

Abstract
The microbiome has been implicated directly in host health, especially host metabolic processes and development of immune responses. These are particularly important in infants where the gut first begins being colonized, and such processes may be modeled in mice. In this investigation we follow longitudinally the urine metabolome of ex-germ-free mice, which are colonized with two bacterial species, Bacteroides thetaiotaomicron and Bifidobacterium longum. High-throughput mass spectrometry profiling of urine samples revealed dynamic changes in the metabolome makeup, associated with the gut bacterial colonization, enabled by our adaptation of non-linear time-series analysis to urine metabolomics data. Results demonstrate both gradual and punctuated changes in metabolite production and that early colonization events profoundly impact the nature of small molecules circulating in the host. The identified small molecules are implicated in amino acid and carbohydrate metabolic processes, and offer insights into the dynamic changes occurring during the colonization process, using high-throughput longitudinal methodology.
The human body is populated by a dense array of microorganisms that constitute a dynamic source of macromolecules and small molecules (metabolites)1. These bacterial-origin metabolites are absorbed continuously by the host and integrated into their systemic circulation. Efforts are in progress to catalogue specific microbial populations in healthy and pathological human states, including the extensive research from the Human Microbiome Project2,3,4,5,6, and new directions in integrating such information for personalized medicine approaches7,8,9. The microbiome has been shown to have multiple connections to human physiology, including the effects of energy modulation and connections to obesity, metabolic disorders and drug efficacy10. The effect of intestinal dysbiosis has been associated with various diseases, including obesity, diabetes, Crohn’s and celiac diseases11,12,13,14,15,16,17,18. The influence of diet on the constitution of the microbiome has been also explored, both in the context of mechanism discovery, as well as effecting physiological changes, such as treatment of disease, obesity reduction as well as modulating drug efficacy and toxicity10,19,20. The study of the microbiome and the implementation of pharmacomicrobiomics20 lends itself to applications relevant to the recent efforts on precision medicine announced by the National Institutes of Health21. The interplay between host-microbiome interactions is being extensively modeled, at all scales, starting from individual biochemical interactions and protein/gene associations to systems-level implementations that predict global effects of host-microbiome modulation, colonization and diet, including genome-scale metabolic models (GEMs)22.
Additional investigations have studied the processes that drive the development of adult-like bacterial composition in the gut, beginning from a “sterile” environment at birth23,24. Studies involving 16 S ribosomal RNA (rRNA) based sequencing25 and metatranscriptomic or proteomic analyses26,27,28 have reported the involvement of conditions at birth and infant diet in the complex development of gut microbiota observed during the first months of life29. The 16 S rRNA-based studies revealed that in the intestinal microbiota of healthy adult humans the majority (>99%) of detected phylotypes belonged to two bacterial divisions: Bacteroides and Firmicutes30. These relative proportions of these bacterial populations have already been implicated in studies contrasting children’s diets31.
The specific mechanisms that select for particular groups of bacteria in the infant gut remain largely unknown. To mimic the development of the bacterial flora in the sterile gut of a newborn, simplified mice models, like germ free (GF) mice, have been employed32 and Gnotobiotic mice (ex-GF mice) colonized with specific sets of microorganisms or full microbial communities have been shown to be useful models in the study of symbiont–host interactions. The dynamics of microbial colonization events early in life upon the host metabolome are also still under investigation. Metabolome changes in feces from germ free mice during the process of acclimatization to non-germ free environment have been previously evaluated33. Martin et al. studied fecal metabolic differences between conventional mice and germ free mice colonized with a mix of seven microbes isolated from baby fecal samples34. Their investigations revealed differences in metabolite content and proportion at individual time points post colonization. We further extend such approaches to the use of time-series correlation trends to follow the effects of bacterial acquisition as described below.
To extract more information about the colonization process new systems biology methodologies are now being implemented, aided by recent advent of metabolomics technologies (e.g. Nuclear Magnetic Resonance (NMR), gas chromatography/mass spectrometry (GC-MS), or ultra-performance liquid chromatography/mass spectrometry (UPLC/MS)35,36), which provide a new toolkit, enabling the profiling and monitoring of all the metabolite components in a given sample. These high-throughput approaches capture the intensities of thousands of components and reveal that host metabolomes are directly impacted by the presence of gut bacteria25. The identity and function of microbes colonizing the gut has a direct impact on the small molecules, metabolites, that are produced34,37. Many of these metabolites, which are still structurally uncharacterized, are taken up into host circulation where they can interact with tissues throughout the body, be co-metabolized by enzymes including those in the liver, and ultimately are excreted through the kidney into urine. Comparison of urine metabolic profiles from conventional and germ free rats and mice38,39 revealed an important contribution of the gut microbiota in liver or kidney metabolism, and a connection to adaptive immune responses40. Other studies have verified how the presence of bacteria impact the colonic luminal metabolome41, as well as some endogenous metabolite levels42.
In this investigation we address the direct response in a germ free environment to bacterial colonization, and evaluate changes in host urine metabolome over time. To mimic the interaction of bacteria-sterile gut we use a gnotobiotic mice model, colonizing germ free mice with Bacteroides thetaiotaomicron (Bt) and Bifidobacterium longum (Bif. longum), commonly present in the infant gut32,43,44. The urine metabolome was profiled over multiple time points (spanning 25 days), once in germ free (GF) used as a reference and every five days in colonized mice, using UPLC-MS technologies. One of the major hurdles in analyzing such time-course data is accounting for the sampling. Missing intensity information for mass features in mass spectrometry spectra leads to unevenness in the sampling, even if the experiment was designed to sample at regular intervals. To allow for this, we applied a method that was initially developed for temporal data in astronomy45,46,47, and later extended to other time series, including biological and omics longitudinal data7,48,49,50,51,52,53. The approach takes the time variable into account, using classification of each signal by autocorrelation and then pattern matching to reveal the underlying collective behavior and is generalizable to longer time series and other omics (Yusufaly and Mias in preparation). The complex data spectra were analyzed in order to identify metabolites and their temporal trends, to identify and classify the various patterns corresponding to the process of bacterial colonization. Interesting features that show the response to microbiota introduction in the host metabolome include both continuous trends and sudden increases and decreases in metabolite intensity levels with a range of metabolic pathways observed (related to immune responses, amino acid and carbohydrate processing pathways).
Results
To evaluate changes due to bacterial colonization in germ free mice, the study followed the global changes in metabolomics from urine samples in Swiss-Webster germ free (GF) mice, after colonization. The urine samples were essentially used as an output signature of changes caused by the bacterial colonization. The small molecules in mouse urine were profiled using mass spectrometry over the course of 25 days (Fig. 1a) at five days intervals.
Figure 1. Inoculation of Germ Free Mice.

(a) Germ free mice were inoculated on Day 0 with Bacteriodes. thetaiotaomicron and Bifidobacterium longum by oral gavage ingestion. Urine and fecal samples were processed for metabolome and colony profiling respectively from these mice on days 5, 10, 15, 20, 25. Principal Component Analysis in both Positive (b) and Negative (c) acquisition mode full metabolome results reveal that the mice samples aggregate into groups, with the Germ Free and Day 5 groups well separated. The components account for 81% and 80% of the variances in Positive and Negative modes respectively. The corresponding normalized distributions are shown on the right panels displaying similar profiles, where intensities IM have been standardized (with median and median absolute deviation), indicating symmetry in approach in positive and negative modes. See also Supplementary Fig. S1, Supplementary Table S1.
The mice, initially all GF, were placed inside gnotobiotic isolators. One group of GF mice (n = 3) was used as a reference for comparison. The other group of initially GF mice (n = 4) was colonized on Day 0 using oral gavage with 108 cfu of Bt (VPI-5482) and 108 cfu of Bif. longum (NCC2705). Urine samples (used for metabolome profile) and feces (used for verifying the colonization composition) from GF mice were collected on Day 0, and similarly from the bi-associated mice on Days 5, 10, 15, 20 and 25 after their colonization. The bacterial composition in the bi-associated mice was determined by plating assays of fecal samples, and did not fluctuate after Day 5 (Supplementary Fig. S1), showing a relatively higher proportion of Bt (75–95% range). The urine samples collected were processed using high affinity liquid chromatography and mass spectrometry (LC-MS), using an Exactive (Thermo Fisher) mass spectrometer, with the electrospray operated in both positive and negative ion modes. The mass spectra were aligned in both retention times and masses (see Methods), resulting in the overall detection of 3245 mass features of interest (1555 and 1692 detected in negative and positive mode respectively).
The processed mass spectra were then analyzed to obtain underlying trends (as outlined in Supplementary Fig. S2). Namely, datasets were: (1) analyzed using Principal Components Analysis (PCA) to assess variation between the GF mice and the bi-associated mice on different days, (2) spectrally analyzed to obtain longitudinal patterns and classified into significance categories based on their profile over time, and (3) potential biological significance assessed through the assignment of putative mass identities and pathway analysis (see Methods). In particular:
(1) The PCA of the aligned comprehensive data revealed a clear separation of the mice into three major sets: (i) The GF mice group and (ii) the Day 5 group, which are distinctly separated from (iii) the remaining groups (Days 10–25) which are rather intermixed as one set (Fig. 1b). The corresponding normalized distributions of metabolites of GF mice and each of the bi-associated mice timepoints remained similar across all measurements, both in negative and positive modes, indicative of the robustness of the normalization procedure. The PCA analysis results can account for most of the variability between the mice groups. In particular the total variance accounted by the three components shown in Fig. 1b was ~80% for the positive mode data (variance from [P1,P2,P3]≈[55%,19%,6%]) and similarly ~81% for negative mode data (variance from [P1,P2,P3]≈[59%,18%,4%]).
(2) The normalized data were used to construct time series signals. For each detected mass feature, a time series was constructed using the GF mice dataset as a stable point reference39 - an effective “day zero” data point. For a given mass feature, the data from the time points measured in the bi-associated mice (Day 5-Day 20 after inoculation) were all compared to the same corresponding GF entry. Each resulting signal of relative metabolite changes was classified and assigned to one of three classes, if it displayed one of the following significant temporal trends: (I) autocorrelated at lag one (at p < 0.05; bootstrap distribution; n = 100,000 with replacement) where the signal displays correlated behavior (essentially linear) between each sequential time point in the signal; or as spike trends showing maxima (II) or minima (III), i.e. aberrantly high or low levels respectively, as compared to the signal baseline with random fluctuations, (at p < 0.05; based on n = 100,000 bootstrap simulation with replacement).
The above outlined classification approach (see Methods) was also tested in simulations, and found to perform well for six time-points (directly applicable to this project, Fig. 2). The simulations assessed robustness and reconstruction of temporal trends in known linear signals. The linear signals were perturbed through the addition of random noise, either 5% or 10% and also combined with random signals. Each signal set was allowed to have up to one time point missing (except the first time point in all series, which was used as a reference point, in analogy of using the GF mice as a comparison reference point). In the simulations, two filters were used to explore the efficiency of the algorithm, (i) a strict p < 0.05 cutoff for autocorrelation at lag one (Filter S) and (ii) a relaxed filter (Filter R), in which the p-value was relaxed until the entire set of linear trends was recovered by the classification. The simulation results suggested modest false discovery rates (FDR). In a typical example shown in Fig. 2, for the case of 5% error in the signals, Filter S had FDR 0.026–0.041 and Filter R recovered all linear signals at p < 0.14 and FDR 0.026–0.08. For 20% errors in the signals, Filter S had FDR 0.026–0.048 and Filter R recovered all linear signals at p < 0.22 and FDR 0.021–0.101. The heatmaps and clustering corresponding to the simulation results are shown in Fig. 2b.
Figure 2. Simulation of Random Time Series.

(a) Simulations of the autocorrelation classification methodology were performed to assess robustness and reconstruction of temporal trends in known linear signals. The example illustrates a random realization that corresponds to a simulation of: 12,000 linear signals, of equal numbers of positive (+) or negative slope/trend (−), combined with a randomly generated equivalent set of 6000 random signals, and in randomized order. The numbers in each set were equally distributed between having zero or one time point missing (excluding the first one which is used as a reference). Additionally a simulation of 100,000 random signals was used as a background of bootstrap simulation. In particular, for simulations of linear signals with 5% random error per timepoint, Filter S corresponds to a strict p < 0.05 cutoff for autocorrelation at lag 1 (random series bootstrap, n = 100,000). In Filter R the p-value is relaxed until the entire set of linear trends is recovered. As seen, this results in modest false discovery rates. The simulation was repeated for linear signals with 20% error per timepoint for both filters. The corresponding heatmaps and clustering are shown in (b). (c) The reconstruction of autocorrelations from the periodogram, and the exact autocorrelations are shown for a straight signal, for a series of n = 6 and n = 100 time points. For n = 100 the reconstruction is indistinguishable from the exact calculation in the figure.
The classification analyses of the experimental urine metabolome data assigned a total of 576 molecules to the different time trend classes (334 autocorrelated, 106 spike maxima, 136 spike minima). 45 of these molecules were considered to be high interest identifications based on their uniqueness of mass, or identity verification through the use of standards using follow-up mass spectrometry experiments (Table 1, Supplementary Table S1 for full data). Hierarchical clustering within each temporal class revealed distinct trends in the metabolite compositions, corresponding to the colonization of the GF mice (see Methods) (Fig. 3, left). The autocorrelated trends revealed two distinct groups, showing contrasting trends – one increasing constantly following colonization (A2 in Fig. 3, which included validated compounds such as tyramine, L-homocysteine, and estriol), with the other decreasing constantly (clustering group A5 in Fig. 3, including validated compounds such as 5 hydroxy-L-tryptophan and N-acetyl-L-methionine). The spike maxima and minima also displayed various trends, with the most prominent spike occurring on Day 5 (e.g. L-phenylalanine in clustering group Min 4, Fig. 3).
Table 1. Possible Metabolites (Higher Priority List).
| Compound | KEGG ID | Mass (amu) | Class Group |
|---|---|---|---|
| 11-Deoxytetrodotoxin | C20026 | 302.11 | A1 |
| Kasugamycin | C17968 | 378.16 | A1 |
| *Inosine | C00294 | 267.07 | A1 |
| N-Methyl-(R,S)-tetrahydrobenzylisoquinoline | C05314 | 238.15 | A2 |
| Amantadine hydrochloride | C07939 | 186.11 | A2 |
| Glycinoeclepin A | C08765 | 433.25 | A2 |
| Dikegulac | C18825 | 275.11 | A2 |
| Longifolonine | C09569 | 296.10 | A2 |
| Silafluofen | C18412 | 407.19 | A2 |
| Mitoxantrone | C11195 | 445.20 | A2 |
| Naltrindole | C18128 | 415.19 | A2 |
| 4,4-Difluoro-17beta-hydroxyandrost-5-en-3-one propionate | C15112 | 381.22 | A2 |
| Lycomarasmine B | C08496 | 276.09 | A2 |
| SR95531 | C13796 | 288.13 | A2 |
| 9alpha-Fluoro-16alpha-hydroxyhydrocortisone | C14638 | 397.19 | A2 |
| *L-Homocysteine | C00155 | 136.04 | A2 |
| *Tyramine | C00483 | 138.09 | A2 |
| *Estriol | C05141 | 287.17 | A2 |
| (2-Butylbenzofuran-3-yl)(4-hydroxyphenyl)ketone | C15049 | 295.13 | A4 |
| Agaritine | C01550 | 266.12 | A4 |
| GA | C11484 | 298.15 | A4 |
| Metconazole | C18476 | 320.15 | A4 |
| 1-Carbazol-9-yl-3-(3,5-dimethylpyrazol-1-yl)-propan-2-ol | C11560 | 320.17 | A4 |
| L-alpha-Acetyl-N,N-dinormethadol; dinor-LAAM | C16662 | 326.20 | A5 |
| Hypoglycin B | C08280 | 271.12 | A5 |
| Dihydrozeatin riboside | C16447 | 352.17 | A5 |
| *5-Hydroxy-L-tryptophan | C00643 | 221.09 | A5 |
| *N-Acetylmethionine | C02712 | 192.07 | A5 |
| SN-38 carboxylate form | C11366 | 410.14 | Max2 |
| Fenpiclonil | C14268 | 234.99 | Max4 |
| Difenpiramide | C17720 | 289.13 | Max4 |
| Anthopleurine | C16994 | 176.10 | Min1 |
| N-Acetyl-beta-D-glucosaminylamine | C01239 | 219.11 | Min2 |
| Phenylacetylglycine dimethylamide; Ralgin | C12958 | 221.12 | Min2 |
| *L-Alanine | C00041 | 88.04 | Min2 |
| Clomipramine | C06918 | 315.15 | Min3 |
| 3beta-Hydroxy-16-phosphonopregn-5-en-20-one monoethyl ester | C15173 | 425.24 | Min3 |
| Propachlor | C18759 | 210.08 | Min4 |
| Amantadine hydrochloride | C07939 | 186.11 | Min4 |
| dl-Methylephedrine hydrochloride | C13639 | 214.11 | Min4 |
| Streptidine | C00837 | 263.14 | Min4 |
| Penthiopyrad | C18482 | 358.13 | Min4 |
| Benalfocin; SK&F 86466 | C10970 | 194.08 | Min4 |
| *L-Phenylalanine | C00079 | 166.07 | Min4 |
| *Urocanate; Urocanic acid | C00785 | 139.05 | Min4 |
High confidence metabolites that showed significant temporal trends (Fig. 2). Mass features were annotated using the KEGG72 API, with mass tolerance 10ppm. Additional masses of interest were compared against mass spectrometry standards for identification. Masses with unique KEGG IDs or identified through the use of standards were classified as high priority and were used for pathway and network analysis through QIAGEN’s Ingenuity® Pathway Analysis (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity). See also Supplementrary Fig. S2 for analysis framework details and Supplementary Table S3 for full classification data.
*were identified by use of standards using LC-MS.
Figure 3. Temporal Trends and Associated Networks.

On the left the hierarchical clustering per classification (Autocorrelated, Spike Maxima and Spike Minima) is shown. For each trend molecules with unique KEGG ID72 or identified through MS standards were used in QIAGEN’s IPA Ingenuity pathway construction (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity). The network displayed on the right (network score 16, “Cell-mediated Immune Response, Inflammatory Response, Gastrointestinal Disease”) includes 5 molecules, each shown aligned horizontally with its corresponding temporal trend and identified with its group on the left. Significant functions and canonical pathway results that include more than two of the metabolites or network components respectively are also included. See also Supplementary Table S4 for detailed network composition and functional analysis and Supplementary Fig. S3.
(3) The identified metabolites of high-interest are associated with multiple possible pathways (26 molecules were found in QIAGEN’s Ingenuity Pathways (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity), Supplementary Fig. S3, including nine molecules that were verified by MS identification through comparison to standards). Examples with the lowest p-values and with more than two identified molecules involved include Amino Acid Metabolism functions, uptake of L-proline and L-alanine (p < 2.5 × 10−5) [both involve L-alanine and L-phenylalanine], as well as carbohydrate metabolism functions [involving tyramine and L-phenylalanine, and estriol in transport of monosaccharide (p < 4.8 × 10−5)], with the full IPA® analysis output included in Supplementary Table S2. For the identified metabolites in the high interest list, networks were constructed algorithmically using IPA® to identify connections to known gene and pathway associations. The highest scoring network (network score 16, i.e. p < 10−16, by IPA®) is shown in Fig. 3 right panel, and involves Cell-mediated Immune Response, Inflammatory Response, Gastrointestinal Disease. The network includes multiple processes such as transports of monosaccharide and D-glucose, uptake of L-alanine, L-proline, ILK signaling and associated genes, proteins and enzymes, with TNF, Vegf and IRS1 amongst the highly connected nodes (Fig. 3).
Discussion
The microbiome is an intrinsic part of the host and should be considered in conjunction with corresponding changes in host molecular components3,4,10. Research is now focusing on the functional aspects of the microbiome-host interaction54,55,56, and reaching a stage where further progress requires systems level datasets. Such datasets will help address the global molecular interactions and resulting collective dynamic responses in metabolites, genes and proteins, and other associated omics data, and provide support for modeling22.
The longitudinal study presented here is a pilot implementation that has allowed us to follow the temporal changes in urine host metabolites, dynamically following colonization of GF mice. Previous experiments in germ free rats have shown that after fecal inoculation there was an increase of specific metabolites such as benzoic acid or phenylacetic acid33,57. Rather than considering individual metabolites, we have considered a collective set of all chemicals that are detected by mass spectrometry and that correspond to a temporal trend caused by the colonization. Our approach demonstrates that microbiotic changes in the gut have an immediate impact on metabolic processes, and provide readouts as collective behavior observed in urine metabolite level changes. These changes both display immediate punctuated response on the fifth day post colonization, in addition to continuous changes, both as gradual increases and decreases in the relative levels of groups of metabolites over the entire time course, suggesting longer term effects. The reported changes also indicate that gut colonization drastically alters host metabolic pathways, including carbohydrate metabolism and molecular transport, relating to host energy balance that has been previously reported in bi-species colonization studies58,59, which have been also modeled using GEMs60. Such modeling may be carried out for the species considered in the presented study, as well as binary and higher combinations. The overall functional association findings and possible connections to adaptive immunity are consistent with changes observed by others in longitudinal monitoring of mice and rats11. Other studies have verified how the presence of bacteria impact the colonic luminal metabolome41, as well as some endogenous metabolite levels42.
The observed host-microbe interaction suggests that in the absence of additional perturbation, colonization has long lasting effects on metabolic processes, which implicate several proteins/genes that participate in host immune responses. Recent mice conventionalization studies have revealed similar such immune connections, including as well the association of TNF40. The approach in our investigation observed the metabolic changes that resulted from the colonization by two bacteria species. This diet-induced colonization may be extended to study more bacterial species, while the use of urine as an output measure allows for generalization to human studies in a rather non-invasive fashion (compared to other body tissue sampling). Starting from different binary/paired combinations of bacterial species used for colonization, we can envision extending colonization studies systematically to triplets of species and increasingly higher numbers. If conducted in parallel with modeling approaches, this will provide the necessary data to help elucidate how metabolic processes are affected by inter-microbe dependability and how metabolism may be modulated through the introduction or removal of bacterial species.
Additionally, supplementing urine studies with monitoring of blood samples will help identify the corresponding expression changes in blood metabolomes, transcriptomes and proteomes and investigate further the connection to host immune responses. For example, other studies have reported on such omics integration that donor features can be reconstituted in conventionalized mice, and modulated through diet61. Such models allow an investigation of the connection of personalized diet effect mechanism, that may be studied using the few microbe approach, with extensions to pharmacomicrobiomics20. Furthermore, some of the validated compounds in our study such as inosine, L-Alanine and L-Phenylalanine have been reported as members of the human urine metabolome62, suggesting that the study of diet effected colonization in mice might have implications in similar applications to humans and modulation of the microbiotic makeup. The simplified mouse model approaches yield results that may be used to evaluate more mechanistic hypotheses63,64,65,66,67, including validating the algorithmic modeling of microbiome-host interactions at a systems level, for example, acting as output tests for dynamic GEM models22. This will also necessitate more thorough database and annotations of associations of metabolites to known genes, proteins and pathways.
The implementation of a high-throughput methodology presented here, from sample to temporal analysis, is generalizable both in sample scale and number of components and time-points. Addressing uneven sampling is of particular concerns in efforts in personalized medicine7,8,68, where we expect metabolomics to play a crucial role in multimodal omics integration. In mass spectrometry missing data points, which may effectively create uneven sampling are an inherent method limitation. We are able to extract patterns from noisy data (Fig. 2), allowing also for recovery of signals with missing time-points. The computational approach scales even better in longer time series (Fig. 2-c), allowing for more uneven data sampling (and more missing data points), and exploration of lags greater than one, thus extending to periodic phenomena. The generalized implementation of this framework will be included in the multi-omics analysis package MathI-Omica (Yusufaly and Mias, in preparation). Finally, we believe that longitudinal studies may study the different effects of microbiotic changes or the host state, and provide resource metabolite sets that correspond to each change (cf. gene set analysis generalized to small molecules).
Methods
Mice
All experimental protocols were in accordance with and as approved by (protocol 19727) the Administrative Panel on Laboratory Animal Care (APLAC), the Stanford Institutional Animal Care and Use Committee (IACUC). Two groups of Swiss-Webster germ free mice were used in this study. The mice were placed inside gnotobiotic isolators. One group of mice was kept GF (n = 3) and urine and feces were collected from this group. The second group of mice was colonized using oral gavage with 108 cfu of Bt (VPI-5482) and 108 cfu of Bif. longum (NCC2705) (n = 4). Bacteria were cultured under anaerobic conditions at 37oC in tryptone-yeast extract-glucose (TYG) medium and Reinforced Clostridial Medium (RCM, Becton Dickinson and Company, MD, United States). Urine and feces from this second group of bi-associated mice were collected at 5, 10, 15, 20 and 15 days after gavage. All samples were placed in a freezer at −80 °C within 30 minutes of collection, until analysis.
Urine sample preparation and analysis39
Forty μl of 10 mM ammonium formate were added to 20 μl of urine. Five μl of the mix was used in the analysis. A 150 mm × 2.1 mm Kinetex 17 μm C18 column was used for chromatographic separation using a ACQUITY Ultra Performance Liquid Chromatography system (Waters). The flow rate was 0.3 ml/min with solvent A composed of water plus 0.1% formic acid and solvent B composed of acetonitrile plus 0.1% formic acid. The gradient consisted of 3% B for one min, followed by a gradient of 35% B over 15 min, hold at 35% B for 5 min, and 100% B for 2 min. The column was equilibrated at 3% B for 2 min. Mass spectrometry was performed an Exactive (Thermo Fisher) operated in positive and negative electrospray mode and controlled by Xcalibur 2.1 software. The scan range was from 70 to 800 m/z, at 50000 FWMH resolution, using nitrogen. For positive mode (ESI+) we used the following conditions: sheath gas flow rate 40 (arbitrary units), auxilary gas flow rate 8 (arbitrary units), sweep gas flow rate one (arbitrary units), spray voltage 3.5 kV, capillary temperature 275 C, capillary voltage – 60 V, tube lens voltage −100 V, skimmer voltage e −20 V. For negative electrospray mode (ESI-): sheath gas flow rate 30 (arbitrary units), auxiliary gas flow rate 4 (arbitrary units), sweep gas flow rate one (arbitrary units), spray voltage 3.5 kV, capillary temperature 275 C, capillary voltage −60 V, tube lens voltage −100 V, skimmer voltage −21 V.
Metabolomics data processing and time analysis framework
Raw mass spectrometry spectral data were collected for each biological replicate at each time point both in positive and negative MS modes. Each dataset was analyzed as summarized in Supplementary Fig. S2: (1) preprocessed to align and assign mass intensities, (2) spectrally analyzed to obtain temporal patterns and classified into significant categories (3) assessed for assignment of mass identities and potential biological significance through pathway analysis. The primary statistical analyses were performed using Mathematica 9.069 (except as indicated below). In particular, for each step:
Data Preprocessing
Continuous mode raw data from the individual analyses were converted to centered mode .mzXML files with msconvert (part of ProteoWizard70) and subjected to nornlinear data alignment by XCMS70,71. In order to identify specific biomarkers by mass, KEGG application programming interface (API) was used for identified compounds72, with a mass tolerance for identification set at 10 ppm. Masses uniquely identified were considered “high-priority” results.
Spectra from profiling at each time point were obtained with 3 technical replicates and aligned for mass and retention times using XCMS. The aligned spectra/mass data were filtered, and the median and median deviation were computed for replicates sets per mass identified, retaining data with a CV <0.5. For each time-point the log-2 intensities distributions were standardized to the median, scaled by the average median deviation, and only sets with mass intensities at least 2/3 time-points and including the germ-free reference were retained. For statistical computations a non-parametric bootstrap distribution with replacement was computed for 100,000 samples. For each of the constructed and experimental mass data the difference in normalized intensities was computed compared to the germ free datapoint, namely the difference, σΔ = σt-σgerm-free, where σt, is the median deviation of each mass normalized intensity at time-point (t) using its own distribution median, and σgerm-free is the median deviation the corresponding normalized mass intensity at the germ-free datapoint (germ-free mice samples), from its distribution median. The resulting time-series differences set for each mass was constructed into a normalized vector (normalized using a Euclidean distance metric).
Classification of Temporal Response
The normalized time series were classified based on the temporal trends observed during the time-course. To allow for missing data points (inherent in MS analyses) which essentially create uneven sampling in time, we adopted a spectral analysis for computing autocorrelations and subsequent visualization. A periodogram of the data in frequency (Fourier) space was obtained, by oversampling and using a Lomb-Scargle transformation/linear least squares harmonic function fit45,46,47, adapted from astronomy and utilized in biological research, which can aid in accounting for unevenly sampled data sets (e.g. due to missing datapoints)7,48,49,50,51,53,73,74,75,76. Briefly, (see also references), the Lomb-Scargle transformation computes the periodogram 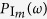 for a time series of intensities
for a time series of intensities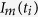 for mass, m, at the ith time point
for mass, m, at the ith time point 
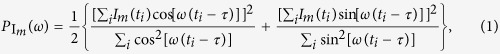 |
where, τ is a parameter computed implicitly,
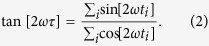 |
The periodogram is used together with an inverse Fourier transform to allow for even resampling and obtain the autocorrelations46,47,48,53. For each mass intensity evenly-(re)sampled time series the autocorrelation at lag one was computed, where the generalized expression
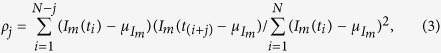 |
is evaluated lag j = 1. A classification scheme of autocorrelated signals and spike signals was implemented7, with signals having statistically significant autocorrelation classified as autocorrelated class (p < 0.05 cutoff, one-tailed, based on autocorrelations distribution of the bootstrap distributions simulated per data-set). The autocorrelated signals were then removed from the dataset, and signals in the remaining set showing spiky behavior of aberrantly high or low high or low normalized intensities at any time-point as compared to a random simulation were classified as spike maxima or minima (p < 0.05, one-tailed by comparison to analysis of randomly simulated distribution of normalized time signals of corresponding length N for each time-series in each class).
Hierarchical Clustering and Annotation
The data in each class were hierarchically clustered in Mathematica 9.069, with (correlation distance measure with average linkage). Groups within each class were determined from the dendrograms, through changes in fusion coefficients. Within each group, the masses were annotated using the KEGG72 API, with a mass tolerance window of 10ppm. Additional masses of interest were compared against mass spectrometry standards for identification. Masses with unique KEGG IDs or identified through the use of standards were classified as higher priority data, and were used for pathway and network analysis through QIAGEN’s Ingenuity® Pathway Analysis (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity), (see Table 1). In particular, each KEGG compound identifier was mapped to its corresponding object in the IPA® Knowledge Base (26 of 45 compounds). These Network Eligible molecules were used for Functional Analysis, to identify biological functions in the Ingenuity Knowledge Base. Right-tailed Fisher’s exact test was used to calculate a p-value determining the probability that each biological function assigned to the data set was due to chance alone. Additionally, canonical pathways from the IPA library that were most significant were ascertained based on p-value (Fisher’s exact test) and ratio of molecules from the data set as compared to the total in the network. Finally, a network (35 molecules) was generated using the Network Eligible molecules as seeds with IPA®’s Network Generation Algorithm. The network score is based on the hypergeometric distribution, calculated as –log[right-tailed Fisher’s Exact Test], (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity).
Additional Information
How to cite this article: Marcobal, A. et al. Metabolome progression during early gut microbial colonization of gnotobiotic mice. Sci. Rep. 5, 11589; doi: 10.1038/srep11589 (2015).
Supplementary Material
Acknowledgments
G.I.M. and research reported in this publication are supported by grants from Michigan State University and the National Human Genome Research Institute of the National Institutes of Health under Award Number 4R00HG007065-02 and previously K99HG007065. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author Contributions A.M. and G.I.M. conceived the study, project planning, did experimental design and work, and statistical analysis and wrote the manuscript. T.Y. was involved in computational work, S.H. was involved in experimental work; M.P.S. was involved in financing and experimental analysis. All authors were involved in manuscript editing. J.L.S. and G.I.M. supervised the project and were involved in project planning, financing, supervision, and manuscript preparation. J.L.S. and G.I.M. are both senior authors.
References
- Whitman W. B., Coleman D. C. & Wiebe W. J. Prokaryotes: the unseen majority. Proc. Natl. Acad. Sci. USA 95, 6578–6583 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Jumpstart Reference Strains, C. et al. A catalog of reference genomes from the human microbiome. Science 328, 994–999, 10.1126/science.1183605 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevers D. et al. The Human Microbiome Project: a community resource for the healthy human microbiome. PLoS Biol. 10, e1001377, 10.1371/journal.pbio.1001377 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice E. A. & Segre J. A. The human microbiome: our second genome. Annu. Rev. Genomics Hum. Genet. 13, 151–170, 10.1146/annurev-genom-090711-163814 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project, C. A framework for human microbiome research. Nature 486, 215–221, 10.1038/nature11209 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project, C. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214, 10.1038/nature11234 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R. et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 148, 1293–1307, 10.1016/j.cell.2012.02.009 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mias G. I. & Snyder M. Personal genomes, quantitative dynamic omics and personalized medicine . Quantitative Biology 1, 71–90 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloissnig S. et al. Genomic variation landscape of the human gut microbiome. Nature 493, 45–50, 10.1038/nature11711 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tralau T., Sowada J. & Luch A. Insights on the human microbiome and its xenobiotic metabolism: what is known about its effects on human physiology? Expert Opin. Drug Metab. Toxicol. 11, 411–425, 10.1517/17425255.2015.990437 (2015). [DOI] [PubMed] [Google Scholar]
- Turnbaugh P. J. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031, 10.1038/nature05414 (2006). [DOI] [PubMed] [Google Scholar]
- Gophna U., Sommerfeld K., Gophna S., Doolittle W. F. & Veldhuyzen van Zanten S. J. Differences between tissue-associated intestinal microfloras of patients with Crohn’s disease and ulcerative colitis. J. Clin. Microbiol. 44, 4136–4141, 10.1128/JCM.01004-06 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadal I., Donat E., Ribes-Koninckx C., Calabuig M. & Sanz Y. Imbalance in the composition of the duodenal microbiota of children with coeliac disease. J. Med. Microbiol. 56, 1669–1674, 10.1099/jmm.0.47410-0 (2007). [DOI] [PubMed] [Google Scholar]
- Vrieze A. et al. The environment within: how gut microbiota may influence metabolism and body composition. Diabetologia 53, 606–613, 10.1007/s00125-010-1662-7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg E. D. et al. Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell 141, 1241–1252, 10.1016/j.cell.2010.05.005 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay-Kumar M. et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328, 228–231, 10.1126/science.1179721 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso G., Gambino R. & Cassader M. Interactions between gut microbiota and host metabolism predisposing to obesity and diabetes. Annu. Rev. Med. 62, 361–380, 10.1146/annurev-med-012510-175505 (2011). [DOI] [PubMed] [Google Scholar]
- Qin J. et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60, 10.1038/nature11450 (2012). [DOI] [PubMed] [Google Scholar]
- Flint H. J., Scott K. P., Louis P. & Duncan S. H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 9, 577–589, 10.1038/nrgastro.2012.156 (2012). [DOI] [PubMed] [Google Scholar]
- ElRakaiby M. et al. Pharmacomicrobiomics: the impact of human microbiome variations on systems pharmacology and personalized therapeutics. OMICS 18, 402–414, 10.1089/omi.2014.0018 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins F. S. & Varmus H. A new initiative on precision medicine. N. Engl. J. Med. 372, 793–795, 10.1056/NEJMp1500523 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoaie S. & Nielsen J. Elucidating the interactions between the human gut microbiota and its host through metabolic modeling. Front. Genet. 5, 86, 10.3389/fgene.2014.00086 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer C., Bik E. M., DiGiulio D. B., Relman D. A. & Brown P. O. Development of the human infant intestinal microbiota. PLoS Biol. 5, e177, 10.1371/journal.pbio.0050177 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggesbo M. et al. Development of gut microbiota in infants not exposed to medical interventions. APMIS 119, 17–35, 10.1111/j.1600-0463.2010.02688.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock G. M. Genomic approaches to studying the human microbiota. Nature 489, 250–256, 10.1038/nature11553 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassens E. S., de Vos W. M. & Vaughan E. E. Metaproteomics approach to study the functionality of the microbiota in the human infant gastrointestinal tract. Appl. Environ. Microbiol. 73, 1388–1392, 10.1128/AEM.01921-06 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishampayan P. A. et al. Comparative metagenomics and population dynamics of the gut microbiota in mother and infant. Genome Biol. Evol. 2, 53–66, 10.1093/gbe/evp057 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig J. E. et al. Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. USA 108 Suppl 1, 4578–4585, 10.1073/pnas.1000081107 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adlerberth I. & Wold A. E. Establishment of the gut microbiota in Western infants. Acta Paediatr. 98, 229–238, 10.1111/j.1651-2227.2008.01060.x (2009). [DOI] [PubMed] [Google Scholar]
- Ley R. E., Peterson D. A. & Gordon J. I. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124, 837–848, 10.1016/j.cell.2006.02.017 (2006). [DOI] [PubMed] [Google Scholar]
- De Filippo C. et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 107, 14691–14696, 10.1073/pnas.1005963107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjursell M. K., Martens E. C. & Gordon J. I. Functional genomic and metabolic studies of the adaptations of a prominent adult human gut symbiont, Bacteroides thetaiotaomicron, to the suckling period. J. Biol. Chem. 281, 36269–36279, 10.1074/jbc.M606509200 (2006). [DOI] [PubMed] [Google Scholar]
- Nicholls A. W., Mortishire-Smith R. J. & Nicholson J. K. NMR spectroscopic-based metabonomic studies of urinary metabolite variation in acclimatizing germ-free rats. Chem. Res. Toxicol. 16, 1395–1404, 10.1021/tx0340293 (2003). [DOI] [PubMed] [Google Scholar]
- Martin F. P. et al. Dietary modulation of gut functional ecology studied by fecal metabonomics. J. Proteome Res. 9, 5284–5295, 10.1021/pr100554m (2010). [DOI] [PubMed] [Google Scholar]
- Lenz E. M. & Wilson I. D. Analytical strategies in metabonomics. J. Proteome Res. 6, 443–458, 10.1021/pr0605217 (2007). [DOI] [PubMed] [Google Scholar]
- Want E. J. et al. Global metabolic profiling procedures for urine using UPLC-MS. Nat. Protoc. 5, 1005–1018, 10.1038/nprot.2010.50 (2010). [DOI] [PubMed] [Google Scholar]
- Wikoff W. R. et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 106, 3698–3703, 10.1073/pnas.0812874106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claus S. P. et al. Systemic multicompartmental effects of the gut microbiome on mouse metabolic phenotypes. Mol. Syst. Biol. 4, 219, 10.1038/msb.2008.56 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcobal A. et al. A metabolomic view of how the human gut microbiota impacts the host metabolome using humanized and gnotobiotic mice. ISME J. 7, 1933–1943, 10.1038/ismej.2013.89 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Aidy S. et al. Temporal and spatial interplay of microbiota and intestinal mucosa drive establishment of immune homeostasis in conventionalized mice. Mucosal Immunol. 5, 567–579, 10.1038/mi.2012.32 (2012). [DOI] [PubMed] [Google Scholar]
- Matsumoto M. et al. Impact of intestinal microbiota on intestinal luminal metabolome. Sci. Rep. 2, 233, 10.1038/srep00233 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. H., An J. H., Park H. M. & Jung B. H. Investigation of endogenous metabolic changes in the urine of pseudo germ-free rats using a metabolomic approach. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 887-888, 8–18, 10.1016/j.jchromb.2011.12.030 (2012). [DOI] [PubMed] [Google Scholar]
- Ley R. E. et al. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 102, 11070–11075, 10.1073/pnas.0504978102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T. et al. Human gut microbiome viewed across age and geography. Nature 486, 222–227, 10.1038/nature11053 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomb N. Least-squares frequency analysis of unequally spaced data. Astrophys. Space Sci. 39, 447–462 (1976). [Google Scholar]
- Scargle J. D. Studies in astronomical time series analysis. II-Statistical aspects of spectral analysis of unevenly spaced data. Astrophys. J. 263, 835–853 (1982). [Google Scholar]
- Scargle J. D. Studies in astronomical time series analysis. III-Fourier transforms, autocorrelation functions, and cross-correlation functions of unevenly spaced data. Astrophys. J. 343, 874–887 (1989). [Google Scholar]
- Hocke K. Phase estimation with the Lomb-Scargle periodogram method. Ann. Geophys. 16, 356–358 (1998). [Google Scholar]
- Van Dongen H. P., Olofsen E., VanHartevelt J. H. & Kruyt E. W. A procedure of multiple period searching in unequally spaced time-series with the Lomb-Scargle method. Biol. Rhythm. Res. 30, 149–177, 10.1076/brhm.30.2.149.1424 (1999). [DOI] [PubMed] [Google Scholar]
- Schimmel M. Emphasizing difficulties in the detection of rhythms with Lomb-Scargle periodograms. Biol. Rhythm. Res. 32, 341–345, 10.1076/brhm.32.3.341.1340 (2001). [DOI] [PubMed] [Google Scholar]
- Van Dongen H. P., Ruf T., Olofsen E., VanHartevelt J. H. & Kruyt E. W. Analysis of problematic time series with the Lomb-Scargle Method, a reply to ‘emphasizing difficulties in the detection of rhythms with Lomb-Scargle periodograms’. Biol. Rhythm. Res. 32, 347–354, 10.1076/brhm.32.3.347.1348 (2001). [DOI] [PubMed] [Google Scholar]
- Glynn E. F., Chen J. & Mushegian A. R. Detecting periodic patterns in unevenly spaced gene expression time series using Lomb-Scargle periodograms. Bioinformatics 22, 310–316, 10.1093/bioinformatics/bti789 (2006). [DOI] [PubMed] [Google Scholar]
- Hocke K. & Kämpfer N. Gap filling and noise reduction of unevenly sampled data by means of the Lomb-Scargle periodogram. Atmos. Chem. Phys. 9, 4197–4206, 10.5194/acp-9-4197-2009 (2009). [DOI] [Google Scholar]
- Holmes E., Li J. V., Athanasiou T., Ashrafian H. & Nicholson J. K. Understanding the role of gut microbiome-host metabolic signal disruption in health and disease. Trends Microbiol. 19, 349–359, 10.1016/j.tim.2011.05.006 (2011). [DOI] [PubMed] [Google Scholar]
- Tremaroli V. & Backhed F. Functional interactions between the gut microbiota and host metabolism. Nature 489, 242–249, 10.1038/nature11552 (2012). [DOI] [PubMed] [Google Scholar]
- Sonnenburg E. D. & Sonnenburg J. L. Starving our microbial self: the deleterious consequences of a diet deficient in microbiota-accessible carbohydrates. Cell Metab. 20, 779–786, 10.1016/j.cmet.2014.07.003 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin B. L., Ruthven C. R. & Sandler M. Gut flora and the origin of some urinary aromatic phenolic compounds. Biochem. Pharmacol. 47, 2294–2297 (1994). [DOI] [PubMed] [Google Scholar]
- Samuel B. S. & Gordon J. I. A humanized gnotobiotic mouse model of host-archaeal-bacterial mutualism. Proc. Natl. Acad. Sci. USA 103, 10011–10016, 10.1073/pnas.0602187103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahowald M. A. et al. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc. Natl. Acad. Sci. USA 106, 5859–5864, 10.1073/pnas.0901529106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoaie S. et al. Understanding the interactions between bacteria in the human gut through metabolic modeling. Sci. Rep. 3, 2532, 10.1038/srep02532 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Aidy S. et al. Gut bacteria-host metabolic interplay during conventionalisation of the mouse germfree colon. ISME J. 7, 743–755, 10.1038/ismej.2012.142 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouatra S. et al. The human urine metabolome. PLoS ONE 8, e73076, 10.1371/journal.pone.0073076 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg J. L., Chen C. T. & Gordon J. I. Genomic and metabolic studies of the impact of probiotics on a model gut symbiont and host. PLoS Biol. 4, e413, 10.1371/journal.pbio.0040413 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel B. S. et al. Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proc. Natl. Acad. Sci. USA 104, 10643–10648, 10.1073/pnas.0704189104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco M. L. et al. Convergence in probiotic Lactobacillus gut-adaptive responses in humans and mice. ISME J. 4, 1481–1484, 10.1038/ismej.2010.61 (2010). [DOI] [PubMed] [Google Scholar]
- Rey F. E. et al. Dissecting the in vivo metabolic potential of two human gut acetogens. J. Biol. Chem. 285, 22082–22090, 10.1074/jbc.M110.117713 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith J. J., McNulty N. P., Rey F. E. & Gordon J. I. Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science 333, 101–104, 10.1126/science.1206025 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanberry L. et al. Integrative analysis of longitudinal metabolomics data from a personal multi-omics profile. Metabolites 3, 741–760 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfram Research, I. Mathematica, Version 9.0. (Wolfram Research, Inc., 2012). [Google Scholar]
- Kessner D., Chambers M., Burke R., Agus D. & Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536, 10.1093/bioinformatics/btn323 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. A., Want E. J., O’Maille G., Abagyan R. & Siuzdak G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 78, 779–787, 10.1021/ac051437y (2006). [DOI] [PubMed] [Google Scholar]
- Kanehisa M. & Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30, 10.1093/nar/28.1.27 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory P. C. Bayesian logical data analysis for the physical sciences: a comparative approach with Mathematica support. (Cambridge Univ Pr, 2005). [Google Scholar]
- Ahdesmaki M., Lahdesmaki H., Gracey A., Shmulevich l. & Yli-Harja O. Robust regression for periodicity detection in non-uniformly sampled time-course gene expression data. BMC Bioinformatics 8, 233 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W., Agyepong K., Serpedin E. & Dougherty E. R. Detecting Periodic Genes from Irregularly Sampled Gene Expressions: A Comparison Study. EURASIP J. Bioinf. Syst. Biol. 2008, 10.1155/2008/769293 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R., Zhang C. & Su Z. LSPR: an integrated periodicity detection algorithm for unevenly sampled temporal microarray data. Bioinformatics 27, 1023–1025 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.