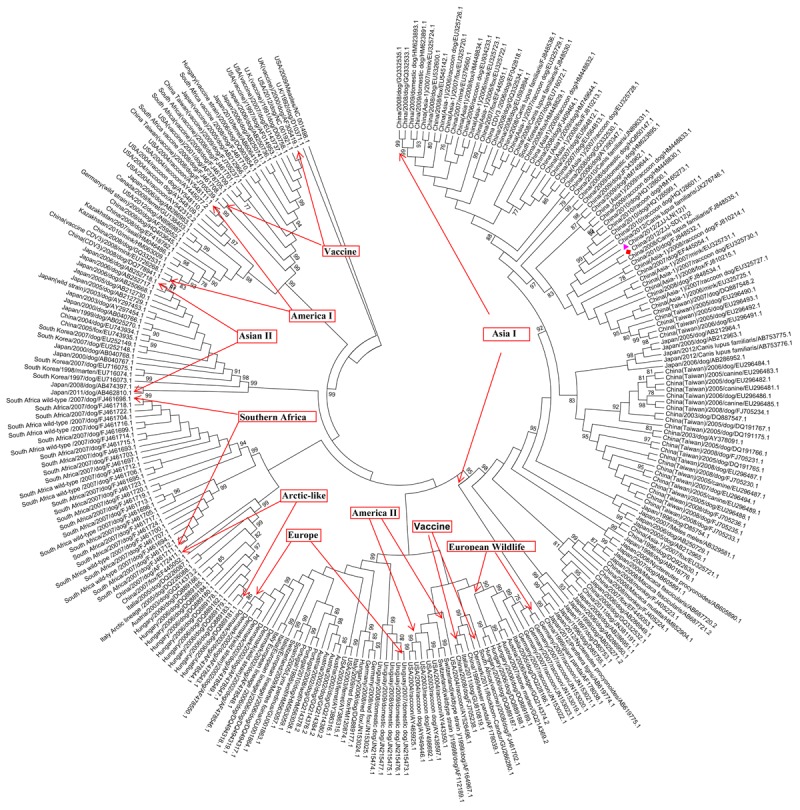
Phylogenetic relationships of canine distemper virus strains obtained from two foxes (presented in bold) caught in China with the published strains based on the sequences of the hemagglutinin gene using the ClustalW program and the MEGA 5.0 software package. Neighbor-joining algorithm was used to generate the tree. Statistical support for nodes was provided by bootstrapping 1000 replicates. Only bootstrap values > 75% were shown. Triangles (▲) (China/2012/ZJJ-LN (12) 1, collected in Liaoning Province) and rhombuses (♦) (China/2012/ZJJ-SD (12) 2, collected in Shandong Province) indicate the two Chinese wild-type CDV strains analyzed in this study. For each strain, the country of origin, year, host species, and GenBank accession number are given. Marked areas indicate strains within lineages. The 262 strains included within each lineage are shown in Table S1.
Analysis of amino acid sites
The aa present at sites 530 and 549 on the HA protein was identified from a total of 262 CDV HA genes. The sequence data from numerous CDV outbreaks in a wide variety of carnivore species and locations would have been ideal. However, available sequence data were strongly biased to CDV strains from domestic dogs, wild canids from Asian fur farms, and multiple sequence data from genetically identical strains from a relatively limited number of hosts. The ratio of amino acid drift in the HA protein at positions 530 and 549 was analyzed. Meanwhile, the genetic drifts of the HA gene in CDV virus strains from different genetic lineages were evaluated.
Results
Complete CDV HA gene sequences were obtained from two domesticated foxes caught in China. Phylogenetic analyses of the HA sequences produced almost identical phylogenetic trees when the neighbor-joining method was used (Figure 1). Phylogenetic analyses revealed that the two strains belonged to the CDV Asia I lineage. The combination of the genetic sequence data with the published data from 260 CDV strains worldwide confirmed the bias of amino acid substitutions in the HA protein at position 549 (Y to H/L/S) in CDV strains from dogs and non-dogs and vaccines (Tables S1 and S2). 3 out of 34 wild canids and 2 out of 13 non-canids have Y549H substitution but at the same time 3 out of 67 domestic dogs have the same substitution for Asia I lineage. The first two genetic variation rates are much higher than the latter. The same is in Europe lineage. Although, strains from dog and non-dog hosts were all exist substitution Y549H or Y549L or Y549S except no data available at all for wild canids and non-canids in Asia II and Southern Africa lineages, wild canids in America I, II and European wild life lineages or non-canids in Arctic lineage. In this paper, site 549 of the HA protein in one of two strains, ZJJ-SD, belonged to the wild canids with drift to H. That means a higher genetic drift ratio Y549H from dog to non-dog strains at least in Asia I and Europe lineages. Site 530 of the HA protein was conserved within CDV lineages from different host species. On the basis of the statistical analysis result (Table S2), site 530 of the HA protein was not directly related to the genetic drift of hemagglutin gene in CVD strains from dog to non-dog species. However, this site has undergone a genetic drift toward A in the Asia I lineage (GenBank No. HM623893.1 and HM623891.1), V in the European wildlife lineage (GenBank No. JN153021.1 and JN153022.1), and K in the Asia II lineage (GenBank No. AY297453.1).
The evaluation results showed that the comparison of 262 CDV strains recovered worldwide illustrated that the residue at site 530 was generally conserved within the different CDV lineages regardless of host species (e.g., G in Asia I and in Europe; D in European wildlife; N in Arctic, South Africa, and America I; and E in Asia II, S in Vaccine) (Table S2). Across the globe, strains from non-canid wildlife species showed five different residues at site 530 (G, D, N, V, and R), whereas the domestic dog strains showed eight residues (G and E to R/D/N/V/A/K in Table S2) at this site. All the CDV strains were included in the Asia I lineage, regardless of host species code for G. In the European wildlife lineage, domestic dogs and non-dog species predominantly code D. The statistical analysis result also indicated that site 530 was not directly related to the genetic drift of hemagglutin gene in CDV strains from different genetic lineages. Site 530 of the genetic variation showed geographical differences.
Global comparison of the aa present at HA gene site 549 in 262 CDV strains revealed a highly significant probability of Y (rather than H) at this site in all the strains, except vaccine strains. Data restriction was observed in areas in which both 549Y and 549H CDV strains have been shown to circulate (Asia I, Asia II, Europe, European wildlife, America I, and America II). A significant bias to 549Y strains recovered from domestic dogs was also observed. Likewise, geographic clade is a variable element. Geographic clade has more significant effect on the existence of H or Y at site 549 compared with host species (e.g., both “Europe” and “European wildlife” have a greater mix of H and Y strains in canids than other clades). These results indicated that dog strains have a high probability to contain 549Y, whereas the non-dog strains showed a bias toward H at site 549. Nevertheless, a trend toward 549Y exists in Asia I and Asia II lineages. The ratio of genetic drift in dogs from Asia I was 4.5% (3/67), whereas that in canids and non canids was 8.8% (3/34) and 15.4% (2/13). The ratio of genetic drift in dogs from Europe was 9.5% (2/21), whereas that in canids and non canids was 8.8% (3/34) and 15.4% (2/13). The difference was highly significant (P < 0.01). Thus, site 549 of HA protein is important to the genetic drift of hemagglutin gene in CDV strains from dog to non-dog species, at least in Asia I and Europe lineages.
Discussion
The strains recovered from the two foxes were identified under the Asia I wildlife lineage through phylogenetic analysis. Most of the HA proteins in the Asia I lineage were found to have residue 549Y. The HA gene of numerous strains in the Asia I and II lineages have been described, and the comparable sequence data from many of the strains in the Asia I and II lineages are available [11,14]. Genotypes of sufficient CDV strains recovered from the foxes in China were also available to provide some general conclusions. The results suggest that a previously unrecognized CDV lineage is circulating in China and that the CDV strains are evolving independently within the area.
Domestic-dog-adapted CDV strains were also found to spread readily to other carnivores. Currently, no evidence presented that carnivores in the Arctic or Southern Africa encountered CDV strains with 549H. These problems, including the small sample sizes and the likely dependence between identical strains, make statistical analyses of residues at these sites challenging. Given the strong effect of lineage on site 530, the diversity of residues at this site was reported only in strains from three categories of host: domestic dogs, wild canids, and non-canids.
One of the key factors influencing the ability of CDV strains to spread from domestic dog hosts to non-dog carnivore hosts is the amino acid substitutions at sites 530 and 549 in the SLAM binding region of the CDV HA protein [6]. The present results covered 260 CDV strains worldwide and confirmed the importance of amino acid substitutions in the HA protein at position 549 (Y to H/L) to the spread of domestic-dog-adapted CDV strains to other animals or vaccine strains. Site 530 of the HA protein in 262 CDV strains was found to be conserved within CDV lineages in different host species. However, this site has undergone genetic drift toward either A in Asia I lineages (HM623893.1 and HM623891.1), V in European wildlife lineages (JN153021.1 and JN153022.1), and K in Asia II lineages (AY297453.1). The HA protein in the two fox strains described had residues 549Y and 549H (Tables S1 and S2). The patterns of 549Y and 549H in wild canid and non-canid species were significantly different. On the basis of the data analysis, the ratio of site 549 genetic drift in the HA gene from dogs and non-dogs have a significant difference. This finding indicates that the virus-host adaptations were dependent on the residue at this site or at least for hosts within the broad categories. The present null hypothesis assumes that domestic dogs, wild-canids, and non-canid hosts will likely encounter strains with Y or H at site 549 and will be equally susceptible to both types of strains. However, most domestic dogs have been infected with CDV strains by other domestic dogs. Therefore, their chance of infection with a 549H strain was low. CDV transmission in wild carnivore species could also occur between individuals within a species and could be influenced by factors such as population size, degree of sociality, and ranging patterns [15]. Strains from non-dog hosts were biased toward the predicted substitution Y549H in Asia I and Europe lineages. An unreported genetic drift was also observed in the HA gene of CDV at position 549, showing Y549H and Y549S in Asia I lineages, Y549L in vaccine strain lineages. The currently available data suggest that both domestic dogs and wild canid species will more likely be infected by CDV strains with 549Y than 549H in the SLAM binding region of the HA protein. However, determining whether the probability of infection of non-canid species will significantly be altered by the presence of either H or Y at site 549 is difficult because of insufficient data. Likewise, CDV strains recovered from wild canid species and non-canid species differed in the prevalence of H or Y at site 549. This finding raises questions on the effect of the residue at this site based on the evaluation of CDV strains to canid versus non-canid host species.
In conclusion, the study revealed that site 530 of the HA protein was not immediately relative to CDV genetic drift from dogs to non-dogs. The analysis indicated that dog strains will more likely have 549Y than 549H, whereas the non-dog strains showed different biases toward H or Y at site 549. Thus, site 549 is considered important in genetic drift from dogs to non-dogs, at least in Asia I and Europe lineages.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (grant: 31402088).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Appel MJ, Summers BA. Pathogenicity of morbilliviruses for terrestrial carnivores. Vet Microbiol. 1995;44:187–191. doi: 10.1016/0378-1135(95)00011-x. [DOI] [PubMed] [Google Scholar]
- 2.Pomeroy LW, Bjornstad ON, Holmes EC. The evolutionary and epidemiological dynamics of the paramyxoviridae. J Mol Evol. 2008;66:98–106. doi: 10.1007/s00239-007-9040-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nikolin VM, Wibbelt G, Michler FU, Wolf P, East ML. Susceptibility of carnivore hosts to strains of canine distemper virus from distinct genetic lineages. Vet Microbiol. 2012;156:45–53. doi: 10.1016/j.vetmic.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 4.Tatsuo H, Ono N, Yanagi Y. Morbilliviruses use signaling lymphocyte activation molecules (CD150) as cellular receptors. J Virol. 2001;75:5842–5850. doi: 10.1128/JVI.75.13.5842-5850.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.von Messling V, Oezguen N, Zheng Q, Vongpunsawad S, Braun W, Cattaneo R. Nearby clusters of hemagglutinin residues sustain SLAM-dependent canine distemper virus entry in peripheral blood mononuclear cells. J Virol. 2005;79:5857–5862. doi: 10.1128/JVI.79.9.5857-5862.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCarthy AJ, Shaw MA, Goodman SJ. Pathogen evolution and disease emergence in carnivores. Proc Biol Sci. 2007;274:3165–3174. doi: 10.1098/rspb.2007.0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.von Messling V, Springfeld C, Devaux P, Cattaneo R. A ferret model of canine distemper virus virulence and immunosuppression. J Virol. 2003;77:12579–12591. doi: 10.1128/JVI.77.23.12579-12591.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martella V, Cirone F, Elia G, Lorusso E, Decaro N, Campolo M, Desario C, Lucente MS, Bellacicco AL, Blixenkrone-Moller M, Carmichael LE, Buonavoglia C. Heterogeneity within the hemagglutinin genes of canine distemper virus (CDV) strains detected in Italy. Vet Microbiol. 2006;116:301–309. doi: 10.1016/j.vetmic.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 9.Panzera Y, Calderon MG, Sarute N, Guasco S, Cardeillac A, Bonilla B, Hernandez M, Francia L, Bedo G, La Torre J, Perez R. Evidence of two co-circulating genetic lineages of canine distemper virus in South America. Virus Res. 2012;163:401–404. doi: 10.1016/j.virusres.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 10.Woma TY, van Vuuren M, Bosman AM, Quan M, Oosthuizen M. Phylogenetic analysis of the haemagglutinin gene of current wild-type canine distemper viruses from South Africa: lineage Africa. Vet Microbiol. 2010;143:126–132. doi: 10.1016/j.vetmic.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 11.Tan B, Wen YJ, Wang FX, Zhang SQ, Wang XD, Hu JX, Shi XC, Yang BC, Chen LZ, Cheng SP, Wu H. Pathogenesis and phylogenetic analyses of canine distemper virus strain ZJ7 isolate from domestic dogs in China. Virol J. 2011;8:520. doi: 10.1186/1743-422X-8-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao JJ, Yan XJ, Chai XL, Martella V, Luo GL, Zhang HL, Gao H, Liu YX, Bai X, Zhang L, Chen T, Xu L, Zhao CF, Wang FX, Shao XQ, Wu W, Cheng SP. Phylogenetic analysis of the haemagglutinin gene of canine distemper virus strains detected from breeding foxes, raccoon dogs and minks in China. Vet Microbiol. 2010;140:34–42. doi: 10.1016/j.vetmic.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 15.Guiserix M, Bahi-Jaber N, Fouchet D, Sauvage F, Pontier D. The canine distemper epidemic in Serengeti: are lions victims of a new highly virulent canine distemper virus strain, or is pathogen circulation stochasticity to blame? J R Soc Interface. 2007;4:1127–1134. doi: 10.1098/rsif.2007.0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.