

Abstract
Haplotypes have gained increasing attention in the mapping of complex-disease genes, because of the abundance of single-nucleotide polymorphisms (SNPs) and the limited power of conventional single-locus analyses. It has been shown that haplotype-inference methods such as Clark’s algorithm, the expectation-maximization algorithm, and a coalescence-based iterative-sampling algorithm are fairly effective and economical alternatives to molecular-haplotyping methods. To contend with some weaknesses of the existing algorithms, we propose a new Monte Carlo approach. In particular, we first partition the whole haplotype into smaller segments. Then, we use the Gibbs sampler both to construct the partial haplotypes of each segment and to assemble all the segments together. Our algorithm can accurately and rapidly infer haplotypes for a large number of linked SNPs. By using a wide variety of real and simulated data sets, we demonstrate the advantages of our Bayesian algorithm, and we show that it is robust to the violation of Hardy-Weinberg equilibrium, to the presence of missing data, and to occurrences of recombination hotspots.
Introduction
Single-nucleotide polymorphisms (SNPs), which are found every 250–350 bp in the human genome (Beaudet et al. 2001), have gained a great popularity in recent years because of their abundance and their utility in the mapping of complex-disease genes and in identifying genetic variants that influence drug response. Owing to its biallelic nature, SNP genotyping is much more amenable to automation and miniaturization than are microsatellite loci. High-throughput genotyping platforms—such as mass spectrometry (Ross et al. 1998), molecular beacon (Tyagi and Kramer 1996), TaqMan assay (Ranade et al. 2001), and high-density oligonucleotide microchips (Hacia et al. 1999)—have been developed concurrently with SNP-discovery efforts, by either experimental methods such as denaturing high-performance liquid chromatography in combination with fluorescence-based DNA sequencing (Niu et al. 2001) or in silico SNP screening in cyberspace (Cox et al. 2001). However, the tremendous amount of SNP data presents a daunting challenge for analysis. Although the simplest and perhaps the most popular way to address the challenge is by the conventional “SNP-centric” approach, linkage disequilibrium (LD) studies by such approaches are unsatisfactory (1) because a single SNP has a relatively low information content and (2) because, for a gene with multiple tightly linked SNPs, not only would the LD information contained in flanking markers be ignored in the single-SNP–based approach but a Bonferroni correction would also be required to protect against an inflated type I error. Thus, the “haplotype-centric” approach, which combines the information of adjacent SNPs into composite multilocus haplotypes, is more desirable. Haplotypes not only are more informative but also capture the regional LD information, which is arguably more robust and powerful (Akey et al. 2001; Daly et al. 2001; Pritchard 2001).
For autosomal loci, when only the multilocus phenotypes (“phenotype” denotes unphased genotype configurations) for each individual are provided, the phase information for those individuals with multiply heterozygous phenotypes is inherently ambiguous. For any individual who has no more than one heterozygous site, the situation is simple, and the individual’s haplotype phase can be resolved with certainty. True resolution for the ambiguous (i.e., multiply heterozygous) phenotypes depends on either molecular haplotyping or typing of close biological relatives. For molecular haplotyping, existing methods such as single-molecule dilution (Ruano et al. 1990), allele-specific long-range PCR (Michalatos-Beloin et al. 1996), isothermal rolling-circle amplification (Lizardi et al. 1998), long-insert cloning (Ruano et al. 1990; Bradshaw et al. 1995), and carbon-nanotube probing (Woolley et al. 2000) are difficult to automate and hence are costly, low throughput, and prone to experimental errors (Judson and Stephens 2001). Although the novel diploid-to-haploid conversion method (Douglas et al. 2001) shows some promise, its technical difficulties and high cost prevent it from being widely adopted in the short run. The typing of close relatives can always reduce the phase ambiguity, but the phase determination is still problematic when the number of loci is only moderately large (Hodge et al. 1999). In the absence of true resolution strategies, in silico haplotype-determination methods become attractive alternatives. We show that these algorithms, especially the algorithms based on explicit statistical models, provide rather robust and accurate explanations of commonly occurring haplotypes in a reasonably sized sample of individuals, even when some of the model assumptions are strongly violated.
There are primarily three categories of algorithms for the inference of haplotype phases of individual genotype data; these categories are exemplified by Clark’s algorithm (Clark 1990), the expectation-maximization (EM) algorithm (Excoffier and Slatkin 1995; Hawley and Kidd 1995; Long et al. 1995; Chiano and Clayton 1998), and a pseudo-Bayesian algorithm (Stephens et al. 2001b). Clark’s parsimony approach attempts to assign the smallest number of haplotypes for the observed genotype data by convoluted updating, starting from phase-unambiguous individuals, of the haplotype list. Albeit simple in nature, Clark’s algorithm has been highly popular and has generated meritorious results in the delineation of the gene-based haplotype variations (Stephens et al. 2001a) and of the genomewide LD in populations with different histories (Reich et al. 2001). The EM algorithm starts with an initial guess of haplotype frequencies and iteratively updates the frequency estimates, to maximize the log-likelihood function. An EM-based haplotype estimation has been applied successfully in the transmission-disequilibrium tests (Zhao et al. 2000) and has been shown to be reasonably accurate under a wide range of parameter settings (Fallin and Schork 2000). Stephens et al. (2001b) employed an iterative stochastic-sampling strategy—the pseudo-Gibbs sampler (PGS)—for the assignment of haplotype phases. They show, by coalescence-based simulations, that the PGS performed better than Clark’s algorithm and the EM algorithm. The improved performance of the PGS is likely due to both the employment of a stochastic search strategy and the incorporation of the coalescence theory into its iteration steps. Although the coalescence model is appropriate for the description of a stable population that has evolved over a long period of time, it may be less suitable for populations with either past gene flow, stratifications, or bottlenecks, all of which are common in real applications. Our study shows that deviations from the basic assumptions of the coalescence model can indeed adversely affect the performance of the PGS.
Despite these previous vigorous endeavors, several challenges for the haplotype inference remain. Specifically, although the treatment of the missing data problem has been previously mentioned (Hawley and Kidd 1995; Stephens et al. 2001b), existing methods cannot handle different types of missing marker data (for details, see the “Methods” section). Furthermore, the handling of a very large number of linked SNPs has not been satisfactorily addressed in Clark’s algorithm or the EM method. Motivated by these challenges, we introduce a robust Bayesian procedure that makes use of the same statistical model used in the EM algorithm and that imposes no assumptions on the population evolutionary history. In this model, each individual’s haplotype pair is treated as two random draws from a pool of haplotypes with unknown population frequencies. By employing two novel techniques, partition ligation (PL) and prior annealing, which improve both the accuracy and capacity, our new method showed improved performance, in comparison to existing methods, in a wide variety of simulated and real data sets. We demonstrate that both the EM and our method performed robustly, providing significantly-more-accurate results than other existing methods, when the Hardy-Weinberg equilibrium (HWE) assumption is violated. Our study also shows that comparison of the performances of different in silico haplotyping methods is subtler than it appears to be—that is, the model underlying the simulation study can greatly affect the conclusion.
Methods
Statistical Model and Maximum-Likelihood Estimation
Consider a sample of n individuals for a local chromosomal region comprising L linked SNPs. Let Y=(y1,…,yn) denote the observed genotype for the n individuals, where yi=(yi1,…,yiL), and let yij denote the genotype for individual i at locus j. Let yij=0, 1, or 2, to denote that individual i is heterozygous, homozygous wild type, or homozygous mutant at SNP marker locus j, respectively. Additional categories are created for two missing alleles (yij=3) and the presence of only one missing allele, when the known allele is either wild type (yij=4) or mutant (yij=5).
Let Z=(z1,…,zn) denote the unobserved haplotype configuration compatible with Y, where zi=(zi1,zi2) designates the assigned haplotype pair for the ith individual. We use the notation zi1⊕zi2=yi to denote that the two haplotypes are compatible with genotype yi. Let Θ=(θ1,…,θM) denote population haplotype frequencies, where M is the number of all possible haplotypes. Suppose that HWE holds true—that is, that the population fraction of individuals with the ordered haplotype pairs (g,h) is θgθh. Then, the likelihood function can be easily expressed as
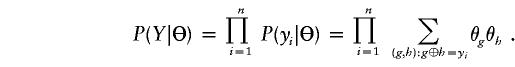 |
By simple algebra, we can show that the maximum-likelihood estimate (MLE) of Θ has to satisfy the estimating equation
 |
where ng is the count of haplotype g in a particular phase configuration Z. Thus, the right-hand side of the equation computes the “expected frequency” of haplotype g by averaging over all compatible Zs. This equation represents the internal consistency of the MLE and gives rise to the following iteration steps for the EM algorithm (Dempster et al. 1977):
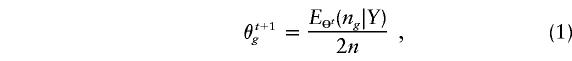 |
where Θt and θt+1g refer to the estimated frequencies at times t and t+1, respectively. A formal EM algorithm iterates equation (1) until Θt does not change much. Individuals’ genotypes can be phased by using the final estimate 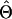 . That is, for a given yi, we find a compatible haplotype pair (g,h) that maximizes
. That is, for a given yi, we find a compatible haplotype pair (g,h) that maximizes 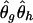 . We can also impute multiple haplotype pairs to reflect the estimation uncertainty.
. We can also impute multiple haplotype pairs to reflect the estimation uncertainty.
Bayesian Inference and Gibbs Sampling
Instead of the MLE approach, we can also seek a Bayesian solution to the problem. Assuming that Θ∼Dirchlet(β) a priori, where β=(β1,…,βM) (see Appendix A), we have
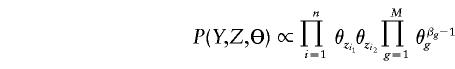 |
for Z compatible with Y, and P(Y,Z,Θ)=0 otherwise. The following iterations constitute a Gibbs sampling algorithm:
- Conditional on Θ, sample a pair of compatible haplotypes for each subject according to
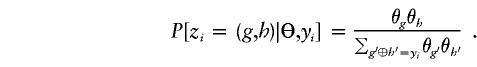
- Conditional on the “imputed” haplotypes Z, update by a random draw from the posterior distribution
where N(Z) is the vector of haplotype counts in Z.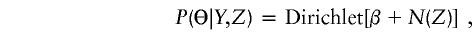
Predictive Updating
The predictive updating strategy (Liu 1994; Chen and Liu 1996) can be applied to further improve the above Gibbs sampling method. That is, we can integrate out Θ explicitly in the joint distribution P(Y,Z,Θ) so that
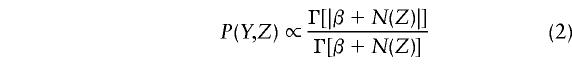 |
where we define Γ(|v|)=Γ(|v1+⋅⋅⋅+vk|) and Γ(v)=j=1kΓ(vj) for a vector v=(v1,…,vk). As a consequence, we obtain a different Gibbs sampler: Pick an individual i at random (or in a certain order) and update his/her haplotype zi by sampling from
where Z-i represents all but the ith person’s haplotypes and where ng and nh are the counts of haplotypes g and h in Z-i, respectively. This strategy gives rise to an intuitive algorithm that is similar in spirit to the Gibbs motif sampler for sequence analysis (Lawrence et al. 1993). Stephens et al. (2001b) also made use of this simple structure in the construction of their PGS algorithm.
PL
The handling of a large number of haplotypes remains a challenging issue for the Gibbs samplers described above. Here, we tackle the problem by PL, a divide-conquer-combine technique. This technique not only allows us to analyze very long SNP sequences but also helps the Monte Carlo algorithm converge more rapidly. In contrast to a Gibbs sampler that deals with the problem by local updating (i.e., updating a few loci of a person, conditional on others [Stephens et al. 2001b]), the PL strategy is more similar in spirit to multigrid Monte Carlo and sequential Monte Carlo methods (Liu 2001). Suppose that a sequence consists of L SNP loci: Without loss of generality, we assume that L=K×M, where K represents the size of each “atomistic unit” (we typically chose K⩽8). The genotype data, Y, and haplotype data, Z, are first partitioned into M subsets each of size K (fig. 1)—that is, Y=(Y1:K,YK+1:2K,…) and Z=(Z1:K,ZK+1:2K,…).
Figure 1.
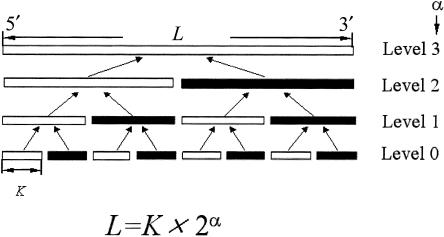
A schematic depicting the PL algorithm. L denotes the total number of loci; K denotes the number of loci in the smallest segment; α is the highest level of the PL pyramidal hierarchy.
Two strategies can be employed for the ligation step: progressive ligation and hierarchical ligation. In both approaches, we first conduct the atomistic-haplotype restoration (i.e., the construction of a set of partial haplotypes implied by each atomistic unit); for example, we process all the possible haplotypes implicated by the first genotype segment Y1:K. Since the number of loci is moderate (typically, K⩽8), we can implement the aforementioned Gibbs samplers. Then, we record the B most probable haplotypes, z11:K,…,zB1:K, which guarantees that Y1:K can be completely “resolved,” together with their estimated population frequencies. Likewise, other atomistic units, YaK+1:aK+K, can be processed to obtain their most probable haplotypes, z1aK+1:aK+K,…,zBaK+1:aK+K. The choice of B (between 40 and 50 in all of our examples) depends on both the sample size and heterozygosity of the data set. It is important to keep B moderately large so as not to discard some haplotype segments that lead to the true haplotypes.
In progressive ligation, we combine the first two atomistic units to form B most probable partial haplotypes of 2K loci, z11:2K,…,zB1:2K, with their estimated frequencies. More precisely, we conduct the Gibbs iterations based on equation (2) by use of only the segments of haplotypes—that is, each person's haplotypes are chosen among the B2 possible combinations of zi1:K,zjK+1:2K. This process is recursively continued by ligation of the next atomistic unit to the current partial haplotype until all the units are ligated to form the whole haplotype.
For an easy description, we assume that L=K×2α in hierarchical ligation (it can be easily modified to apply to other types of L). In hierarchical ligation, similar to progressive ligation, we have 2α atomistic units, each containing K loci. Then, as shown in figure 1, the (2j-1)th segment is ligated with the (2j)th segment for j=1,…,2α-1 to form 2α-1 level 1 “larger” segments. Then, we ligate the adjacent level 1 segments to form 2α-2 level 2 segments, and so forth. The ligation procedure is the same as that described above.
For a data set with n individuals and L linked SNPs, the running time of the PL algorithm is O(nL), provided that K, B, and the numbers of the Gibbs iterations per individual for both the atomistic-haplotype–construction and ligation steps remain constant. This is because the PL needs L/K atomistic restorations and (L/K)-1 ligations per individual.
Prior Annealing
To enable the Gibbs sampler to freely maneuver in the haplotype space, we applied the prior-annealing technique: in the beginning of the iteration, high pseudocounts, β0, that conform to the Dirichlet distribution, are used as the initial prior. As the iteration proceeds, the pseudocounts are dwindled in a fixed rate. To be specific, if we suppose that the pseudocounts for all haplotypes are β0 and βT for the start and the end of the Tth iteration, then the pseudocounts at the tth iteration, βt, are given as follows:
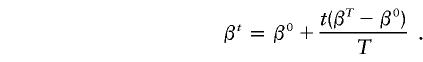 |
Missing Marker Data
The problem of the absence of both alleles of an SNP marker is common owing to PCR dropouts and was first addressed by Hawley and Kidd (1995). However, the “one-allele” problem, in which only one allele for a particular SNP is unscored owing to ambiguity, is also a legitimate concern for a number of SNP-genotyping methodologies, such as oligonucleotide-ligation assay or single-base extension (SBE) coupled with fluorescence-polarization detection. For example, the new high-throughput SNP-genotyping technique known as the “TAG-SBE method” (Fan et al. 2000)—which analyzes allele-specific SBE reactions on standardized high-density oligonucleotide arrays—has a number of advantages, such as parallelism, flexibility, and robustness. However, in ∼1% of genotype calls for heterozygous sites, it can score only one allele unequivocally. In our algorithm, the missing data are classified into three categories: type I, for both alleles missing; type II, for one known wild-type allele; and type III, for one known mutant allele. All of the missing types can be handled by the PL with small modifications to its sampling steps: for type I, all of the different alleles at the locus are considered without constraint; and for types II and III, the sampling choices are partially constrained owing to the presence of the known allele.
Implementation of the Algorithms
We implemented Clark’s algorithm and the PGS algorithm by use of existing software packages. HAPINFERX, which implements Clark’s algorithm, was kindly provided by A. G. Clark. PHASE, which implements the PGS algorithm as described by Stephens et al. (2001b), was downloaded from their Web site (Mathematics Genetics Group). The EM algorithm for haplotype construction was coded by the authors in a program (named “EM-DeCODER”) according to equation (1) and is freely available from our website (Jun Liu's Home Page). We note that the PL idea can also be applied to the EM algorithm, with minor modifications. Our simulations suggest that the PL idea serves not only as an effective computational trick but also as a “regularizer” to prevent the algorithm from being too “greedy.” Our Gibbs sampler with the PL and prior-annealing procedures is generally referred to as “the PL algorithm” and was coded in a software package named “HAPLOTYPER” (for details, see Appendix A).
Results
To illustrate our proposed algorithm and to compare it with existing ones, we analyzed two real data sets and conducted several simulation studies. A distinguishing feature of some of our simulation studies is the use of recently published real haplotypes (e.g., the β2-adrenergic receptor gene, the angiotensin I–converting enzyme [ACE] gene, the cystic fibrosis transmembrane conductance regulator gene, and the 5q31 region). We have also conducted two population-theory–based simulations, one of which follows the coalescence model and the other of which produces artificial populations that experienced a bottleneck effect.
β2-Adrenergic Receptor (β2AR) Gene Data Set
The gene encoding β2AR is devoid of introns within its coding region. Abnormality of the β2AR has been hypothesized to be involved in the physiology of asthma (Reihsaus et al. 1993). Because albuterol, the effective bronchodilator used as a first-line drug for treatment of asthma, acts primarily by binding to β2AR expressed on the airway smooth muscle cells, several studies suggest that molecular variants of this gene may predict patients’ response to β2-agonist (Martinez et al. 1997; Drysdale et al. 2000; Israel et al. 2000). According to the data presented by Drysdale et al. (2000), 10 distinct haplotypes, each with 13 loci (L=13), were found in a population of 121 subjects of European descent. The χ2 test for the data indicates that HWE holds well (P=.32). The EM, PGS, and PL algorithms phased all of the 121 individuals successfully, whereas Clark’s algorithm made two mistakes (i.e., predicted two individuals’ phases incorrectly).
Impact of the HWE Assumption
To assess the sensitivity of the algorithms to the HWE assumption, we took the 12 haplotypes together with their observed frequencies from the β2AR data set and performed simulations by use of five different models to represent different levels of departures from HWE. For each model, 1,000 replications were conducted. In each replication, the genotypes of 15 hypothetical individuals were drawn independently from the space of all ordered pairs of haplotypes according to a probability matrix C=(cij)12×12 (i.e., cij⩾0 , and 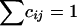 ). That is, we have the probability cij of picking haplotype pair (hi,hj). The matrix C followed distribution Dirichlet(100D) (see Appendix A), where D=(dij)12×12 is also a probability matrix, thereby satisfying the relationship D∝PTWP with P=(p1,…,p12) being the vector of the observed frequencies of the 12 haplotypes. Because dij=wijpipj, letting wij=1 tends to produce samples conforming to HWE. Matrix W can be interpreted as the fitness of individuals with those particular genotypes and can be controlled to reflect the degree of departure from HWE. For simplicity, we let
). That is, we have the probability cij of picking haplotype pair (hi,hj). The matrix C followed distribution Dirichlet(100D) (see Appendix A), where D=(dij)12×12 is also a probability matrix, thereby satisfying the relationship D∝PTWP with P=(p1,…,p12) being the vector of the observed frequencies of the 12 haplotypes. Because dij=wijpipj, letting wij=1 tends to produce samples conforming to HWE. Matrix W can be interpreted as the fitness of individuals with those particular genotypes and can be controlled to reflect the degree of departure from HWE. For simplicity, we let
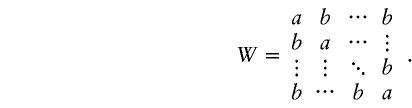 |
Hence, a >b implies that the homozygous state is preferred and vice versa. The five models are (1) neutral, in which a=b=1; (2) moderate heterozygote favoring, in which a=1 and b=2; (3) strong heterozygote favoring, in which a=1 and b=3; (4) moderate homozygote favoring, in which a=2 and b=1; and (5) strong homozygote favoring, in which a=3 and b=1. For each of the five models, the instances of incorrectly inferred haplotype phases, the values of a χ2 statistic (with 9 df) that tests for HWE, and the number of homozygotes among the 15 individuals are recorded.
The impact that HWE violation has on the PL algorithm, Clark's algorithm, the EM algorithm, and the PGS algorithm is demonstrated in figure 2, on the basis of which we made the following observations:
-
1.
A greater extent of the HWE violation due to an excess of heterozygosity leads to a higher error rate for all four algorithms.
-
2.
Clark’s algorithm and the PGS algorithm performed worse across the board and were more vulnerable to the departure from HWE than the EM and PL algorithms; the PL and EM algorithms performed indistinguishably in all the cases.
-
3.
The level of homozygosity correlates more directly with the inference accuracy than the χ2 test statistic (details shown below).
Figure 2.
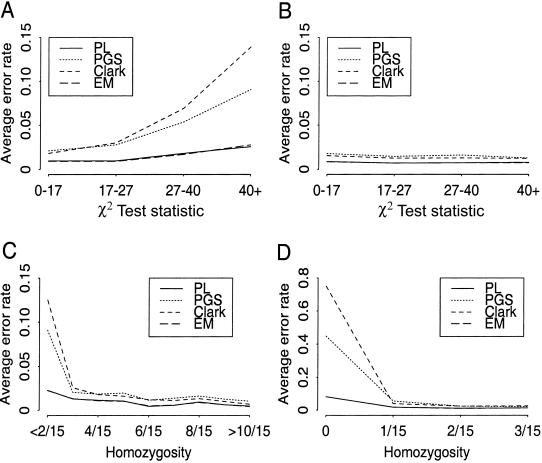
The impact that HWE violation has on the performances of the PL algorithm, the PGS algorithm, Clark's algorithm, and the EM algorithm. The simulation study was conducted under five scenarios, each with 1,000 replications: (1) neutral, (2) moderate heterozygosity, (3) strong heterozygosity, (4) moderate homozygosity, and (5) strong homozygosity. For each trial, a χ2 test statistic for testing HWE (after pooling the categories with small counts, this gives rise to the independence test of a 4×4 table, which has 9 df) was computed, the number of homozygotes was counted, and the error rates of each algorithm were recorded. A, Average error rate (defined as the number of erroneous phase calls divided by the total number of phase calls) of each method versus HWE χ2 test statistic after combining simulations from models (1), (2), and (3). B, Average error rate versus HWE χ2 test statistic after combining simulations from models (1), (4), and (5). Note that the χ2 values of 21.67, 16.92, and 14.68 correspond to the 99th, 95th, and 90th percentiles, respectively. C, Average error rate versus sample haplotype homozygosity after combining all simulations. D, Zoom-in view of panel C at left tail of the homozygosity distribution (i.e., 0/15–3/15).
The same extent of the HWE violation according to the χ2 test can be caused by either a significant excess of homozygosity due to inbreeding (Lander and Botstein 1987) or a significant excess of heterozygosity due to its selective advantage (Schroeder et al. 1995). We observed that, in the case of the former circumstance, the algorithms’ accuracies were not compromised by the HWE violation (fig. 2B), whereas, in the case of the latter circumstance, as the test statistic increases, the number of phasing errors increased monotonically (fig. 2A). Thus, the χ2 test statistic alone confounds two distinct scenarios and is not a good indicator of the “difficulty level” of the data set. In contrast, when the error rates of the phase prediction were stratified according to the number of homozygotes present in the sample, the performances of each algorithm in the five simulation models were similar, thereby implying that the sample homozygosity is a good indicator for the difficulty level of the data set for all the algorithms. Figures 2C and D display the pooled results of all simulations.
It is worthwhile to emphasize that, although the PL and EM methods make an explicit assumption of HWE, these two methods were, in fact, much less affected by strong departures from HWE than was either Clark's algorithm or the PGS algorithm, despite the fact that Clark's algorithm did not make an explicit HWE assumption. Clark’s algorithm underperformed other methods in the case in which too few homozygotes were present in the population—but its performance improved more rapidly than the others with the increase of homozygotes in the sample, and it outperformed the PGS when there was a significant proportion of homozygous individuals.
ACE Data Set
The ACE gene is an intriguing candidate in pharmacogenetic studies of the widely used ACE inhibitors for treatment of essential hypertension and congestive heart failure (Niu et al., in press). The human ACE gene contains 26 exons, with exons 4–11 and 17–24 encoding two homologous domains of the ACE molecule that are highly similar in both size and sequence, indicating the occurrence of a gene duplication event during the evolutionary process. Rieder et al. (1999) completed the genomic sequencing of ACE for 11 subjects. A total of 78 varying sites in 22 chromosomes were identified over a genomic region of >15 kb, and data on 52 biallelic markers are available (Rieder et al. 1999).
To test the stability of the algorithms, we performed 100 runs for each algorithm, which are presented in table 1. Since the EM algorithm is limited in the number of heterozygous loci allowable in the genotype data (the upper limit is ∼15 segregating loci), it was excluded from the comparison. Among the three algorithms, on average, the PL algorithm yielded the lowest error rate, and the PGS yielded the highest error rate (table 1). The high error rate yielded by the PGS algorithm was perhaps because the coalescence model may not be suitable for the heterogeneous genotype data for both European American and African American subjects.
Table 1.
Comparison of Average Error Rates of the PL Algorithm, the PGS Algorithm, and Clark’s Algorithm, for Two Real Data Sets
|
Average Error Rate (Standard Error) forb |
||
| Algorithma | ACEc (L = 52) | CFTRd (L = 23) |
| PL | .19 (.003) | .39 (.008) |
| PGS | .36 (.004) | .48 (.009) |
| Clark's | .27 (.000) | .47 (.018) |
The EM algorithm was excluded from the comparison because it cannot handle more than 15 heterozygous loci in the data.
Average error rates were defined as the number of erroneous phase calls divided by the total number of phase calls.
Average error rates were obtained by 100 independent runs of each algorithm.
Average error rates were for 100 data sets generated by randomly permuting 56 of the 57 complete haplotypes reported by Kerem et al. (1989).
Analysis with Incomplete Marker Information
To assess the accuracy of the PL algorithm in the presence of missing data, we simulated 100 data sets. Each data set was almost identical to the genotype data of the 11 individuals from the ACE data set, except that, for every marker of every individual, there is a 1% or 2% probability, respectively, of missing values. Among all the markers with missing values, 50% miss two alleles, and 50% miss one allele. The average number of incorrectly phased individuals was 3.2 when there was a 1% probability that a marker was missing and 4.0 when there was a 2% probability that a marker was missing, in comparison to 2.1, 4.0, and 3.0—for the PL algorithm, the PGS algorithm, and Clark's algorithm, respectively—when there was no missing data. The results suggested that the PL algorithm performs stably in the presence of missing data, but extra caution should be exercised. Markers with nonrandom patterns of typing failures should be redesigned or should be dropped from the genotyping set.
Cystic Fibrosis Transmembrane-Conductance Regulator (CFTR) Gene Data Set
Cystic fibrosis is one of the most common autosomal recessive disorders affecting whites, with an incidence of 1 case per 2,000 births. A 3-bp deletion in the open reading frame (ΔF508) has been identified in the CFTR gene on chromosome 7q31, and this mutation accounts for >60% of all affected individuals. Kerem et al. (1989) collected data on 23 SNPs in a 1.8-Mb candidate region on chromosome 7q31 from affected individuals and from healthy control subjects, and this data set has been analyzed by many haplotype-based LD methods (for more references, see Liu et al. [2001]). We took the subset of 57 haplotypes with no missing data from the 94 experimentally identified disease haplotypes in Kerem et al. (1989). These haplotypes were randomly permuted 100 times to form 100 data sets of 28 hypothetical individuals. The PL algorithm, the PGS algorithm, and Clark's algorithm were applied to each of the data sets. The average error rates determined by the three algorithms are shown in table 1. On average, the PL algorithm produced a significantly lower error rate than the other two algorithms applied, although all mean error rates were >30%. To illustrate how each algorithm performed in each simulated data set, figure 3 also presents a box plot for the error differences between the PL algorithm and other algorithms. A reason for the poor performances of the three algorithms is presumably the excessive number (29) of distinct haplotypes in a small population (only 28 individuals).
Figure 3.
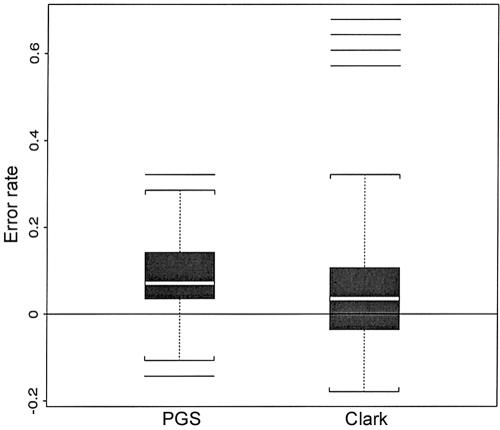
Box plots of δA=EA-EPL, where EA and EPL denote numbers of erroneous phase calls made by algorithm A (the PGS algorithm or Clark’s algorithm) and the PL algorithm, respectively, in each data set. The higher the value the worse algorithm A is in comparison to the PL algorithm. One hundred data sets were simulated; each set consisted of 28 hypothetical individuals whose genotypes were generated by randomly permuting 56 of the 57 complete haplotypes of the 23 linked SNPs near the CFTR gene provided by Kerem et al. (1989).
5q31 Data Set and Recombination Hot Spot
A subset of the haplotype data from the study by Daly et al. (2001) at 5q31 were used in our simulations to mimic the presence of recombination hotspots in the region. Between block 3 (with 9 component loci) and block 4 (with 11 component loci), shown in figure 2 of the study by Daly et al. (2001), there is a recombination hotspot with an estimated haplotype exchange rate of 33%. By using the site of the hotspot as the reference point, we generated new haplotypes with 20 loci by picking the left segment (i.e., block 3) and the right segment (i.e., block 4) independently with the frequencies reported by Daly et al. (2001). For convenience, we discarded rare haplotypes from these two blocks and normalized their common haplotype frequencies to 1. For each trial, we generated 40 haplotypes this way and formed 20 individuals. We are interested in seeing whether the PL method can, by use of the partition intentionally directed at the hotspot, perform better than that using the regular (default) partition. Among the 1,000 repeated simulations, the hot-spot cut (9|11 partition) outperformed the regular partition (10|10 partition) in 199 cases; the regular partition performed better in 42 cases; and the two methods produced identical results in the remaining 759 cases. The total number of incorrect predictions was reduced by 5% by use of the hot-spot cut. This shows that the PL method is insensitive to the presence of hotspots as long as they do not produce too many candidate partial haplotypes. In other words, the regular partition procedure can only lose accuracy if some “good” partial haplotypes are discarded prematurely owing to an overwhelmingly large number of possibilities. In such extreme circumstances, perhaps no algorithms can work well.
Population-Theory–Based Simulations
Simulation of the bottleneck effect.—In this scheme, we simulated genotype data sets to mimic the bottleneck phenomenon (Reich et al. 2001). In the simulation, the population was evolved from 30 common ancestors ∼1,000 years ago. During evolution, each individual’s two haplotypes were randomly selected from its parental haplotype population, allowing for the occurrences of recombination and mutational events (see the legend for fig. 4). The parameters used in our model were set to be comparable to those estimated from the European population (Liu et al. 2001). As shown in figure 4, the PL algorithm performed the best across various numbers of loci. The results obtained by use of Clark’s algorithm exhibited substantial variations in performance, whereas the PGS algorithm yielded the highest average error rates when the total number of loci was large (e.g., L=80 or 160).
Figure 4.
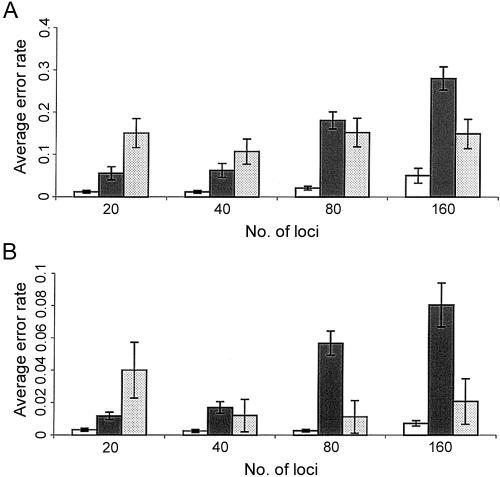
Histograms of average error rates (number of erroneous phase calls divided by the total number of phase calls) for simulations based on the bottleneck model. We generated 100 independent data sets, each of which consisted of n pairs of unphased chromosomes with L linked SNPs. The chromosomes in each data set are drawn randomly from a simulated population of the 102d-generation descendants of a founder group of 30 ancestors (with mutation rate 10-5 and crossover rate 10-3 per generation). The growth rate for the first two generations was 2.0, and that for the remaining generations was 1.05. The error bars are shown as ±1 standard error. The error rates of the PL algorithm (open bars), of the PGS algorithm (shaded bars), and of Clark’s algorithm (dotted bars), for L=20, 40, 80, 160 and for n=20 (A) and n=40 (B), respectively.
Simulation based on coalescence model.—In this scheme, samples of gametes were generated according to a neutral mutation-drift model with recombination (Hudson 1991; Long Lab Web site). For each simulated data set, the number of gametes, the number of polymorphic sites, and the size of the region being considered in units of 4Nc were explicitly specified (see the legend for fig. 5), and a total of 100 replications were made for each parameter setting. Because the data were generated according to the coalescence model on which the iterative update formula of the PGS is based, the PGS of Stephens et al. (2001b) performed the best among the four algorithms tested. The PL algorithm was a close second (fig. 5).
Figure 5.
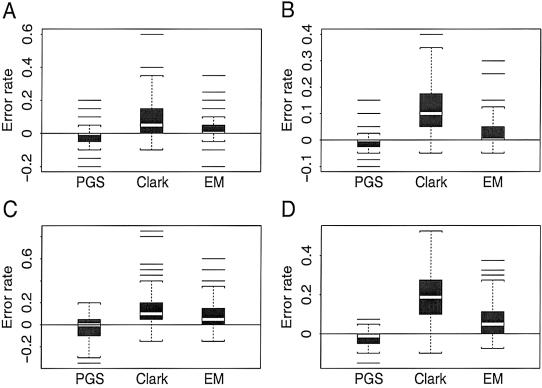
Box plots of δA=EA-EPL, where EA and EPL refer to the numbers of erroneous phase calls made by algorithm A (the PGS algorithm, Clark’s algorithm, or the EM algorithm) and the PL algorithm, respectively, for each simulated data set. All the simulated data sets were based on the coalescence model and were obtained from the Simulation Gametes program of the Long Lab. A total of 100 replications were performed for a regional size of 10 units of 4Nc, each of which consisted of n pairs of unphased chromosomes with L linked SNP loci. A, L=8, and n=20. B, L=8, and n=40. C, L=16, and n=20. D, L=16, and n=40.
Discussion
The mapping of genes that contribute to complex diseases such as breast cancer, diabetes, osteoporosis, and hypertension will be a major challenge during the postgenome era (Risch 2000). It is becoming more and more clear to researchers that, instead of testing SNPs one at a time, the haplotype-centric approach is crucial to the detection of susceptibility genes, especially when allelic heterogeneity is present (Daly et al. 2001; Pritchard 2001). The determination of haplotypes for a large number of linked SNPs by experimental methods can be very expensive, if not infeasible. With the growing speed and efficiency of SNP identification and profiling, computational methods are perhaps the only practical means for large-scale haplotype determinations, and they will continue to play an essential role in the mapping of complex traits.
The existing algorithms have strengths and weaknesses. Despite its simplicity and its dependence on the order of the individuals in the data set, Clark's parsimony algorithm is intuitively appealing and effective when the data set to which it is applied contains a sufficient number of homozygous individuals. The EM algorithm has been shown to be accurate in the inference of common haplotypes (Tishkoff et al. 2000; Zhang et al. 2001), but it cannot handle a large number of SNPs. The PGS algorithm updates each person's haplotype pair, zi, by drawing from Pi(zi|yi,Z-i), a distribution crafted on the basis of the coalescence theory. As mentioned by Stephens et al. (2001b), the Pis do not correspond to the conditionals of a proper joint distribution (which is why the method is named the “pseudo-Gibbs sampler”). Therefore, although the induced Markov chain in the PGS is still geometrically convergent, its stationary distribution may depend on the order of the yis (Gelman and Speed 1993), and the pseudoposterior probabilities attached to the constructed haplotypes become difficult to interpret. Although the PGS provides a guide for each locus in the haplotype, whether this position is difficult to infer, it lacks a measure of the overall goodness of the constructed haplotypes, thereby making it difficult to compare outputs generated from multiple runs. Nevertheless, the PGS provides an appealing strategy for the incorporation of evolution effects in haplotype construction. Our simulations showed that the PGS is advantageous when the population conforms to the coalescence assumption.
We proposed a novel Bayesian Monte Carlo method with the underlying statistical model similar to that of the EM. Two computational techniques—prior annealing and PL—were utilized in our algorithm. Prior annealing helps the algorithm escape from a local maximum. PL helps construct the whole haplotype by recursively stacking haplotype segments. This “block-by-block” strategy not only allows the handling of a large number of SNPs, but also deals with the local-mode problem better than the “piece-by-piece” strategy of the PGS. The resulting PL algorithm was compared with the PGS algorithm, the EM algorithm, and Clark's algorithm, by use of both real data and data simulated under different scenarios. Overall, the PL algorithm is robust; it outperformed other algorithms in all real data applications and was second to the PGS algorithm only in the coalescence-based simulation. The PL algorithm also performed adequately in the presence of a small percentage of missing marker data.
The PL algorithm, similar to the EM algorithms, assumes HWE and random mating, which is appropriate to populations of large sizes that have existed for a long period of time. Both Stephens et al. (2001b) and Fallin and Schork (2000) performed assessments of their algorithms when HWE was violated and concluded that the impact on their algorithms was not dramatic. We compared the performances, under five different levels of HWE violations, of the PL algorithm, the PGS algorithm, Clark's algorithm, and the EM algorithm and found that the PL and EM algorithms exhibited stronger algorithmic stability than the PGS algorithm and Clark's algorithm. Contrary to some common wisdom, Clark’s algorithm was most vulnerable to the violation of HWE, although there is no HWE assumption in its derivation. In a study of the effect of population mixture (another way in which HWE may be violated), we simulated 100 data sets, each consisting of three independent subgroups of 10 individuals generated from a coalescence model under HWE. The PL algorithm performed marginally better than the PGS, despite that the PGS is rooted for the coalescence model (data not shown).
Our simulations based on the 5q31 data suggested that the partitioning step was not sensitive to the presence of recombination hotspots, although knowing and partitioning at the hotspot can yield marginal improvement. Daly et al. (2001) showed that, despite the presence of local hotspots, there is still clear long-range LD among blocks, thereby suggesting that the majority of the recombination hotspots are moderate. Thus, the PL algorithm should perform even more robustly in real cases than in our simulations. Indeed, Farrall et al. (1999) reported that the ACE 52-locus haplotype analyzed in the “Results” section has a recombination hotspot located between loci 9 and 12. The partition sites of the PL algorithm in the vicinity of this recombination hotspot, however, follow immediately after marker loci 6 and 13. As shown by our results, the accuracy of the PL algorithm was not compromised. Since the recombination hotspots are generally not known in advance, it is of interest to develop an automated procedure to simultaneously screen for “stable” blocks of low haplotype diversity and conduct PL.
The PL algorithm was implemented in American National Standards Institute C++ and was compiled on the Red Hat Linux operating system by use of a Dell PowerEdge 4400 server with twin 866-MHz Pentium Xeon processors. The PL algorithm runs about three times faster than the PGS algorithm (implemented as PHASE with a moderate number of iterations). In our simulation studies of the bottleneck effect, for L=20, 40, 80, and 160 loci and n=20, the central-processing-unit times were ∼2.3, ∼6.1, ∼11.6, and ∼25.9 s, respectively; for L=20, 40, 80, and 160 loci and n=40, the central-processing-unit times were ∼3.8, ∼9.2, ∼18.9, and ∼36.5 s, respectively. It is noted that the data complexity for the same number of SNPs can still vary tremendously, since the amount of LD present across the genomic region can vary dramatically. For a sample of 100 individuals, our software currently can handle 256 SNPs; for a sample of 1,000 individuals, our software can handle 50 SNPs. Our software outputs not only the putative haplotypes but also measures of their accuracies, as well as the overall likelihood. The user can conduct multiple runs and select the result with the highest likelihood. Nevertheless, statistical methods are exploratory tools, and, especially for those individuals with large posterior uncertainties, it would be prudent to validate the haplotypes inferred by algorithms by use of molecular-haplotyping techniques.
Once haplotypes are constructed, various statistical methods can be applied to detect haplotype-disease associations and to cluster/classify patients. These include the χ2 test, the likelihood-ratio test (Fallin et al. 2001), logistic regression (Wallenstein et al. 1998), cladistic analysis (Templeton 1995; Heng and Low 2000), and the haplotype pattern-mining method (Toivonen et al. 2000). We believe that, by coupling with some haplotype-based LD analysis, the utility of our method may have significant implications in positional cloning for complex traits.
Acknowledgments
We thank A. G. Clark and J. Long, for providing their haplotype-estimation software, and the two anonymous reviewers, for their suggestions. We are grateful to J.-B. Fan, A. Speak, P. Attie, and Z. Hu, for helpful discussions and comments. This work was supported in part by National Institutes of Health grant 1R01 HL/AI 56371-01A1 and National Science Foundation grants DMS-9803649 and DMS-0094613.
Appendix A
Dirichlet Distribution
We say that a random vector X=(X1,…,Xn) follows the Dirichlet distribution Dirichlet(β1,…,βn) if its density is of the form
and Γ(β) is the Γ function. Thus, it is necessary that Xi⩾0 and X1+⋅⋅⋅+Xn=1, thereby implying that it is a random probability vector. A simple property of this distribution is that 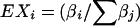 .
.
Software Programs
HAPLOTYPER implements the PL Gibbs sampling method as described in this article. Sample input and output files for HAPLOTYPER are provided in figure A1.
Figure A1.

A, Input file format for HAPLOTYPER. Each line in the input file represents the marker data for each subject; in each line, each SNP occupies one space, and no white spaces are allowed between the neighboring loci. For each SNP, “0” denotes heterozygote, “1” denotes homozygous wild type, “2” denotes homozygous mutant, “3” denotes that both alleles were missing, “4” denotes that only the wild-type allele—“(A,*)”—was known (in the notation, “A” denotes the wild-type allele, and “*” denotes the unknown allele), and “5” denotes that only the mutant allele was known. B, Output file format for HAPLOTYPER. The output file consists of two parts: The first part lists the two predicted haplotypes with their individual identification designations and the associated posterior probabilities. The second part is the summary of the overall haplotype frequency estimated from this sample. If the number of SNPs is >20, we also included a haplotype code (shown in parentheses), which is a decimal number converted from the binary sequence of the haplotype configuration (e.g., haplotype 101 is converted to 22+20=5).
EM-DeCODER implements the EM algorithm for haplotype constructions as described in this article. Its input and output file formats are the same as those of HAPLOTYPER.
HaplotypeManager is a graphical user interface for displaying the haplotype data and for visualizing the haplotype distributions that were implemented using Java Development Kit v1.0.
Example data files and documentation for HAPLOTYPER, EM-DeCODER, and HaplotypeManager are available from Jun Liu's Home Page.
Electronic-Database Information
URLs for data in this article are as follows:
- Jun Liu's Home Page, http://www.people.fas.harvard.edu/~junliu/ (for example data files and documentation for HAPLOTYPER, EM-DeCODER, and HaplotypeManager)
- Long Lab, http://hjmuller.bio.uci.edu/~labhome/coalescent.html (for coalescent-process tools)
- Mathematics Genetics Group, http://www.stats.ox.ac.uk/mathgen/software.html (for PHASE)
References
- Akey J, Jin L, Xiong M (2001) Haplotypes vs single marker linkage disequilibrium tests: what do we gain? Eur J Hum Genet 9:291–300 [DOI] [PubMed] [Google Scholar]
- Beaudet L, Bedard J, Breton B, Mercuri RJ, Budarf ML (2001) Homogeneous assays for single-nucleotide polymorphism typing using AlphaScreen. Genome Res 11:600–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw MS, Bollekens JA, Ruddle FH (1995) A new vector for recombination-based cloning of large DNA fragments from yeast artificial chromosomes. Nucleic Acids Res 23:4850–4856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Liu JS (1996) Predictive updating methods with application to Bayesian classification. J R Stat Soc Ser B 58:397–415 [Google Scholar]
- Chiano MN, Clayton DG (1998) Fine genetic mapping using haplotype analysis and the missing data problem. Ann Hum Genet 62:55–60 [DOI] [PubMed] [Google Scholar]
- Clark AG (1990) Inference of haplotypes from PCR-amplified samples of diploid populations. Mol Biol Evol 7:111–122 [DOI] [PubMed] [Google Scholar]
- Cox D, Boillot C, Canzian F (2001) Data mining: efficiency of using sequence databases for polymorphism discovery. Hum Mutat 17:141–150 [DOI] [PubMed] [Google Scholar]
- Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES (2001) High-resolution haplotype structure in the human genome. Nat Genet 29:229–232 [DOI] [PubMed] [Google Scholar]
- Dempster AP, Laird NM, Rubin DB (1977) Maximum likelihood from incomplete data via EM algorithm. J R Stat Soc Ser B 39:1–38 [Google Scholar]
- Douglas JA, Boehnke M, Gillanders E, Trent JM, Gruber SB (2001) Experimentally-derived haplotypes substantially increase the efficiency of linkage disequilibrium studies. Nat Genet 28:361–364 [DOI] [PubMed] [Google Scholar]
- Drysdale CM, McGraw DW, Stack CB, Stephens JC, Judson RS, Nandabalan K, Arnold K, Ruano G, Liggett SB (2000) Complex promoter and coding region β2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. Proc Natl Acad Sci USA 97:10483–10488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Slatkin M (1995) Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol 12:921–927 [DOI] [PubMed] [Google Scholar]
- Fallin D, Cohen A, Essioux L, Chumakov I, Blumenfeld M, Cohen D, Schork NJ (2001) Genetic analysis of case/control data using estimated haplotype frequencies: application to APOE locus variation and Alzheimer's disease. Genome Res 11:143–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallin D, Schork NJ (2000) Accuracy of haplotype frequency estimation for biallelic loci, via the expectation-maximization algorithm for unphased diploid genotype data. Am J Hum Genet 67:947–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan JB, Chen X, Halushka MK, Berno A, Huang X, Ryder T, Lipshutz RJ, Lockhart DJ, Chakravarti A (2000) Parallel genotyping of human SNPs using generic high-density oligonucleotide tag arrays. Genome Res 10:853–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrall M, Keavney B, McKenzie C, Delépine M, Matsuda F, Lathrop GM (1999) Fine-mapping of an ancestral recombination breakpoint in DCP1. Nat Genet 23:270–271 [DOI] [PubMed] [Google Scholar]
- Gelman A, Speed TP (1993) Characterizing a joint probability distribution by conditionals. J R Stat Soc Ser B 55:185–188 [Google Scholar]
- Hacia JG, Fan JB, Ryder O, Jin L, Edgemon K, Ghandour G, Mayer RA, Sun B, Hsie L, Robbins CM, Brody LC, Wang D, Lander ES, Lipshutz R, Fodor SP, Collins FS (1999) Determination of ancestral alleles for human single-nucleotide polymorphisms using high-density oligonucleotide arrays. Nat Genet 22:164–167 [DOI] [PubMed] [Google Scholar]
- Hawley ME, Kidd KK (1995) HAPLO: a program using the EM algorithm to estimate the frequencies of multi-site haplotypes. J Hered 86:409–411 [DOI] [PubMed] [Google Scholar]
- Heng CK, Low PS (2000) Cladistic analysis: its applications in association studies of complex diseases. Ann Acad Med Singapore 29:313–321 [PubMed] [Google Scholar]
- Hodge SE, Boehnke M, Spence MA (1999) Loss of information due to ambiguous haplotyping of SNPs. Nat Genet 21:360–361 [DOI] [PubMed] [Google Scholar]
- Hudson RR (1991) Gene genealogies and the coalescent process. In: Futuyma D, Antonovics J (eds) Oxford surveys in evolutionary biology, vol 7. Oxford University Press, Oxford, pp 1–44 [Google Scholar]
- Israel E, Drazen JM, Liggett SB, Boushey HA, Cherniack RM, Chinchilli VM, Cooper DM, Fahy JV, Fish JE, Ford JG, Kraft M, Kunselman S, Lazarus SC, Lemanske RF, Martin RJ, McLean DE, Peters SP, Silverman EK, Sorkness CA, Szefler SJ, Weiss ST, Yandava CN (2000) The effect of polymorphisms of the β2-adrenergic receptor on the response to regular use of albuterol in asthma. Am J Respir Crit Care Med 162:75–80 [DOI] [PubMed] [Google Scholar]
- Judson R, Stephens JC (2001) Notes from the SNP vs haplotype front. Pharmacogenomics 2:7–10 [DOI] [PubMed] [Google Scholar]
- Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, et al (1989) Identification of the cystic fibrosis gene: genetic analysis. Science 245:1073–1080 [DOI] [PubMed] [Google Scholar]
- Lander ES, Botstein D (1987) Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science 236:1567–1570 [DOI] [PubMed] [Google Scholar]
- Lawrence CE, Altschul SF, Boguski MS, Liu JS, Neuwald AF, Wootton JC (1993) Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science 262:208–214 [DOI] [PubMed] [Google Scholar]
- Liu JS (1994) The collapsed Gibbs sampler in Bayesian computations with applications to a gene regulation problem. J Am Stat Assoc 89:958–966 [Google Scholar]
- Liu JS (2001) Monte Carlo strategies in scientific computing. Springer-Verlag, New York [Google Scholar]
- Liu JS, Sabatti C, Teng J, Keats BJ, Risch N (2001) Bayesian analysis of haplotypes for linkage disequilibrium mapping. Genome Res 11:1716–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizardi PM, Huang X, Zhu Z, Bray-Ward P, Thomas DC, Ward DC (1998) Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nat Genet 19:225–232 [DOI] [PubMed] [Google Scholar]
- Long JC, Williams RC, Urbanek M (1995) An E-M algorithm and testing strategy for multiple-locus haplotypes. Am J Hum Genet 56:799–810 [PMC free article] [PubMed] [Google Scholar]
- Martinez FD, Graves PE, Baldini M, Solomon S, Erickson R (1997) Association between genetic polymorphisms of the β2-adrenoceptor and response to albuterol in children with and without a history of wheezing. J Clin Invest 100:3184–3188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalatos-Beloin S, Tishkoff SA, Bentley KL, Kidd KK, Ruano G (1996) Molecular haplotyping of genetic markers 10 kb apart by allele-specific long-range PCR. Nucleic Acids Res 24:4841–4843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu T, Chen X, Xu X. Angiotensin converting enzyme gene insertion/deletion polymorphism and cardiovascular disease: therapeutic implications. Drugs (in press) [DOI] [PubMed] [Google Scholar]
- Niu T, Seielstad M, Zeng X, Apffel A, Li G, Hahnenberger K, Xu X (2001) Detection of novel ALAD gene polymorphisms using denaturing high-performance liquid chromatography. Hum Biol 73:429–442 [DOI] [PubMed] [Google Scholar]
- Pritchard JK (2001) Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet 69:124–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranade K, Chang MS, Ting CT, Pei D, Hsiao CF, Olivier M, Pesich R, Hebert J, Chen YD, Dzau VJ, Curb D, Olshen R, Risch N, Cox DR, Botstein D (2001) High-throughput genotyping with single nucleotide polymorphisms. Genome Res 11:1262–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich DE, Cargill M, Bolk S, Ireland J, Sabeti PC, Richter DJ, Lavery T, Kouyoumjian R, Farhadian SF, Ward R, Lander ES (2001) Linkage disequilibrium in the human genome. Nature 411:199–204 [DOI] [PubMed] [Google Scholar]
- Reihsaus E, Innis M, MacIntyre N, Liggett SB (1993) Mutations in the gene encoding for the β2-adrenergic receptor in normal and asthmatic subjects. Am J Respir Cell Mol Biol 8:334–339 [DOI] [PubMed] [Google Scholar]
- Rieder MJ, Taylor SL, Clark AG, Nickerson DA (1999) Sequence variation in the human angiotensin converting enzyme. Nat Genet 22:59–62 [DOI] [PubMed] [Google Scholar]
- Risch NJ (2000) Searching for genetic determinants in the new millennium. Nature 405:847–856 [DOI] [PubMed] [Google Scholar]
- Ross P, Hall L, Smirnov I, Haff L (1998) High-level multiplex genotyping by MALDI-TOF mass spectrometry. Nat Biotechnol 16:1347–1351 [DOI] [PubMed] [Google Scholar]
- Ruano G, Kidd KK, Stephens JC (1990) Haplotype of multiple polymorphisms resolved by enzymatic amplification of single DNA molecules. Proc Natl Acad Sci USA 87:6296–6300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder SA, Gaughan DM, Swift M (1995) Protection against bronchial asthma by CFTR ΔF508 mutation: a heterozygote advantage in cystic fibrosis. Nat Med 1:703–705 [DOI] [PubMed] [Google Scholar]
- Stephens JC, Schneider JA, Tanguay DA, Choi J, Acharya T, Stanley SE, Jiang R, et al (2001a) Haplotype variation and linkage disequilibrium in 313 human genes. Science 293:489–493 [DOI] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P (2001b) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templeton AR (1995) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping or DNA sequencing. V. Analysis of case/control sampling designs: Alzheimer's disease and the apoprotein E locus. Genetics 140:403–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff SA, Pakstis AJ, Ruano G, Kidd KK (2000) The accuracy of statistical methods for estimation of haplotype frequencies: an example from the CD4 locus. Am J Hum Genet 67:518–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toivonen HT, Onkamo P, Vasko K, Ollikainen V, Sevon P, Mannila H, Herr M, Kere J (2000) Data mining applied to linkage disequilibrium mapping. Am J Hum Genet 67:133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi S, Kramer FR (1996) Molecular beacons: probes that fluoresce upon hybridization. Nat Biotechnol 14:303–308 [DOI] [PubMed] [Google Scholar]
- Wallenstein S, Hodge SE, Weston A (1998) Logistic regression model for analyzing extended haplotype data. Genet Epidemiol 15:173–181 [DOI] [PubMed] [Google Scholar]
- Woolley AT, Guillemette C, Li Cheung C, Housman DE, Lieber CM (2000) Direct haplotyping of kilobase-size DNA using carbon nanotube probes. Nat Biotechnol 18:760–763 [DOI] [PubMed] [Google Scholar]
- Zhang S, Pakstis AJ, Kidd KK, Zhao H (2001) Comparisons of two methods for haplotype reconstruction and haplotype frequency estimation from population data. Am J Hum Genet 69:906–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Zhang S, Merikangas KR, Trixler M, Wildenauer DB, Sun F, Kidd KK (2000) Transmission/disequilibrium tests using multiple tightly linked markers. Am J Hum Genet 67:936–946 [DOI] [PMC free article] [PubMed] [Google Scholar]