

Abstract
Binding of transcription factors (TFs) to regulatory sequences is a pivotal step in the control of gene expression. Despite many advances in the characterization of sequence motifs recognized by TFs, our ability to quantitatively predict TF binding to different regulatory sequences is still limited. Here, we present a novel experimental assay termed BunDLE-seq that provides quantitative measurements of TF binding to thousands of fully designed sequences of 200 bp in length within a single experiment. Applying this binding assay to two yeast TFs, we demonstrate that sequences outside the core TF binding site profoundly affect TF binding. We show that TF-specific models based on the sequence or DNA shape of the regions flanking the core binding site are highly predictive of the measured differential TF binding. We further characterize the dependence of TF binding, accounting for measurements of single and co-occurring binding events, on the number and location of binding sites and on the TF concentration. Finally, by coupling our in vitro TF binding measurements, and another application of our method probing nucleosome formation, to in vivo expression measurements carried out with the same template sequences serving as promoters, we offer insights into mechanisms that may determine the different expression outcomes observed. Our assay thus paves the way to a more comprehensive understanding of TF binding to regulatory sequences and allows the characterization of TF binding determinants within and outside of core binding sites.
Deciphering the binding determinants of transcription factors (TFs) is fundamental to understanding the mechanisms underlying the formation of robust and timely gene expression patterns. Beginning with early studies of the lac operon and the discovery of a motif recognized and bound by the Lac repressor (Jacob and Monod 1961), much research has been focused on the identification and characterization of short sequences to which TFs bind, commonly referred to as TF core binding sites. Great advances in the characterization of such sites were made in recent years, with the development of platforms for high-throughput and accurate in vitro binding measurements of TFs to thousands of short sequences (Berger and Bulyk 2009; Fordyce et al. 2010; Nutiu et al. 2011; Jolma et al. 2013). However, the complementary development of protocols for genome-wide in vivo TF binding measurements (e.g., ChIP-chip and ChIP-seq) revealed that, although some binding events are well accounted for by the underlying presence of an in vitro–deduced binding site, many gaps still remain in deciphering TF binding (Levo and Segal 2014; Slattery et al. 2014). These include differential in vivo binding to various occurrences of the same motif (White et al. 2013), as well as cases of structurally related TFs that were found to have highly similar binding site preferences yet showed distinct binding patterns in vivo, with crucial implications on the formed gene expression patterns (Gordan et al. 2013). These observations demonstrate the need for a more comprehensive understanding of the various factors influencing TF binding to regulatory sequences, going beyond the characterization of core binding sites.
Several recent studies that aim to address this gap employed in vitro–based methods (e.g., DIP-seq [Liu et al. 2006], PB-seq [Guertin et al. 2012], gcPBM [Siggers et al. 2011; Wong et al. 2011; Gordan et al. 2013], EMSA-seq [Wong et al. 2011], SELEX-seq [Slattery et al. 2011; Jolma et al. 2013], MITOMI [Maerkl and Quake 2007; Fordyce et al. 2010], HiTS-FLIP [Nutiu et al. 2011]) and identified various mechanisms that affect TF binding, including chromatin accessibility (Liu et al. 2006; Guertin et al. 2012), cofactors that influence binding specificity (Siggers et al. 2011; Slattery et al. 2011), TF dimer interactions (Wong et al. 2011; Jolma et al. 2013), and the effect of sequences flanking the core TF binding site (TFBS) (Maerkl and Quake 2007; Nutiu et al. 2011; Gordan et al. 2013; Jolma et al. 2013; Rajkumar et al. 2013) that can be mediated through DNA shape.
Here, we present a novel experimental approach, termed BunDLE-seq (Binding to Designed Library, Extracting, and sequencing), which allows the quantitative investigation of several determinants of TF binding within a single experiment. Specifically, the assay provides quantitative TF binding measurements to large-scale libraries of fully designed sequences. Our assay is unique in its ability to study thousands of long and systematically designed sequences, as well as in its capacity to isolate different states of TF binding to these sequences. We show that these attributes allow us to study several aspects of TF binding, including sequence determinants outside of the core TFBS (e.g., constructing predictive TF-specific binding specificity models based on sequences flanking the core binding site), the likelihood of co-occurring TF binding events (e.g., characterizing the dependency on TFBS multiplicity), and the propensity of each of the examined sequences to form nucleosomes. Our results demonstrate that BunDLE-seq can assess the differential contribution of various mechanisms to TF binding, paving the way to a more refined and comprehensive understanding of TF binding to regulatory sequences.
Results
Quantitative measurements of TF binding to thousands of designed, long DNA sequences
To study different determinants of TF binding, we established a new experimental assay, BunDLE-seq, which enables quantitative measurements of binding to a pool of thousands of designed sequences in a single experiment. In this assay, we design a library of sequence variants up to 200 bp in length that is then synthesized on Agilent programmable microarrays (LeProust et al. 2010). Next, we incubate the obtained DNA with a buffer alone (no protein present) or with different concentrations of the examined TF, run the products of this incubation on gel, and extract the DNA from each of the bands detected, corresponding to either naked DNA or DNA bound to different numbers of TF molecules. We amplify the DNA with a unique barcode marking the originating band, join all samples together, and send them to high-throughput sequencing (Fig. 1). For each tested sequence, the sequencing data provide the frequency of its occurrence in each of the bands. From these measurements we compute a binding score that captures the observed versus expected frequency of each sequence in each binding state (represented by a different band) under each of the experimental conditions tested.
Figure 1.

Measurements of TF binding to thousands of long, designed DNA sequences. Schematic illustration of our experimental assay, BunDLE-seq. A library of thousands of fully designed DNA sequences at lengths of 150 or 200 bp was synthesized and cleaved from Agilent programmable microarrays. These sequences differ, for instance, in their general context or their TFBS composition (with binding sites for the TF with which the experiment is carried out colored in purple, as the illustrated TF is colored, while binding sites for other TFs are colored in cyan or green). The pool of DNA sequences was incubated with buffer alone (“DNA only”) or with different concentrations of either Gcn4 or Gal4. The DNA was then run on a gel (an example of a band corresponding to DNA bound by a single TF is marked in blue, while a band corresponding to DNA bound by two TFs is marked in red), extracted from each band, amplified with a barcode marking the originating band, and sent to high-throughput sequencing. Based on the sequencing results, we computed the binding score as the ratio of the observed frequency of each sequence in each binding state (each band) versus the expected frequency (based on the “DNA only” sample). The sorted binding scores computed for a single-TF binding band (in blue) and a two-TF binding band (in red) are shown with a schematic illustration of some of the sequences that were found to be enriched in each of these bands. Filled squares represent TFBSs; filled ovals, TFs. TTTTTT represents poly(dA:dT) tracts.
We found that our computed binding scores are extremely robust, as demonstrated by the high reproducibility (R2 = 0.97) (Supplemental Fig. S1) obtained across experimental replicates (with binding, isolation, amplification of the DNA, and sequencing performed independently). Notably, as we show, the particular score chosen facilitates easy comparison of our binding measurements to expression measurements (Supplemental Section B).
We applied our assay to more than 10,000 sequences containing variations to the content, multiplicity, location, and genomic context of the TFBSs of two yeast TFs, Gcn4 and Gal4. Notably, the selected TFs are structurally distinct and are representatives of the two most abundant yeast TF families (basic leucine zipper [bZIP] class and zinc cluster domain class, respectively) (Hahn and Young 2011). We performed the assays under several concentrations of these TFs and found that increased concentrations of the TF increases the intensity of the band representing the bound state and that an additional band, likely corresponding to more than one binding event, appears at the highest concentration (Fig. 1, gel).
We also chose these sequences for our study since 6500 of them recently served as promoters in a high-throughput reporter assay in yeast cells (hereafter referred to as “expression measurements”) (Sharon et al. 2012). In such an application, BunDLE-seq can shed light on the TF's “readout” of the tested regulatory sequences and thereby provide insights into mechanisms underlying the corresponding expression.
The effect of binding determinants within the core binding site
We first used our assay to examine the dependence of binding on the TFBS core sequence content. We started by measuring the binding of Gcn4 and Gal4 to ∼6500 sequences, including ∼1800 sequences containing one to seven Gcn4 binding sites and ∼1200 sequences containing one to five Gal4 binding sites. Binding was highly specific, with high binding scores for sequences containing TFBSs for the TF with which the experiment was carried out, low binding score for sequences containing TFBSs for the other TF, and an increase in the score as the number of TFBSs for the respective TF increased (Fig. 2A,B). Moreover, we observed stronger binding for sequences containing a previously characterized strong site (Hill et al. 1986; Oliphant et al. 1989; Nutiu et al. 2011) compared with sequences containing a weak site, across tens of pairs of sequences differing in the strength of the Gcn4 or Gal4 binding site.
Figure 2.

TF binding depends on sequence determinants both within and outside core TFBSs. (A, left) The sorted binding scores computed for ∼6000 sequences in an experiment with Gcn4 are shown in dark blue. The number of strong Gcn4 TFBSs, weak Gcn4 TFBSs, and Gal4 TFBSs in each corresponding sequence is shown in light blue in the following panels. (B) Same as in A, but for an experiment carried out with Gal4. (C) For sequences with either no mutations or with single, double, and triple mutations in the Gcn4 core binding site, expression measurements (left) (adapted from Sharon et al. 2012 with permission from Nature Publishing Group © 2012) and the Gcn4 binding score (right) are shown. (D) The binding score for a sequence with a strong Gcn4 site (top) and for a corresponding sequence with a single mutation in the binding site (bottom) is shown in dark blue. The binding score of more than 1000 sequences with the same single strong binding site, differing in the location and context in which the site is embedded, is shown in gray (sequences are sorted by their score), spanning the range attained by a single destructive mutation within the site.
To further study the effect of the nucleotide content within the core TFBSs on TF binding, we used a set of ∼40 sequences containing the 7-bp consensus Gcn4 binding site TGACTCA (Hill et al. 1986; Oliphant et al. 1989; Nutiu et al. 2011), either with no mutation or with single, double, or triple bp mutations. The binding to the consensus site and its reverse complement was substantially stronger than the binding to any of the other variants (Fig. 2C, binding score). This is consistent with previous in vitro characterizations of Gcn4 binding site affinities carried out with different experimental systems (Hill et al. 1986; Oliphant et al. 1989; Nutiu et al. 2011). Additionally, the pronounced difference in the binding score of the sequences with the consensus site compared with other examined sequences was recapitulated in the expression measurements performed with the exact same sequences (Fig. 2C, left; Sharon et al. 2012). Together, these results demonstrate that TF binding in our system occurs in a highly specific manner, and further indicate the ability of our system to provide a characterization of binding dependency on TFBS nucleotide content that is highly relevant to our quantitative understanding of expression levels.
The effect of binding determinants outside the core binding site
Whereas the nucleotide content of the TFBS is known to be a major determinant of TF binding, genome-wide TF binding patterns cannot be explained solely by this effect (Liu et al. 2006; Guertin and Lis 2010; Zhou and O'Shea 2011; Gordan et al. 2013; White et al. 2013). In fact, for the same TFBS, occurrences in different genomic locations were found to display differential binding (Liu et al. 2006; Guertin and Lis 2010; Gordan et al. 2013; White et al. 2013). An intriguing possibility is that the effect of surrounding sequences may not be solely mediated by direct interactions with other proteins. Our measurements support this idea as we observed that even with no additional proteins present, a set of more than 1000 sequences with an identical single strong binding site for Gcn4 embedded in different sequence contexts or at different locations along each context spanned a considerable range of binding scores comparable to that obtained by mutations within the TFBS (Fig. 2D). Intriguingly, since for Gcn4 even a single mutation within the site commonly reduces binding affinity significantly, almost abolishing binding, changes in sequence context outside of the core binding site seem to offer means for obtaining more gradual changes in binding affinity (as was recently suggested also for Pho4) (Rajkumar et al. 2013).
Notably, we observed pronounced fluctuations in binding even in a simple case where the same consensus TFBS, either for Gcn4 or for Gal4, was placed in different locations along a single sequence context (derived either from the HIS3 native promoter, a known target of Gcn4, or from the GAL1-10 context, a known target of Gal4) (Fig. 3A–D, blue lines), with the lowest values almost equivalent to those obtained with a weak binding site (Supplemental Fig. S2A). The pattern observed was highly reproducible across several TF concentrations tested (Supplemental Fig. S3), and more importantly, it is TF- and context-specific, indicating that it does not stem from an inherent property of the binding in our assay, such as the relative location of the binding site within the DNA fragment. Although in vivo various mechanisms can contribute to differential binding or expression from different TFBS locations, we found that our in vitro binding measurements were correlated with in vivo expression measurements carried out using the same sequences (Pearson's correlations of 0.62 and 0.5 for Gcn4 with HIS3- and GAL1-10–derived contexts, and 0.47 and 0.73 with Gal4, respectively) (Supplemental Fig. S4).
Figure 3.
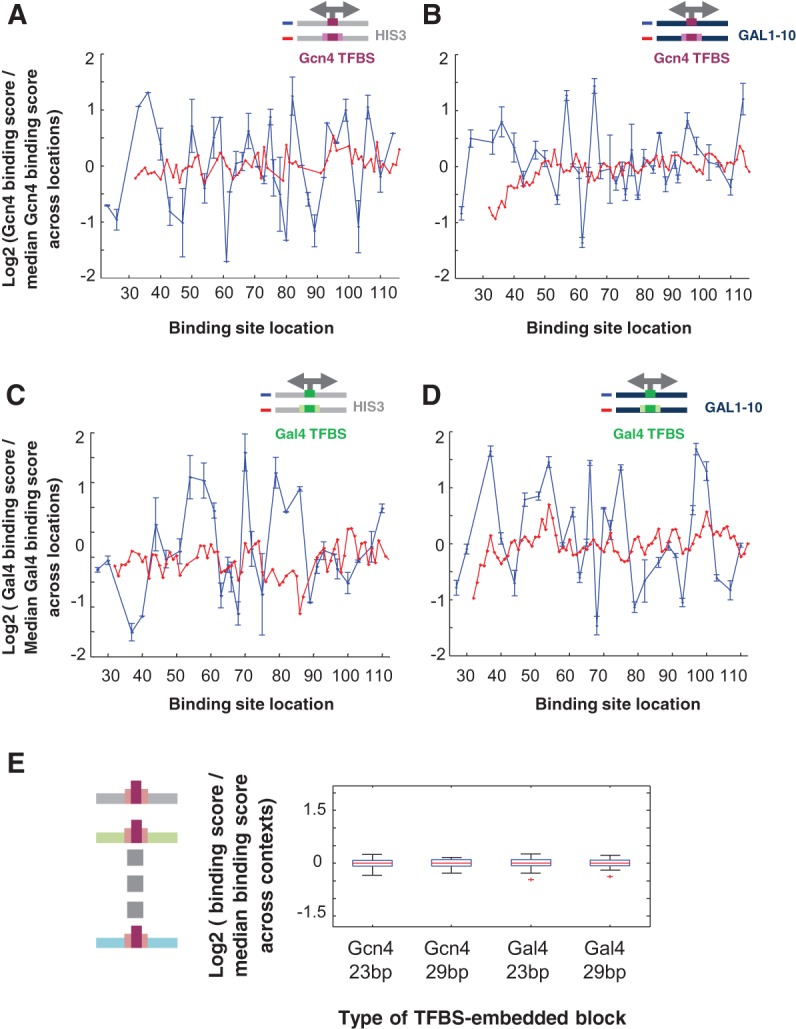
The flanking sequences surrounding the core TFBS affect binding. For a set of sequences in which a single strong binding site was placed at different locations within a specific sequence context, the plot shows the log2 of the ratio of the binding score attained by each sequence (with the x-coordinate marking the location of the center of the site) divided by the median binding score across all sequences in this set. (A) Gcn4 TFBS composed of 9 bp (in blue) or 23 bp, including fixed-flanks site (in red) placed along the HIS3-derived context. (B) Gcn4 TFBS of 9 bp (in blue) or 23 bp, including fixed-flanks site (in red) placed along the GAL1-10–derived context. (C) Gal4 TFBS of 17 bp (in blue) or 23 bp, including fixed-flanks site (in red) placed along the HIS3-derived context. (D) Gal4 TFBS of 17 bp (in blue) or 23 bp, including fixed-flanks site (in red) placed along the GAL1-10–derived context. (E) For each type of binding site, a boxplot shows a log2 ratio of the binding score for sequences containing a site with fixed flanks within different sequence contexts divided by the median score across all contexts.
Thus, the sequences flanking the core TFBS can have a pronounced effect on TF binding; this is evident even when the overall sequence content remains the same, as in the case of a TFBS that is differentially located along a single sequence context. Moreover, our results suggest that such effects can contribute to a corresponding differential binding and expression in vivo.
Flanking sequences of core binding sites affect the binding of TFs
Different locations of a TFBS, even along a single sequence, differ in both proximal and distal nucleotides, and our system allows us to compare the contribution of these different flanking regions to overall TF binding. We first hypothesized that the proximal environment of the binding site (defined here as the 3- to 7-bp immediate flanks) will bear a more significant effect on binding compared with distal regions. To test this hypothesis, we compared the magnitude of differential binding observed when the core TFBS was placed in different locations along a sequence (Fig. 3A–D, blue line) to that observed when placing the core site, now flanked by fixed proximal base pairs (bp). We found that for both Gcn4 and Gal4, fixing the flanks resulted in substantially smaller binding fluctuations on both the HIS3-derived and GAL1-10–derived contexts (Fig. 3A–D, red line) and on an additional ∼40 random contexts (Supplemental Fig. S5). Importantly, varying the more distal flanks resulted in smaller binding fluctuations, as demonstrated by placing a site with fixed proximal flanks in a single location in different sequence contexts (Fig. 3, A–D, blue line, vs. E).
These results prompted us to delve deeper into the effect of proximal flanking sequences and characterize the number of influential nucleotides and the TF-specific, quantitative dependency of binding on these nucleotides. For this purpose, we constructed several linear regression models with binary features (i.e., zero or one) corresponding to the occurrence of any possible 1- to 4-mer at each position within differently sized windows of flanking bp. As such, a direct count of sequence content yields many features, and we employed a LASSO algorithm (Friedman et al. 2010), attempting to construct more concise models with a sparser number of features. We applied this approach in a 10-fold cross-validation scheme to a set of all sequences with the same single strong 9-bp Gcn4 binding site that has unique 15-bp flanking sequences (412 sequences) (see illustration in Fig. 4A) and to a set of all sequences with the same single strong 17-bp Gal4 binding site with unique 15-bp flanking regions (315 sequences). We started by accounting for the entire base content within the 15-bp flanks and found that this resulted in good predictions of our binding measurements on the test set (for Gcn4, a 1mer + 2mer model resulted in R2 = 0.74 averaged across the cross-validation runs, and for Gal4, a 1mer + 2mer model resulted in R2 = 0.87) (see Supplemental Fig. S6A). Incorporating 3-mers or 4-mers into the models did not significantly improve the results. Models learned and tested on various subsets of the sequences also performed fairly well, with common features receiving the highest weights (see Supplemental Fig. S6B–D).
Figure 4.

Computational models based on the flanking sequences of the core TFBS successfully predict differential binding to sequences that contain the same core binding site. (A) Shown are 412 sequences that contain the same strong Gcn4 site sorted by their binding score. We show the core TFBSs (in blue) and the identity of each nucleotide (color-coded) within 15-bp flanks upstream of and downstream from the site. Examples of specific flanking sequences that are enriched among the low or high scoring sequences are highlighted by colored squares. (B) Scatter plot of Gcn4 binding scores versus model predictions, with 3-bp flanks, based on the 1mer + 2mer model. (C) Same as B but for Gal4. (D) Feature weights for the top 15 sequence features for the model in B. (E) Feature weights for the top 15 sequence features for the model in C. (F) Scatter plot of Gcn4 binding versus model predictions, with 15-bp flanks, DNA-shape-based model. DNA shape features are minor groove width, roll, propeller twist, and helix twist (Yang et al. 2014). For each of these features, the model includes a value computed per bp (minor groove width and propeller twist) or bp step (roll and helix twist) derived from a 5-bp window surrounding that bp using DNAshape (Zhou et al. 2013), a mean value across the 15-bp downstream flanks, the 15-bp upstream flanks, and the concatenated 30-bp flanks. (G) Same as F but for Gal4 binding. (H) Boxplots of log2 of the binding score (left) or expression levels (right) of sequences with 15-bp poly(dA:dT) tracts, at different distances from a strong Gcn4 or Gal4 TFBS, divided by the binding score/expression levels of the same sequence without the poly(dA:dT) tract.
Notably, although the models based on the entire 15-bp flanking sequences performed well, the top ranking features represented proximal flanks (Supplemental Fig. S6A). We therefore tested the ability of models that only use proximal positions to explain the data; whereas a model accounting only for 1-bp flanks results in an extreme binning of the data (Supplemental Fig. S6G), in a model accounting for 3-bp flanks, this binning is less pronounced (Fig. 4B,C) and the model performance is comparable to that of the model accounting for 15-bp flanks (for Gcn4, a 1mer + 2mer model results in R2 = 0.74, and for Gal4, a 1mer + 2mer + 3mer model results in R2 = 0.9, with a ±SE overlap between these two models). A model accounting for three distal positions (positions 4–6 upstream of and downstream from the core TFBS) instead of the three proximal positions performs poorly (see Gcn4 in Supplemental Fig. S6H), which is consistent with previous reports highlighting the importance of proximal flanks over distal ones (Gordan et al. 2013; Rajkumar et al. 2013).
Notably, the identity of preferred flanking sequences, as elucidated by our models (see examples in Fig. 4D,E), agrees with previous studies whose characterization of TFBSs included some preferences for bp beyond the 9-bp core binding site of Gcn4 (Hill et al. 1986; Zhu et al. 2009; Nutiu et al. 2011) or the 17-bp core target of Gal4 (Zhu et al. 2009). These reports include a recent study that specifically highlighted the importance of the 2 bp flanking a 7mer Gcn4 consensus site by performing in vitro affinity measurements to several 11–12mers (Nutiu et al. 2011) in which these flanks were varied. Our models, accounting for either 2 or 3 bp flanking the Gcn4 core binding site (that is, addressing 13- or 15-bp target sites) or accounting for 3 bp flanking the Gal4 core binding site (that is, addressing 23-bp target sites), extend these previous characterizations and best explain the differential binding captured in our measurements. (For an alternative representation of the flanking sequences preferences observed in our measurements in the form of a PWM, see Supplemental Fig. S7.)
Taken together, whereas sequences sharing the well-characterized (Hill et al. 1986; Oliphant et al. 1989; Nutiu et al. 2011) strong binding site for either Gcn4 or Gal4 show pronounced differences in binding, a simple TF-specific model accounting for 3-bp flanks successfully predicts these differences.
DNA shape features provide a mechanistic explanation for the effect of flanking sequences
One possible mechanism that might mediate the effect of flanking sequences on TF binding involves the intrinsic three-dimensional DNA structure (Rohs et al. 2009). Specifically, recent work suggested that local DNA shape properties, such as minor groove width and helical parameters, can contribute to differential binding of different TFs to various DNA sequences (Slattery et al. 2011; Gordan et al. 2013; Yang et al. 2014). Intriguingly, a model based solely on DNA shape features (i.e., minor groove width, roll, propeller twist, and helix twist), derived from the DNAshape method (Zhou et al. 2013) and computed over a window of 15 bp 5′ and 3′ of the Gcn4 or Gal4 core binding sites, instead of the explicit nucleotide-content features, indeed possesses a predictive power with respect to our binding measurements (Gcn4, R2 = 0.4; Gal4, R2 = 0.72) (see Fig. 4F,G).
The different performance of these models for Gcn4 and Gal4 suggests distinct DNA recognition mechanisms used by the two TFs. Gcn4 binds DNA as a bZIP homodimer mainly through an intensive network of hydrogen bonds with the major groove edges of the central 7 bp of its binding site (Ellenberger et al. 1992). The nucleotide composition of the Gcn4 core-binding site is therefore highly conserved (as was indeed demonstrated by the effect of mutations to the core binding site) (see Fig. 2C), and flanking sequences are expected to only fine-tune the binding specificity, as previously observed for the binding of bHLH TFs to E-boxes (Gordan et al. 2013). The consensus binding site of the Gal4 homodimer, however, is much longer (17 nucleotides), yet only the CGG triplets at the 5′ and 3′ ends are directly contacted by the protein and are thus highly conserved (Marmorstein et al. 1992), while the 11 bp between these two triplets are somewhat variable (Morozov and Siggia 2007). The inner 11-bp core is crucial for the correct positioning of the outer CGG triplets to enable Gal4 contacts, which likely requires a high conservation in DNA shape despite variable sequence (Morozov and Siggia 2007), as opposed to the strict sequence composition of the core displayed by Gcn4. As a consequence, the variation of flanking sequences, as mediated by DNA shape features, might have a larger impact on the more variable Gal4 consensus site compared with the more conserved Gcn4 consensus site (Supplemental Fig. S8); providing a possible explanation for the higher predictive power of models including the flanks, and particularly the shape-based ones, in the case of Gal4 binding compared with Gcn4 binding.
The effect of poly(dA:dT) tracts adjacent to TFBSs
One particular type of flanking sequence that seems to affect TF binding is poly(dA:dT) tracts, with DNA shape again suggested to be one of the mechanisms mediating this effect (Rohs et al. 2009, 2010). In a recent in vitro characterization of human TF binding specificities with HT-SELEX, the core site of many TFs was found to be flanked by 3–5 A/T bp (Jolma et al. 2013), and this was also previously shown for the yeast TF Gcn4 examined here (Hill et al. 1986). As even longer poly(dA:dT) tracts are highly prevalent in eukaryotic promoters (Segal and Widom 2009b) and are thus often found in the vicinity of TFBSs, we sought to utilize the ability of our system to examine longer flanks and specifically test how the presence of such tracts influences TF binding.
Notably, we found that the presence of a 15-bp poly(dA:dT) tract can affect TF binding. Sequences with a poly(dA:dT) tract immediately adjacent to a Gcn4 binding site show higher binding scores than corresponding sequences lacking this tract. A smaller effect is observed when the tract is placed 1 bp away from the binding site, and it seems to diminish when the tract is placed even further away (Fig. 4H). Notably, a 15-bp poly(dA:dT) tract placed 1 bp away from a Gal4 binding site, but not a tract that was located further away, reduced Gal4 binding (Fig. 4H).
As discussed in previous studies, a poly(dA:dT) tract leads to a narrow minor groove (Alexeev et al. 1987; Rohs et al. 2009) and possibly facilitates the binding to the adjacent major groove. This can account for the observed contribution of these tracts to Gcn4 binding as this TF binds to the major groove (Supplemental Fig. S8), and the tract might enhance the DNA bending that was observed for Gcn4 binding sites (Keller et al. 1995). In contrast, Gal4 binding relies on the direct contacts to the outer CGG triplets of the binding site, and as common for GC-rich regions (Rohs et al. 2010), the minor groove in these regions was reported to be rather wide (Marmorstein et al. 1992). Narrowing of the minor groove by a tract immediately adjacent to the Gal4 core site will likely compromise Gal4 contacts (an effect that will fade with additional nucleotides separating the tract from the CGG triplet). Thus, the opposite effects of the poly(dA:dT) tract in the flanking regions of these TFs suggest TF-specific binding mechanisms that relate to DNA shape features preferred by either TF.
In addition, we found that the in vitro–observed effects of poly(dA:dT) tracts on TF binding agree with the nature of the effects observed when expression measurements were carried out with the same set of sequences (Fig. 4H, cf. expression to binding). Notably, the effect of poly(dA:dT) tracts in vivo can reflect a combination of several mechanisms, including a direct effect of the tract on TF binding affinity, as captured by our measurements, and a nucleosome-mediated effect (likely manifested in increased accessibility of the DNA to a TF, conferred by the nucleosome disfavoring nature of these tracts) (Raveh-Sadka et al. 2012). While a nucleosome-mediated effect of the poly(dA:dT) tract is likely to increase as the tract is closer to the TFBS (Raveh-Sadka et al. 2009, 2012), we observe a decrease in the tract's effect on expression when the tract is separated by 1 bp from the Gal4 site compared to when it is located further away (Wilcoxon rank-sum P = 0.0091). This observation agrees with our measured negative effect of the closely located poly(dA:dT) tract on Gal4 binding, which diminished when the tract was separated by additional bp. Our results thus suggest that the in vivo effect of a poly(dA:dT) tract directly adjacent to the binding site might stem from a direct, TF-specific effect of this sequence element on TF binding, in addition to other effects, as those involving additional proteins, including the formation of nucleosomes.
The effect of multiple TFBSs
TF binding to sequences with two TFBSs
Eukaryotic regulatory sequences typically contain multiple TFBSs (Lelli et al. 2012). However, even in the simple case where these sites are bound by the same TF (commonly referred to as “homotypic TFBS cluster”), a quantitative understanding of the dependency of TF binding and, consequently, the expression outcome on the multiplicity and arrangement of putative sites is still lacking (Levo and Segal 2014). As our assay includes long sequences that can contain multiple binding sites and can also isolate different binding states (e.g., distinguishing between a single TF binding event and two co-occurring binding events) (Supplemental Fig. S9), it allows for the characterization of this dependency.
We first examined a set of sequences with two binding sites, where one site resides at a fixed location and the second site is placed at different locations (as those shown in Fig. 3A–D). If binding to the two sites occurs in an independent manner and the differential location of the core TFBS has an effect on TF binding (as evident from our measurements of the single-site containing sequences) (Fig. 3A–D), then a sequence in which the second site is placed at an unfavorable location will show lower binding propensity by the two TFs compared to a sequence in which the second site is placed at a favorable location. This situation generally recapitulates the pattern observed for a single TF binding event to sequences with a differentially located single site. We found that this is indeed generally the case for both Gcn4 and Gal4 in two examined sequence contexts (Fig. 5A; Supplemental Fig. S10A). Notably, we found that a deviation from this trend can occur when the second site is placed in very close proximity to the first site (e.g., immediately adjacent or separated by a single bp) (Fig. 5A; Supplemental Fig. S9A). It is likely that in such close proximity, the binding to one site might interfere with the binding to the other.
Figure 5.

A simple thermodynamic model of TF binding as a function of TF concentration. TFBS multiplicity accounts for the binding measurements. (A) A set of sequences with a strong Gcn4 site placed at different locations along a specific sequence context, either in the presence of an additional strong Gcn4 site located in a fixed location (with the pink rectangle marking the location of the center of this site) (in red) or without this additional site (in blue). Shown is the log2 of the ratio of the binding score attained by each sequence (with the x-coordinate marking the location of the center of the site) divided by the median binding score across all sequences in this set. The binding score for the sequences with a single site is computed based on a band representing a single TF binding (see the band marked by a blue square in Fig. 1), while the binding score for sequences with two sites is computed based on the band representing two TF binding events (see the band marked by a red square in Fig. 1). The black arrow points to a sequence where the 9-bp sites are separated by a single bp. Sequences with Gcn4 TFBSs of 9 bp placed along the HIS3-derived context (left panel) and along the GAL1-10–derived context (right panel). (B) For a set of sequences with all possible combinations of one to seven binding sites for Gcn4 in seven predefined locations, the average frequency of sequences in different bands (“binding states”) is shown as a function of the number of sites within the sequence (in blue). The graphs correspond to the bands displayed in the gel in the right bottom corner. A detailed description of the plotted “normalized sequence frequency” measure can be found in Supplemental Section C. The predictions of these dependencies based on a simple thermodynamic model assuming multiple TF binding events are independent (for detailed description, see Supplemental Section C) and are also plotted (in black). (C) For the single parameter in the thermodynamic model, which represents the weight contribution of a TF binding event and is expected to be proportional to the TF concentration, the value used in the model that best fits the measured data is plotted against a measure of the concentration of Gcn4 that was actually used in the experiments presented here.
Analysis of another set of ∼600 sequences, in which both sites are differentially located farther apart, demonstrates this, as sequences in which the sites are located close together generally show reduced binding by the two TFs (after normalizing for the specific sites' locations) compared to sequences where the sites are further separated (Supplemental Fig. S10B,C). Interestingly, the sequences in which the sites are located extremely close together and for which lower binding by two TFs was observed display relatively low expression (Supplemental Fig. S10D–G). This suggests that the lower TF binding strength may contribute (possibly in concert with other mechanisms, such as a reduced capacity to promote expression from sites in close proximity) to the measured expression.
Thus, for each of the examined TFs, our results suggest two regimes: one that applies when the sites are located in close proximity, in which case binding of the TF to one of the sites likely interferes with the binding to the other site, and another that applies when the sites are located further apart from each other, in which case TF binding to the two sites seems to be largely independent.
General dependency on site multiplicity under different TF concentrations
To obtain a more general and quantitative understanding of the dependence of TF binding on the number of sites, we examined a set of sequences containing all possible combinations of one to seven available sites for Gcn4 at seven locations within two distinct sequence contexts. For each examined context, we produced a graph describing the relationship among the average sequence frequency in each of the bands formed on the gel (i.e., representing naked DNA, DNA bound by a single TF, and DNA bound by two TFs) as a function of the number of sites within the sequence (averaging over the different locations of the site). This was done for eight experiments, carried out with different Gcn4 concentrations (Fig. 5B, blue curves; for detailed description, see Supplemental Section C). As expected, there was generally an increase in binding as the number of binding sites increased. We note that in experiments where a band representing the binding of more than one TF also appeared, we found a decline in binding by a single TF to sequences with a high number of sites, presumably because these sequences were more prevalent in this additional band (Fig. 5; Supplemental Fig. S9).
To examine our quantitative understanding of these trends, we employed a simple thermodynamic model that assumes that TF binding to different sites is independent (Raveh-Sadka et al. 2009; Segal and Widom 2009a). This model allows us to predict the probability of different states corresponding to those captured on the gel as a function of the number of TFBSs in the sequence (see Supplemental Section C). The model has a single parameter that represents the weight contribution of a TF binding event (termed w) that can be defined as a product of the TF concentration and affinity. Although the units of this parameter are arbitrary, its value is expected to be proportional to the concentration of the respective TF. We scanned a range of values for this parameter for each of the eight binding experiments performed, extracting the value that produced the best fit to all measured curves. We found that the model accounts for the measured data (Fig. 5B), and the values for the w parameter yielding the best-fitting curves were highly correlated with the TF concentrations that were actually used in these experiments (Pearson's correlation = 0.978) (Fig. 5C). Thus, without introducing any explicit data on the relative TF concentrations, we were able to extract this information by applying a simple thermodynamic model to our measured data.
Overall, the successful predictions of the model suggest that its underlying assumptions are applicable to the measured binding dynamics, namely, the assumption of a thermodynamic equilibrium and the generally independent nature of binding events to multiple binding sites. Furthermore, as the measurements are carried out for numerous, systematically manipulated sequences, they reveal deviations from these general predictable trends as those discussed above with regard to sites that are located in close proximity. This refined quantitative understanding thus provides a step forward in our ability to predict binding, and consequently expression, to complex regulatory sequences.
Measuring nucleosome formation on the designed library of sequences
TF binding to regulatory sequences in vivo is influenced by the presence of other proteins, with histones being one such prominent example, as they occupy most of the eukaryotic DNA. As our assay allows the examination of binding events to sequences >147 bp (the length occupied by a single nucleosome), it allows us to examine the propensity for nucleosome formation on the same set of sequences for which TF binding and expression measurements were carried out.
To test this capability, we applied BunDLE-seq to our library of sequences for the binding to histone octamers rather than TF molecules (Fig. 6A) and reassuringly found that known nucleosome sequence preferences were captured by our assay. Specifically, we recapitulated across thousands of sequences the intrinsically nucleosome disfavoring nature of poly(dA:dT) tracts (Struhl and Segal 2013) that was previously deduced only from a handful of direct in vitro tests comparing nucleosome formation on sequences with or without such a tract (Anderson and Widom 2001; Bao et al. 2006) and mostly from the generally low nucleosome occupancy of regions enriched with these tracts genome-wide (Field et al. 2008; Kaplan et al. 2009; Zhang et al. 2009). We found that sequences lacking a 15-bp poly(dA:dT) tract generally show a higher nucleosome binding score than sequences with such a tract present, which in turn show a higher binding score than sequences with two such tracts present (Fig. 6B). We further found that in 93% of ∼2000 pairs of sequences examined, differing only in the presence of a 15-bp poly(dA:dT) tract, a sequence lacking this tract showed a higher nucleosome binding score compared to a corresponding sequence with the poly(dA:dT) tract present (Fig. 6C; for dependency of this effect on the tract length, see Supplemental Fig. S11A).
Figure 6.

Nucleosome sequence preferences captured by BunDLE-seq suggest additional mechanisms underlying expression differences between different regulatory sequences. (A) Schematic illustration of the application of BunDLE-seq with histones. (B) Sequences were sorted according to the nucleosome binding score (computed based on the histones-bound band). (Right) For each sequence, we show the number of 15-bp poly(dA:dT) tracts that it contains. (C) Scatter plots of nucleosome binding scores for sequences with a 15-bp poly(dA:dT) tract versus the binding score of the corresponding sequences lacking this tract [among 1972 pairs tested, for 1843 the sequence without the poly(dA:dT) tracts showed a higher binding score than a corresponding sequence with the tract]. (D) Scatter plot of the expression measurements of sequences with the HIS3-derived context versus the corresponding sequences (i.e., containing the same set of regulatory elements placed at the same locations along the sequence) with the GAL1-10–derived context (with 71% below the line). (E) Scatter plots of nucleosome binding scores for the same sequences shown in D (with 80% above the line).
Notably, in the expression measurements carried out with our library, sequences with a 15-bp poly(dA:dT) tract were found to generally have higher expression compared to corresponding sequences lacking this tract (Supplemental Fig. S10B; Sharon et al. 2012). As discussed above, for sequences in which the tract is adjacent to the TFBS, expression might be influenced both from the “direct” effect of the tract on TF binding, as captured in our TF binding measurements, as well as from the nucleosome disfavoring nature, captured by our histone binding measurements. When the tract is located further away from the site, the effect on TF binding affinity diminishes and it is likely that the elevated expression observed is dominated by the nucleosome-mediated effect (Figs. 4A, 6C; Supplemental Fig. S11C,D).
Our binding measurements also suggest the involvement of nucleosomes in mediating the higher expression that was observed for sequences based on the HIS3-derived context compared to sequences based on the GAL1-10–derived context (Fig. 6D; Sharon et al. 2012). We found that for the majority of 2600 examined sequence pairs, in which the same set of regulatory elements was placed in either of these two contexts, the nucleosome binding score for the sequence based on the GAL1-10–derived context was higher than that of the sequence based on the HIS3-derived context (Fig. 6E). Other mechanisms may clearly contribute to the differential expression for these two contexts, yet the higher nucleosome occupancy, likely hindering the accessibility of TFs and the transcription machinery on the sequence context that displays the lower expression, supports the intriguing possibility of nucleosome contributions to the resulting expression.
Thus, by coupling TF binding and nucleosome formation measurements to expression measurements performed on the same set of sequences, we suggest the differential involvement of these DNA-binding proteins in mediating the sequence effects (e.g., the presence of poly(dA:dT) tracts and general sequence context) on the expression outcomes.
Discussion
The means by which regulatory sequences direct TF binding are still not fully understood. Here we studied TF binding determinants both within the TFBS and outside of it. To this end, we introduced BunDLE-seq, a novel experimental assay that allows high-throughput measurements of TF binding to long, fully designed sequences.
Notably, we found that a multitude of binding levels can be attained even when the core binding site is fixed. We accounted for these pronounced differences by devising TF-specific models based on the sequence content of the proximal flanking sequences of the core TFBS. Interestingly, models based only on DNA shape features of the TFBS flanks also performed well and suggested different modes of DNA recognition employed by Gcn4 and Gal4, which are structurally distinct TFs (i.e., differing in the extent to which they rely on base-readout vs. shape-readout mechanisms) (Rohs et al. 2010).
As our assay provides quantitative information both on single TF binding events and multiple co-occurring events, it offers a unique opportunity to test current mechanistic models that produce explicit predictions on the occurrence of such binding events based on the composition of the sequence and the TF concentration. While we are able to account for the general relationship between binding and site multiplicity with a simple thermodynamic model that assumes independent binding, we found that for binding sites in close proximity, the binding of one TF seems to influence the binding of another. Future applications of our assay to probe binding to sequences specifically designed to densely sample different distances between sites can offer a high-throughput approach for characterizing finer patterns of the dependency between two binding events (e.g., extending on the recently reported periodic effect that one bound TF can have on another, possibly due to deformation in terms of DNA shape) (Kim et al. 2013).
A desirable goal when performing in vitro binding measurements is to be able to analyze these with respect to in vivo binding or expression measurements, as the former can offer insights as to the mechanisms contributing to the latter, by identifying aspects that are in agreement between such data sets and by revealing differences. However, such comparisons are often far from trivial. One difficulty emerges when the in vitro investigation aims to isolate and systematically characterize the role of a particular parameter (e.g., the effect of sequences flanking the core binding site), as is the case in this work, yet the vast majority of in vivo measurements are carried out on genomic sequences in which variation in this parameter naturally occurs in concert with variations in other parameters (e.g., in the composition or multiplicity of core sites). Nevertheless, several approaches can be taken to facilitate such comparisons; for instance, future applications of BunDLE-seq can be performed on sequences derived from native genomes (e.g., testing whether differential TF binding to genomic regions sharing a similar TFBS composition [Liu et al. 2006; White et al. 2013] measured in vivo can be recapitulated in vitro).
Alternatively, as we chose to do in this work, BunDLE-seq can be employed as a complementary method for the rapidly emerging protocols for high-throughput reporter assays (Levo and Segal 2014), thus allowing the same library of sequence variants to serve as input to both in vitro binding and in vivo expression measurements. A quantitative comparison of our binding measurements to expression measurements obtained with the same sequences serving as promoters in yeast cells (see Supplemental Section D; Supplemental Fig. S12; Sharon et al. 2012) reveals that differences in the affinity to the core binding site that are captured by BunDLE-seq have pronounced effects on the expression in vivo (Fig. 2C; Supplemental Fig. S12E,F,K,L). More intriguingly, we observe an agreement between these data sets with respect to the effect of placing the same core binding site in different locations along a specific context (Supplemental Fig. S4), the effect of a poly(dA:dT) tract immediately flanking the TFBS (Fig. 4), and the effect of closely located TFBSs relative to each other (Supplemental Fig. S9). Differential expression that cannot be accounted for by our TF binding measurements suggests the involvement of other components within the cell, with additional binding measurements, performed with possible candidates, offering means to characterize their role. Our binding measurements carried out with histones provide such an example, suggesting that a variable propensity to form nucleosomes contributes to differential expression in the presence of a poly(dA:dT) tract or different sequence contexts (Fig. 6; Supplemental Fig. S11). These results thus demonstrate the capability of BunDLE-seq to offer mechanistic insights into in vivo expression differences, even in seemingly complex situations where the composition of TFBSs in the corresponding regulatory sequence is similar.
Although our assay already provides means to widen the scope of TF binding studies from a local, site-oriented perspective to a regulatory, sequence-based perspective, it should be noted that the technology employed for the synthesis of DNA variants currently imposes some limitations as to the length and number of examined sequences (see Supplemental Section A).
However, an appealing direction for future applications of BunDLE-seq entails expanding the type of DNA-binding proteins, in addition to varying the targeted sequences. Different TFs, histones, and components of the transcription machinery can be assayed either separately or together (possibly allowing isolation from the gel of different combinations of binding events) and under different conditions (e.g., varying the protein concentration, adding cofactors or chromatin remodelers). Thus, our assay provides means to introduce various aspects to classical in vitro–based investigation of protein binding (at the level of the DNA and proteins involved), building toward the complexity of the in vivo environment while gaining a quantitative understanding of different individual and combined effects.
Methods
Library description and preparation
A library of 6500 sequences of 150 bp in length, as described previously (Sharon et al. 2012), was used as input for binding measurements. Among these sequences, ∼3800 contained at least one binding site for either Gcn4 or Gal4 or served as controls. An additional library of 13,000 sequences of length 200 bp was also used. Among these sequences, ∼7700 contained sites for Gcn4 or Gal4 with fixed flanks.
Each library was synthesized by Agilent (LeProust et al. 2010) and cloned into the pKT103-based plasmid as described elsewhere (Sharon et al. 2012). Input sequences for BunDLE-seq were then produced by PCR amplification from the plasmid following by purification form gel (for details, see Supplemental Section A).
Proteins used
Gcn4
GST-His-GCN4 (1–109, Saccharomyces cerevisiae) was cloned into pET-TevH plasmid and expressed in Bl21(DE3) bacteria. The cells were lysed, and the GST-Gcn4 protein was pulled down using glutathion beads. The GST tag was then cleaved by using TEV protease. For additional details about the Gcn4 purification, see Supplemental Section A.
Gal4
Gal4 (1–147, S. cerevisiae) + α helix was purchased from Abcam.
Description of BunDLE-seq
The reaction buffer (0.15 M NaCl, 0.5 mM PMSF [Sigma], 1 mM BZA [Sigma], 0.5 × TE, and 0.16 μg/μL PGA [Sigma]) was incubated at room temperature for 2 h in low binding tubes (Sorenson). When Gcn4 at a different protein/DNA molar ratio (see table in Supplemental Section A) was used as a binding TF, the tubes were cooled for 30 min at 4°C, and then 0.067 μg/μL BSA (Sigma) was added before adding the Gcn4 protein. Two hundred nanograms of DNA were then added, and the protein and DNA were incubated for 1 h at 4°C. When Gal4 was used, BSA and then the Gal4 protein at a different protein/DNA molar ratio (see table in Supplemental Section A) were added, and after the addition of 200 ng DNA the protein and DNA were incubated for 30 min at 30°C.
When chicken histone octamers (kindly supplied by the Widom laboratory) were used as binding molecules, histone octamers at 3:1 protein/DNA molar ratio were incubated with the reaction buffer (see above) at room temperature for 2 h in low binding tubes (Sorenson). The tubes were then cooled for 30 min at 4°C, the DNA (200 ng) was added, and the protein and DNA were incubated for 1 h at 4°C.
When either of the DNA-binding molecules was used, the reaction mix was run with Ficoll (Sigma) in 7.5% acrylamide gel in cold 0.25 × TBE buffer. The samples were loaded while the gel was running in order to minimize the time of incubation of the samples in the wells and thus reduce detachment of protein–DNA in the presence of the high salt-containing running buffer. The gel was stained for 30 min with GelStar (Lonza), and the bands were cut under UVIblue blue light transilluminator (UVITEC). The DNA was eluted from the gel using electroelution Midi GeBAflex tubes (Gene Bio-Application), precipitated with 1 volume isopropanol, 1/10 volume 3 M NaOAc (pH 5.2), and 1 μg/μL glycogen (Fermentas) overnight at −20°C, and resuspended in 1 × TE buffer. The DNA from each band was diluted to 0.1 ng/μL, and 1 ng from each band was taken for eight cycles of PCR amplification using 3′ primer that was common to all bands (5′-NNNNNTTATGTG ATAATGCCTAGGATCGC-3′, where N's represent random nucleotides) and 5′ primer with a unique upstream 5-bp barcode sequence (underlined) specific to each band (5′-XXXXXGGGGA CCAGGTGCCGTAAG-3′, where Xs represent the band unique sequence).
A detailed experimental protocol for BunDLE-seq is available as Supplemental Material and at http://genie.weizmann.ac.il/data/factor_binding.html.
Sequencing and mapping
The DNA collected from each experiment, with barcodes marking the band from which the DNA was excised, were joined. Ten nanograms were used in library preparation for sequencing (protocol adopted from Blecher-Gonen et al. 2013). The DNA was amplified using 14 amplification cycles and sequenced on a 50-bp, single-read flowcell on an Illumina HiSeq 2000 sequencer at the Israel National Center for Personalized Medicine (INCPM) unit at the Weizmann Institute of Science. The reads were separated according to the band barcode and mapped to the designed library based on a 10-bp barcode (found 19 bp from the read start) (Sharon et al. 2012). Quality controls on sequence mapping were applied, and sequences with more than two mismatches discarded from further analysis.
Sequences represented by less than 100 reads in the no protein band were also discarded from further analysis.
Extraction of measures from sequencing data
For each tested sequence, the sequencing data provide its frequency in each of the bands. An additional sample, treated as all the others except with no exposure to the TF (DNA only), served to estimate the frequency of each sequence in the initial pool. The “binding score” computed per sequence per band is the frequency of that sequence among the sequences extracted from that band divided by its frequency in the initial library. For more details on the extracted measures, see Supplemental Section B.
Binding scores obtained with BunDLE-seq are available as Supplemental Material and at http://genie.weizmann.ac.il/data/factor_binding.html.
Data access
Raw and processed data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE66143.
Acknowledgments
This work is dedicated to the late Jonathan Widom who inspired this project and greatly assisted us in its development. We thank Shira Albeck and Yoav Peleg from the Israel Structural Proteomics Center (ISPC) at the Weizmann Institute of Science for producing the Gcn4 protein, Irene K. Moore from the Widom laboratory for purifying the histone octamers, and Ghil Jona for fruitful discussions. This work was supported by the European Research Council and the National Institutes of Health to E.S. and grants R01GM106056 and U01GM103804 to R.R. R.R. is an Alfred P. Sloan Research Fellow. M.L. thanks the Azrieli Foundation for the award of an Azrieli Fellowship.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.185033.114.
References
- Alexeev DG, Lipanov AA, Skuratovskii I. 1987. Poly(dA).poly(dT) is a B-type double helix with a distinctively narrow minor groove. Nature 325: 821–823. [DOI] [PubMed] [Google Scholar]
- Anderson JD, Widom J. 2001. Poly(dA-dT) promoter elements increase the equilibrium accessibility of nucleosomal DNA target sites. Mol Cell Biol 21: 3830–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Y, White CL, Luger K. 2006. Nucleosome core particles containing a poly(dA.dT) sequence element exhibit a locally distorted DNA structure. J Mol Biol 361: 617–624. [DOI] [PubMed] [Google Scholar]
- Berger MF, Bulyk ML. 2009. Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat Protoc 4: 393–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blecher-Gonen R, Barnett-Itzhaki Z, Jaitin D, Amann-Zalcenstein D, Lara-Astiaso D, Amit I. 2013. High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nat Protoc 8: 539–554. [DOI] [PubMed] [Google Scholar]
- Ellenberger TE, Brandl CJ, Struhl K, Harrison SC. 1992. The GCN4 basic region leucine zipper binds DNA as a dimer of uninterrupted α helices: crystal structure of the protein-DNA complex. Cell 71: 1223–1237. [DOI] [PubMed] [Google Scholar]
- Field Y, Kaplan N, Fondufe-Mittendorf Y, Moore IK, Sharon E, Lubling Y, Widom J, Segal E. 2008. Distinct modes of regulation by chromatin encoded through nucleosome positioning signals. PLoS Comput Biol 4: e1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordyce PM, Gerber D, Tran D, Zheng J, Li H, DeRisi JL, Quake SR. 2010. De novo identification and biophysical characterization of transcription-factor binding sites with microfluidic affinity analysis. Nat Biotechnol 28: 970–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, Hastie T, Tibshirani R. 2010. Regularization paths for generalized linear models via coordinate descent. J Stat Softw 33: 1–22. [PMC free article] [PubMed] [Google Scholar]
- Gordan R, Shen N, Dror I, Zhou T, Horton J, Rohs R, Bulyk ML. 2013. Genomic regions flanking E-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell Rep 3: 1093–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin MJ, Lis JT. 2010. Chromatin landscape dictates HSF binding to target DNA elements. PLoS Genet 6: e1001114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin MJ, Martins AL, Siepel A, Lis JT. 2012. Accurate prediction of inducible transcription factor binding intensities in vivo. PLoS Genet 8: e1002610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn S, Young ET. 2011. Transcriptional regulation in Saccharomyces cerevisiae: transcription factor regulation and function, mechanisms of initiation, and roles of activators and coactivators. Genetics 189: 705–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DE, Hope IA, Macke JP, Struhl K. 1986. Saturation mutagenesis of the yeast his3 regulatory site: requirements for transcriptional induction and for binding by GCN4 activator protein. Science 234: 451–457. [DOI] [PubMed] [Google Scholar]
- Jacob F, Monod J. 1961. Genetic regulatory mechanisms in the synthesis of proteins. J Mol Biol 3: 318–356. [DOI] [PubMed] [Google Scholar]
- Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, Morgunova E, Enge M, Taipale M, Wei G, et al. 2013. DNA-binding specificities of human transcription factors. Cell 152: 327–339. [DOI] [PubMed] [Google Scholar]
- Kaplan N, Moore IK, Fondufe-Mittendorf Y, Gossett AJ, Tillo D, Field Y, LeProust EM, Hughes TR, Lieb JD, Widom J, et al. 2009. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature 458: 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller W, Konig P, Richmond TJ. 1995. Crystal structure of a bZIP/DNA complex at 2.2 A: determinants of DNA specific recognition. J Mol Biol 254: 657–667. [DOI] [PubMed] [Google Scholar]
- Kim S, Brostromer E, Xing D, Jin J, Chong S, Ge H, Wang S, Gu C, Yang L, Gao YQ, et al. 2013. Probing allostery through DNA. Science 339: 816–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelli KM, Slattery M, Mann RS. 2012. Disentangling the many layers of eukaryotic transcriptional regulation. Annu Rev Genet 46: 43–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeProust EM, Peck BJ, Spirin K, McCuen HB, Moore B, Namsaraev E, Caruthers MH. 2010. Synthesis of high-quality libraries of long (150mer) oligonucleotides by a novel depurination controlled process. Nucleic Acids Res 38: 2522–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levo M, Segal E. 2014. In pursuit of design principles of regulatory sequences. Nat Rev Genet 15: 453–468. [DOI] [PubMed] [Google Scholar]
- Liu X, Lee CK, Granek JA, Clarke ND, Lieb JD. 2006. Whole-genome comparison of Leu3 binding in vitro and in vivo reveals the importance of nucleosome occupancy in target site selection. Genome Res 16: 1517–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maerkl SJ, Quake SR. 2007. A systems approach to measuring the binding energy landscapes of transcription factors. Science 315: 233–237. [DOI] [PubMed] [Google Scholar]
- Marmorstein R, Carey M, Ptashne M, Harrison SC. 1992. DNA recognition by GAL4: structure of a protein-DNA complex. Nature 356: 408–414. [DOI] [PubMed] [Google Scholar]
- Morozov AV, Siggia ED. 2007. Connecting protein structure with predictions of regulatory sites. Proc Natl Acad Sci 104: 7068–7073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutiu R, Friedman RC, Luo S, Khrebtukova I, Silva D, Li R, Zhang L, Schroth GP, Burge CB. 2011. Direct measurement of DNA affinity landscapes on a high-throughput sequencing instrument. Nat Biotechnol 29: 659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliphant AR, Brandl CJ, Struhl K. 1989. Defining the sequence specificity of DNA-binding proteins by selecting binding sites from random-sequence oligonucleotides: analysis of yeast GCN4 protein. Mol Cell Biol 9: 2944–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkumar AS, Denervaud N, Maerkl SJ. 2013. Mapping the fine structure of a eukaryotic promoter input-output function. Nat Genet 45: 1207–1215. [DOI] [PubMed] [Google Scholar]
- Raveh-Sadka T, Levo M, Segal E. 2009. Incorporating nucleosomes into thermodynamic models of transcription regulation. Genome Res 19: 1480–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveh-Sadka T, Levo M, Shabi U, Shany B, Keren L, Lotan-Pompan M, Zeevi D, Sharon E, Weinberger A, Segal E. 2012. Manipulating nucleosome disfavoring sequences allows fine-tune regulation of gene expression in yeast. Nat Genet 44: 743–750. [DOI] [PubMed] [Google Scholar]
- Rohs R, West SM, Sosinsky A, Liu P, Mann RS, Honig B. 2009. The role of DNA shape in protein-DNA recognition. Nature 461: 1248–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohs R, Jin X, West SM, Joshi R, Honig B, Mann RS. 2010. Origins of specificity in protein-DNA recognition. Annu Rev Biochem 79: 233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Widom J. 2009a. From DNA sequence to transcriptional behaviour: a quantitative approach. Nat Rev Genet 10: 443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Widom J. 2009b. Poly(dA:dT) tracts: major determinants of nucleosome organization. Curr Opin Struct Biol 19: 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon E, Kalma Y, Sharp A, Raveh-Sadka T, Levo M, Zeevi D, Keren L, Yakhini Z, Weinberger A, Segal E. 2012. Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat Biotechnol 30: 521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siggers T, Duyzend MH, Reddy J, Khan S, Bulyk ML. 2011. Non-DNA-binding cofactors enhance DNA-binding specificity of a transcriptional regulatory complex. Mol Syst Biol 7: 555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery M, Riley T, Liu P, Abe N, Gomez-Alcala P, Dror I, Zhou T, Rohs R, Honig B, Bussemaker HJ, et al. 2011. Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins. Cell 147: 1270–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery M, Zhou T, Yang L, Dantas Machado AC, Gordan R, Rohs R. 2014. Absence of a simple code: how transcription factors read the genome. Trends Biochem Sci 39: 381–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl K, Segal E. 2013. Determinants of nucleosome positioning. Nat Struct Mol Biol 20: 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MA, Myers CA, Corbo JC, Cohen BA. 2013. Massively parallel in vivo enhancer assay reveals that highly local features determine the cis-regulatory function of ChIP-seq peaks. Proc Natl Acad Sci 110: 11952–11957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong D, Teixeira A, Oikonomopoulos S, Humburg P, Lone IN, Saliba D, Siggers T, Bulyk M, Angelov D, Dimitrov S, et al. 2011. Extensive characterization of NF-kappaB binding uncovers non-canonical motifs and advances the interpretation of genetic functional traits. Genome Biol 12: R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Zhou T, Dror I, Mathelier A, Wasserman WW, Gordan R, Rohs R. 2014. TFBSshape: a motif database for DNA shape features of transcription factor binding sites. Nucleic Acids Res 42: D148–D155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Moqtaderi Z, Rattner BP, Euskirchen G, Snyder M, Kadonaga JT, Liu XS, Struhl K. 2009. Intrinsic histone-DNA interactions are not the major determinant of nucleosome positions in vivo. Nat Struct Mol Biol 16: 847–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, O'Shea EK. 2011. Integrated approaches reveal determinants of genome-wide binding and function of the transcription factor Pho4. Mol Cell 42: 826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Yang L, Lu Y, Dror I, Dantas Machado AC, Ghane T, Di Felice R, Rohs R. 2013. DNAshape: a method for the high-throughput prediction of DNA structural features on a genomic scale. Nucleic Acids Res 41: W56–W62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Byers KJ, McCord RP, Shi Z, Berger MF, Newburger DE, Saulrieta K, Smith Z, Shah MV, Radhakrishnan M, et al. 2009. High-resolution DNA-binding specificity analysis of yeast transcription factors. Genome Res 19: 556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]