

Abstract
In eukaryotes, damaged or unneeded proteins are typically degraded by the ubiquitin-proteasome system. In this system, the protein substrate is often first covalently modified with a chain of ubiquitin polypeptides. This chain serves as a signal for delivery to the 26S proteasome, a 2.5 MDa, ATP-dependent multisubunit protease complex. The proteasome consists of a barrel-shaped 20S core particle (CP) that is capped on one or both of its ends by a 19S regulatory particle (RP). The RP is responsible for recognizing the substrate, unfolding it, and translocating it into the CP for destruction. Here we describe simple, one-step purifications scheme for isolating the 26S proteasome and its 19S RP and 20S CP subcomplexes from the yeast Saccharomyces cerevisiae, as well as assays for measuring ubiquitin-dependent and ubiquitin-independent proteolytic activity in vitro.
Keywords: proteasome, ubiquitin, ATPase, purification, proteolytic activity
INTRODUCTION
In eukaryotes, 80 – 90% of short-lived regulatory, misfolded or damaged intracellular proteins are degraded by the 26S proteasome (Goldberg, 2003). As the proteasome is involved in degrading a wide variety of substrates, it influences almost every cellular process (Tomko and Hochstrasser, 2013). Thus, the proteasome provides an attractive drug target for treating various diseases (Schmidt and Finley, 2014). In fact, several proteasome inhibitors either have been approved for clinical use or are currently in clinical trials (Zhang et al., 2013).
A source of pure and active proteasomes is often essential to study substrate degradation mechanisms, to dissect proteasome-regulated cellular processes, to screen for proteasome modulators, or to examine interactions of small molecules with proteasomes. Given the exceptional conservation of the proteasome from yeasts to humans, and the relative ease and low cost of yeast cell culture, the budding yeast Saccharomyces cerevisiae has often been the organism of choice for the preparation of large quantities of pure 26S proteasomes and its subcomplexes.
This unit describes methods to isolate enzymatically active 26S proteasomes and proteasome subcomplexes from S. cerevisiae by one-step affinity purification. These methods can be easily adapted to purify other proteasome subcomplexes, such as the RP subcomplexes known as lid and base, or proteasome assembly intermediates, by placing the epitope tag onto different subunits and/or by combining with additional chromatography steps. Basic Protocols 1, 2 and 3 describe one-step affinity-purification procedures for the 26S proteasome, 19S RP and 20S CP. The general approach is the same except that buffer conditions used to isolate each proteasomal species are different. Basic Protocol 4 describes how to monitor 26S and 20S CP peptidase activities in nondenaturing polyacrylamide gels. Basic Protocol 5 describes how to measure the 26S and 20S CP peptidase activities in solution. Basic Protocol 6 describes how to measure the ATP- and ubiquitin-dependent degradation activity of the 26S proteasome. Support Protocol 1 provides a general method to prepare concentrated cellular material derived from frozen yeast cells that have been ground to a fine powder, which is used for native protein purification, and Support Protocol 2 provides a method to regenerate the 3xFLAG resin used for purifying 26S proteasomes and its various subcomplexes for reuse.
BASIC PROTOCOL 1: Purification of active 26S proteasomes
Active 26S proteasomes are purified by a one-step affinity procedure that takes advantage of a triplicated FLAG peptide affinity tag (3xFLAG) that has been placed on one of the proteasome subunits. The Rpn11 RP subunit is most commonly used in yeast because C-terminal tagging of Rpn11 causes no discernable effect on proteasome function or organismal health. It is worth noting that several salt-sensitive proteins have been found associated with the yeast 26S proteasome, and this protocol will result in the loss of some (or potentially all) of these factors. If retention of these proteins is desired, we point the reader toward other protocols (Leggett et al., 2005).
Materials
Cell powder from Saccharomyces cerevisiae strain MHY5841 (RPN11-6xGly-3xFLAG:kanMX6) or similar
Buffer A (see recipe)
10x ATP regenerating system (see recipe)
ATP, Grade I (≥99%), Sigma catalog #A2383
BCA Assay Kit, Pierce catalog #23227
Anti-FLAG M2 affinity gel, Sigma catalog #A2220
3xFLAG peptide, Sigma catalog #F4799
Gelcode Blue Stain, Thermo Scientific catalog #24592
High speed centrifuge, 4 °C
40 mL high-speed centrifuge tubes and rotor
Bench-top centrifuge, 4 °C
Microcentrifuge, 4 °C
50 mL disposable polypropylene conical centrifuge tubes
Rotator or nutator for conical vials
Vivaspin 500 centrifugal concentrator (100 kDa molecular weight cutoff), Sartorius catalog #VS0141
Mini gel electrophoresis system (Bio-Rad) or similar
G-Box (Syngene) or similar gel documentation system
Protocol steps
Prepare cell powder from a 2 L YPD culture of MHY5841 as described in Support Protocol 1 (below).
Add one volume of ice cold Buffer A containing 1 mM ATP and 1x ATP-regenerating system to the cell powder. Vortex the sample vigorously to completely resuspend the cell powder in the buffer.
Incubate the mixture on ice for 10–15 minutes. Vortex the polypropylene tube at five-minute intervals for 10 seconds each during this incubation to maximize protein extraction.
Transfer the mixture to two 40 mL high-speed centrifuge tubes that have been pre-cooled on ice. Centrifuge at 30,000 × g for 20 minutes to pellet cell debris.
-
Decant the supernatant to a fresh 50 mL conical tube.
After centrifugation, there will often be a yellow-whitish layer of lipids at the top of the supernatant. If this is apparent, the supernatant should be decanted through 2–3 thicknesses of cheesecloth to remove the lipids, which may otherwise interfere with downstream steps.The cheesecloth will absorb some yeast lysate. After passing the lysate through the cheesecloth, it can be gently wrung out into the 50 mL tube to recover the absorbed lysate while retaining the lipids. -
Determine the protein concentration of the extract using the BCA assay kit.
Typically, the protein concentration will be between 5 and 20 mg/mL, depending on the quality of the cell powder and the culture density at harvest. We typically obtain 500 – 1000 mg of crude protein from 2 L of saturated yeast culture. -
Calculate the total amount of protein in the extract. Add to the extract 600 μL of a 50% slurry of anti-FLAG M2 affinity gel (equivalent to 300 μL of packed resin) per 400 mg of protein present.
Be sure to thoroughly resuspend the resin prior to pipetting it, as it is extremely viscous due to the high glycerol content, but avoid high-speed vortexing. We typically use a large-bore pipette tip when transferring the resin to avoid clogging. Incubate the tube on an end-over-end rotator for one hour at 4 °C to allow binding of proteasomes in the extract to the anti-FLAG antibodies on the resin.
After one hour, centrifuge the mixture at 1500 × g for 2 minutes at 4 °C to pellet the resin.
Carefully decant the supernatant. Resuspend the resin in 25 mL of Buffer A containing 1 mM ATP and 1x ATP-regenerating system. Incubate with end-over-end mixing at 4 °C for five minutes to remove nonspecifically bound materials.
Centrifuge mixture at 1500 × g for 2 minutes at 4 °C to pellet the resin.
Repeat steps 10 and 11 for a second wash.
After decanting the supernatant, resuspend the resin in 1 mL of Buffer A plus 1 mM ATP and 1x ATP-regenerating system, and split evenly between two 1.5 mL microcentrifuge tubes. Centrifuge the tubes at 1500 × g for 30 seconds to pellet resin, and carefully pipette out the supernatant.
-
Add 3 resin volumes of Buffer A plus 1 mM ATP and 1x ATP-regenerating system containing 100 μg/mL 3xFLAG peptide to each tube, and place on an end-over-end rotator at 4 °C for 45 minutes.
It is important to use 3xFLAG peptide rather than FLAG peptide; the 3xFLAG-tagged protein cannot be competed efficiently from the resin by the single FLAG peptide. -
Centrifuge microcentrifuge tubes at 1500 × g for 30 seconds at 4 °C to pellet resin. Collect and combine the eluates from each tube into a fresh microcentrifuge tube on ice using a Pipetteman.
It is important to avoid collecting any of the resin during this step, as this will reduce the purity of the final preparation. We often will centrifuge the eluate again after transfer to the fresh 1.5 mL microcentrifuge tube, and transfer the supernatant to a second 1.5 mL microcentrifuge tube to assure that no resin beads remain in the eluate. Transfer the eluate to a 100 kDa-cutoff Vivaspin 500 centrifugal concentrator tube. If the total volume of eluate is greater than 500 μL, then transfer 500 μL at a time to the concentrator, concentrate by centrifugation at 10,000 × g for 15 minutes at 4 °C, discard the flow-through, and add up to 500 μL of the remaining eluate to the concentrator. Repeat until all of the eluate has been added to the concentrator. Invert the concentrator to mix the concentrated retentate with the newly added eluate between concentration runs. This minimizes the chance of precipitation of the proteins during concentration. Concentrate the total eluate to approximately 50–100 μL.
-
Once the entire eluate has been concentrated, transfer the retentate to a pre-chilled 1.5 mL microcentrifuge tube.
It is important to thoroughly mix the retentate by pipetting up and down carefully several times prior to pipetting from the concentrator housing to maximize recovery of concentrated proteasomes. Avoid foaming, which damages proteins. To estimate the final concentration of proteasomes, run 0.5 μL of the purified proteasomes on a 12% SDS-PAGE gel made as Table 1 with a dilution series of bovine serum albumin (BSA) standards of 750, 500, 250, and 125 ng and stain the gel with Gelcode Blue (Figure 1). Quantify the BSA band intensities and the intensity of a band from an individual subunit of the proteasome. For 26S proteasome or RP purifications, we use the Rpn3 RP subunit because it has a unique migration at ~60 kDa that is well separated from other subunits; for 20S CP purifications we usually choose the Pre10/α7 CP subunit (~32 kDa) for this purpose. The concentration can then be derived from the equation of the line formed by the BSA standards, the molecular mass of the chosen subunit, and the known volume of proteasomes added to the gel.
-
Dilute proteasomes to the desired concentration using Buffer A containing 1 mM ATP. Make 5–10 μL aliquots, and snap-freeze in liquid nitrogen. Store proteasome aliquots at −80°C.
Dilution of proteasomes to 1 μM is convenient for degradation assays (See BASIC Protocol 6).
Table 1.
Recipe to make two 12% 1 mm SDS-PAGE gels.
| Resolving gel, 12% acrylamide | Stacking gel, 4% acrylamide | ||||
|---|---|---|---|---|---|
| solution | [Stock] | 15 mL | solution | [Stock] | 5 mL |
| H2O | 4.23 mL | H2O | 3.541 mL | ||
| Tris-HCl, pH 8.8 | 1.5 M | 3.75 mL | Tris-HCl, pH 6.8 | 1 M | 625 μL |
| acrylamide | 40% | 4.38 mL | acrylamide | 40% | 487 μL |
| bis-acrylamide | 2% | 2.4 mL | bis-acrylamide | 2% | 267 μL |
| SDS | 20% | 75 μL | SDS | 20% | 25 μL |
| APS | 10% | 150 μL | APS | 10% | 50 μL |
| TEMED | 15 μL | TEMED | 5 μL | ||
Figure 1.

Purified yeast 20S CP and 26S proteasome subunits resolved by electrophoresis through a 12% SDS-polyacrylamide gel. The 20S CP and 26S proteasomes were purified as described in Basic Protocol 3 and Basic Protocol 1, respectively. The gel was stained with Gelcode Blue at room temperature for one hour. Lane 1 contains molecular weight markers. Lanes 2 to 5 are BSA standards with, respectively, 750 ng, 500 ng, 250 ng and 125 ng BSA loaded per lane. Lane 6 is loaded with 2.6 μg of purified 20S proteasome (CP). Lane 7 is loaded with 7.8 μg of purified 26S proteasomes. The 20S proteasome is purified through Pre1 (β4)-3xFLAG, which has a molecular weight shift from 22.5 kDa to 25.3 kDa. It migrates together with other subunits at around 26 kDa, shown in Lane 6 as a dark band that poorly separated. The 26S proteasome is purified through Rpn11–3xFLAG, which has a molecular weight shift from 34.4 kDa to 37.2 kDa. It migrates together with Rpn8 and Rpn9 and can not be distinguished in a 12% SDS-PAGE gel as shown in Lane7. Rpn3 (molecular weight 60377.3 Da) and Pre10 (α7, molecular weight 31521.3 Da) are labeled as our choice of the subunits for quantification.
SUPPORT PROTOCOL 1: Growth of yeast strains and production of yeast cell powder
This protocol is used to prepare the concentrated frozen yeast cell powder for use in all the proteasome complex purifications.
Materials
Saccharomyces cerevisiae strain MHY5841 (RPN11-6xGly-3xFLAG:kanMX6), MHY6952 (PRE1-6xGly-3xFLAG:kanMX6) or similar
YPD agar plate (see recipe)
YPD liquid medium (see recipe)
Ice-cold deionized water (diH2O)
Shaker-incubator, set to 30 °C
High speed centrifuge, 4 °C
Bench-top centrifuge, 4 °C
Ceramic mortar, 80 mm × 130 mm, VWR catalog #89038-152 or equivalent
Ceramic pestle, 194 mm, VWR catalog #89038-168 or equivalent
Liquid nitrogen
Hammer
Spatula or Spoon
Large (~500 mL) and small (~40 mL) high-speed centrifuge tubes and rotors
50 mL disposable polypropylene conical centrifuge tubes
Protocol steps
Revive strain MHY5841 (for 26S proteasome and 19S RP purification) or MHY6952 (for 20S CP purification) and grow by streaking from a glycerol stock onto a YPD plate and incubating for 2–3 days at 30 °C until colonies arise.
Inoculate 20 mL of YPD liquid medium with a single yeast colony and grow overnight in a shaker-incubator at 30 °C with agitation at 200 rpm.
Prepare two 4L flasks each containing 1 L of YPD liquid medium. Inoculate each flask with 10 mL of the overnight culture. Grow for approximately 48 hours to saturation in a shaker at 30 °C with agitation at 200 rpm.
Divide the culture among the requisite number of 500 mL centrifuge tubes, and harvest the culture via centrifugation in a high-speed centrifuge at 4 °C for five minutes at 5000 × g. Decant the supernatants.
Resuspend the cell pellets and combine them in a total of 25 mL of ice-cold diH2O, and transfer to a labeled, pre-chilled (4 °C) 50 mL conical vial.
Centrifuge the cells in a benchtop centrifuge at 4 °C for 5 minutes at 5000 × g. Decant the supernatant.
Resuspend the cell pellet again in 25 mL of ice-cold diH2O, and centrifuge in a benchtop centrifuge at 4 °C for 5 minutes at 5000 × g. Decant the supernatant.
-
Store the cells by snap-freezing the conical vial in liquid nitrogen, followed by transfer to −80 °C.
The frozen cell pellet can be stored at −80 °C for at least 3 months with no appreciable loss of quality or yield. -
Chill the mortar and pestle by placing the pestle in the mortar and pouring liquid nitrogen into the mortar until it is approximately half full. The liquid nitrogen will boil rapidly until the mortar cools sufficiently.
It is important for the mortar, pestle, and any tools that will contact the frozen cells to be pre-chilled and remain at near-liquid nitrogen temperature, or the cells will stick to the warm surface. Retrieve the cell pellet from the polypropylene tube by wrapping the liquid-nitrogen frozen tube in a paper towel, and hitting with a hammer to shatter the tube. Pick the pieces of the cell pellet out of the tube with a pre-chilled spatula and place into the pre-chilled mortar.
-
Using gentle pressure and a rapid rotation of the pestle, mill the frozen cells into a fine powder with the consistency of flour. Cool the mortar approximately every two minutes (or as needed) to keep the sample cold.
Finely ground powder maximizes cell lysis and thus protein extraction. It is often helpful to use the hammer to reduce the size of large cell pellet fragments prior to milling in the mortar. -
Once a finely milled powder is obtained, transfer the powder to a 50 mL conical tube pre-chilled in liquid nitrogen using a pre-chilled Scoopula or spoon. Note the resultant volume of cell powder (see below).
The cell powder can be stored at −80 °C indefinitely.
SUPPORT PROTOCOL 2: Regeneration of anti-FLAG M2 affinity gel
The anti-FLAG M2 affinity gel can be used for purification at least five times if regenerated and stored as directed below. The procedure can be carried out in batch mode or on a column; we provide a batch-mode procedure for regeneration. To regenerate the gel, the resin is washed briefly in a solution of glycine, pH 3.5 to release the 3xFLAG peptide from the antibody. The resin is then washed extensively with Buffer A to neutralize the pH and is stored at 4 °C until use.
Materials
Buffer A (see recipe)
0.1 M glycine, pH 3.5
1% (w/v) NaN3 in diH2O
Protocol steps
Resuspend the affinity gel with three resin volumes of Buffer A and transfer to a 15 mL conical polypropylene vial.
Centrifuge at 1500 × g for 2 minutes to pellet the resin and aspirate the supernatant.
-
Resuspend the resin in three resin volumes of 0.1 M glycine, pH 3.5 and incubate on the benchtop for 15 minutes.
It is important not to exceed 15 minutes in the 0.1 M glycine, pH 3.5, or the FLAG antibody will begin to permanently denature. Centrifuge the resin as in step 2, and again aspirate the supernatant.
Resuspend the resin in three resin volumes of Buffer A and repeat centrifugation as in step 2.
-
Aspirate the supernatant, and resuspend resin in one resin volume of Buffer A. Test the pH of the buffer by placing a small drop on a pH strip. It should register approximately pH 7.5.
The pH of Buffer A increases with decreasing temperature. If using refrigerated Buffer A, the pH may read closer to 8.5 using pH strips; this is acceptable. If the pH is ~7.5, then proceed to step 8. If it is less than this, then repeat steps 5 and 6 until pH 7.5 is achieved.
-
Add 1% NaN3 to a final concentration of 0.01% as a preservative. Store the resin at 4°C.
Alternatively, the resin can be resuspended in Tris-buffered saline with 50% glycerol, and stored at −20°C.
BASIC PROTOCOL 2: Purification of the 19S regulatory particle (RP)
The RP can be purified using a variation of the protocol for purification of the 26S proteasome. The lysis, purification and wash steps are performed in a buffer containing 500 mM NaCl. The high salt concentration disrupts the interaction between the RP and the CP, allowing the RP to be purified away from CP. Note that the isolated RP does not appear to have appreciable deubiquitinating or unfolding activity toward the polyubiquitinated Sic1-PY substrate protein, at least when isolated under these conditions (See Basic Protocol 6); these activities might be dependent upon the CP in yeast. The purification procedure is identical to that for the 26S proteasome (Basic Protocol 1) for steps 1 to 12, except that Buffer A500 (supplemented with 1 mM ATP and 1x ATP-regenerating system) is substituted for Buffer A for all steps until the elution of the bound complexes, at which point Buffer A supplemented with ATP and ATP-regenerating system is used to lower the final NaCl concentration prior to storage.
Materials
Cell powder from Saccharomyces cerevisiae strain MHY5841 (RPN11-6xGly-3xFLAG:kanMX6) or similar
Buffer A (see recipe)
Buffer A500 (see recipe)
10x ATP regenerating system (see recipe)
ATP, Grade I (≥99%), Sigma catalog #A2383
BCA Assay Kit, Pierce catalog #23227
Anti-FLAG M2 affinity gel, Sigma catalog #A2220
3xFLAG peptide, Sigma catalog #F4799
Gelcode Blue Stain, Thermo Scientific catalog #24592
High speed centrifuge, 4 °C
40 mL high-speed centrifuge tubes and rotor
Bench-top centrifuge, 4 °C
Microcentrifuge, 4 °C
50 mL disposable polypropylene conical centrifuge tubes
Rotator or nutator for conical vials
Vivaspin 500 centrifugal concentrator (100 kDa molecular weight cutoff), Sartorius catalog #VS0141
Mini gel electrophoresis system (Bio-Rad) or similar
G-Box (Syngene) or similar gel documentation system
Protocol steps
Prepare cell powder from a 2 L YPD culture of MHY5841 as described in Support Protocol 1 (above).
Follow steps 1 through 12 from Basic Protocol 1, substituting Buffer A500 plus 1 mM ATP and 1x ATP-regenerating system for Buffer A, to extract, bind, and wash 19S RP complexes.
-
Resuspend the resin in 1 mL of Buffer A supplemented with 1 mM ATP and 1x ATP-regenerating system, and split evenly between two 1.5 mL microcentrifuge tubes.
This step serves to reduce the total NaCl concentration from 500 mM to ~150 mM in the sample prior to elution with the 3xFLAG peptide by resuspension of the resin in Buffer A rather than Buffer A500. Centrifuge microcentrifuge tubes at 1500 × g for 30 seconds to pellet the resin, and pipette out the supernatant carefully.
-
Add 3 resin volumes of Buffer A plus 1 mM ATP and 1x ATP-regenerating system containing 100 μg/mL 3xFLAG peptide to each tube, and place on an end-over-end rotator at 4 °C for 45 minutes.
It is important to use 3xFLAG peptide rather than FLAG peptide; 3xFLAG tagged proteins cannot be competed off of the resin efficiently by single FLAG peptide. -
Centrifuge microcentrifuge tubes at 1500 × g for 30 seconds at 4 °C to pellet the resin. Collect and combine the 3xFLAG eluates from each tube into a fresh 1.5 mL microcentrifuge tube on ice.
It is important to avoid collecting any of the resin during this step, as this will reduce the purity of the final preparation. We often will centrifuge the eluate again after transfer to the fresh 1.5 mL microcentrifuge tube, and transfer the supernatant to a second 1.5 mL microcentrifuge tube to assure that no resin beads remain in the eluate. Transfer the eluate to a 100 kDa-cutoff Vivaspin 500 centrifugal concentrator tube. If the total volume of eluate is greater than 500 μL, then transfer 500 μL at a time to the concentrator, concentrate by centrifugation at 10,000 × g for 15 minutes at 4 °C, discard the flow-through, and add up to 500 μL of the remaining eluate to the concentrator. Repeat until all of the eluate has been added to the concentrator. Invert the concentrator to mix the concentrated retentate with the newly added eluate between concentration runs. This minimizes the chance of precipitation of the proteins during concentration. Concentrate the total eluate to approximately 50–100 μL by centrifugation at 4 °C.
-
Once the entire eluate has been concentrated, transfer the retentate to a pre-chilled 1.5 mL microcentrifuge tube.
It is important to thoroughly mix the retentate by pipetting up and down carefully several times prior to pipetting from the concentrator housing to maximize recovery of concentrated proteasomes. Avoid foaming, which damages proteins. To estimate the final concentration of RP, run 1 μL of the purified RP on an SDS-PAGE gel with a dilution series of BSA standards of 750, 500, 250, and 125 ng and stain the gel with Gelcode blue. Quantify the BSA band intensities and the intensity of a band from an individual subunit of the RP. For 26S proteasome or RP purifications, we use the Rpn3 RP subunit because it has a unique migration at ~60 kDa that is well separated from other subunits; for 20S CP purifications we usually choose the Pre10/α7 CP subunit (~32 kDa) for this purpose. The concentration can then be derived from the equation of the line formed by the BSA standards, the molecular mass of the chosen subunit, and the known volume of RP added to the gel.
-
Dilute the RP to the desired concentration using Buffer A containing 1 mM ATP. Make 5 μL aliquots, and snap-freeze in liquid nitrogen. Store RP aliquots at -80°C.
It is often preferable to dilute the purified RP to a convenient concentration for biochemical assays. We routinely dilute purified RP to 1 μM using Buffer A before snap-freezing and storage.
BASIC PROTOCOL 3: Purification of active 20S CP
In contrast to purification of the 26S proteasome or the 19S RP, purification of the 20S CP is typically performed on extracts of yeast expressing 3xFLAG-tagged Pre1/β4 in a buffer lacking ATP and glycerol. These components are omitted because they help to stabilize the interaction between the 19S RP and the 20S CP. Inclusion of 500 mM NaCl in the lysis and washing buffers disrupts the association of the RP and CP, allowing selective retention of 20S CP on the resin.
Materials
Cell powder from Saccharomyces cerevisiae strain MHY6952 (PRE1-6xGly-3xFLAG:kanMX6) or similar
Buffer A (see recipe)
20S lysis buffer (see recipe)
20S washing buffer (see recipe)
BCA Assay Kit, Pierce catalog #23227
Anti-FLAG M2 affinity gel, Sigma catalog #A2220
3xFLAG peptide, Sigma catalog #F4799
Gelcode Blue Stain, Thermo Scientific catalog #24592
High speed centrifuge, 4 °C
40 mL high-speed centrifuge tubes and rotor
Bench-top centrifuge, 4 °C
Microcentrifuge, 4 °C
50 mL disposable polypropylene conical centrifuge tubes
Rotator or nutator for conical vials
Vivaspin 500 centrifugal concentrator (100 kDa molecular weight cutoff), Sartorius catalog #VS0141
Mini gel electrophoresis system (Bio-Rad) or similar
G-Box (Syngene) or similar gel documentation system
Protocol steps
Prepare cell powder from a 2 L YPD culture of MHY6952 as described in Support Protocol 1 (above).
Follow steps 2 through 9 from Basic Protocol 1, substituting 20S lysis buffer for Buffer A, to extract 20S CP and bind it to the anti-FLAG resin.
After centrifuging the resin at 1500 × g for 2 minutes at 4 °C, carefully decant the supernatant. Resuspend the resin in 25 mL of 20S washing buffer. Incubate with end-over-end mixing at 4 °C for five minutes to remove nonspecifically bound materials.
Centrifuge the mixture at 1500 × g for 2 minutes at 4 °C to pellet the resin.
-
Repeat steps 3 and 4 for a second wash with 20S lysis buffer.
In contrast to 20S washing buffer, 20S lysis buffer does not contain Triton X-100 detergent. Step 5 minimizes the detergent in the following steps. After decanting the supernatant, resuspend the resin in 1 mL of Buffer A, and split evenly between two 1.5 mL microcentrifuge tubes. Centrifuge the tubes at 1500 × g for 30 seconds at 4 °C to pellet resin, and carefully pipette out the supernatant.
-
Add 3 resin volumes of Buffer A containing 100 μg/mL 3xFLAG peptide to each tube, and place on an end-over-end rotator at 4 °C for 45 minutes.
It is important to use 3xFLAG peptide rather than FLAG peptide; the 3xFLAG-tagged protein cannot be competed efficiently from the resin by the single FLAG peptide. -
Centrifuge microcentrifuge tubes at 1500 × g for 30 seconds 4 °C to pellet resin. Collect and combine the eluates from each tube into a fresh microcentrifuge tube on ice using a Pipetteman.
It is important to avoid collecting any of the resin during this step, as this will reduce the purity of the final preparation. We often will centrifuge the eluate again after transfer to the fresh 1.5 mL microcentrifuge tube, and transfer the supernatant to a second 1.5 mL microcentrifuge tube to assure that no resin beads remain in the eluate. Transfer the eluate to a 100 kDa-cutoff Vivaspin 500 centrifugal concentrator tube. If the total volume of eluate is greater than 500 μL, then transfer 500 μL at a time to the concentrator, concentrate by centrifugation at 10,000 × g for 15 minutes at 4 °C, discard the flow-through, and add up to 500 μL of the remaining eluate to the concentrator. Repeat until all of the eluate has been added to the concentrator. Invert the concentrator to mix the concentrated retentate with the newly added eluate between concentration runs. This minimizes the chance of precipitation of the proteins during concentration. Concentrate the total eluate to approximately 50–100 μL by centrifugation at 4 °C.
-
Once the entire eluate has been concentrated, transfer the retentate to a pre-chilled 1.5 mL microcentrifuge tube.
It is important to thoroughly mix the retentate by pipetting up and down carefully several times prior to pipetting from the concentrator housing to maximize recovery of concentrated proteasomes. Avoid foaming, which damages proteins. To estimate the final concentration of the 20S CP, run 1 μL of the purified CP on a 12% SDS-PAGE gel made as Table 1 with a dilution series of BSA standards of 750, 500, 250, and 125 ng and stain with Gelcode Blue (Figure 1). Quantify the BSA band intensities and the intensity of a band from an individual subunit of the 20S CP. We choose the Pre10/α7 CP subunit because it has a unique migration at ~32 kDa (Figure 1). The concentration can then be derived from the equation of the line formed by the BSA standards, the molecular mass of Pre10/α7 (31521.3 g/mol), and the known volume of CP added to the gel.
-
Dilute CP to the desired concentration using Buffer A. Make 5–10 μL aliquots, and snap-freeze in liquid nitrogen. Store proteasome aliquots at −80°C.
Dilution of CP to 1 μM is convenient for activity assays (See Basic Protocols 4–6).
BASIC PROTOCOL 4: In-gel peptidase activity assay for 20S CP and 26S proteasomes
Proteolytic activities of the 20S CP and 26S proteasomes can be measured by the cleavage of fluorogenic peptide substrates specific for each of the three different CP active sites. For example, proteasome chymotrypsin-like activity can be monitored by the fluorescence of aminomethylcoumarin (AMC) released from the succinyl-Leu-Leu-Val-Tyr-AMC (suc-LLVY-AMC) substrate. For general quantification of proteasome proteolytic activity, the chymotrypsin-like activity is most often assayed, as it is the most potent. However, proteasome trypsin-like and caspase-like activities can also be monitored with substrates bearing their preferred cleavage sites (for example, ac-Arg-Leu-Arg-AMC and Z-Leu-Leu-Glu-AMC, respectively). In this protocol and Basic Protocol 5, we use the suc-LLVY-AMC substrate.
Based on their size, charge and shape, different proteasomal species, including doubly-capped 26S (RP2CP), singly-capped 26S (RPCP) and free 20S (CP) proteasomes, can be resolved by electrophoresis through a native polyacrylamide gel. Their peptidase activities can be visualized in the gel under a UV illuminator after immersing the gel in a buffer containing a fluorogenic peptide substrate. The advantage of the in-gel peptidase assay is that it can resolve the activity contributed by multiple proteasomal species present in the same protein sample. Although the N-terminal tails of the CP outer-ring subunits typically form a “gate” that precludes entry of substrates into the isolated CP, this gate can be artificially opened with a low concentration (0.02% w/v) of SDS detergent, allowing measurement of free CP activity.
Materials
0.9 M Tris-Boric acid, pH 8.3 (see recipe)
1 M MgCl2 (see recipe)
1 M ATP (see recipe)
25% (w/v) sucrose
75% (v/v) glycerol
40% acrylamide, Bio-Rad catalog #161-0140
2% bis-acrylamide, Bio-Rad catalog #161-0142
10% (w/v) ammonium persulfate (APS) in diH2O
20% (w/v) sodium dodecyl sulfate (SDS) in diH2O
N,N,N′,N′-tetramethylethylenediamine (TEMED), Sigma catalog #T7024
1x native gel running buffer (see recipe)
5x native gel loading buffer (see recipe)
Purified 20S CP and/or 26S proteasomes, 1 μM
Buffer A (see recipe)
Developing buffer (see recipe)
10 mM suc-LLVY-AMC, Sigma #S6510 (see recipe)
Gel releasers, Bio-Rad catalog #165-3320, or similar tools for the system of choice
Gel tray, GenHunter catalog #B107 or similar
Mini gel electrophoresis system (Bio-Rad) or similar
G-Box (Syngene) or a similar gel-documentation system
Protocol steps
-
Prepare at least 500 mL of native gel running buffer, an amount sufficient for an electrophoretic separation using a single mini-gel box, and pre-cool it to 4 °C.
Native gel running buffer can be made in advance without ATP and stored at 4 °C. -
Prepare a 4% native gel containing 1 mM ATP as described in Table 2. The stacking gel is made with 3% polyacrylamide and also contains 1 mM ATP.
Wash glass plates with H2O and rinse with ethanol. Let them air dry. Don’t use paper towels to dry glass plates as they leave small fibers on the plates and will interfere with the fluorescence imaging for the in-gel peptidase activity assay.Always use freshly prepared native gels, as ATP tends to hydrolyze quickly.If only the 20S CP is being analyzed, ATP can be omitted from both the polyacrylamide gel and the running buffer, as it is not required for CP activity or stability. -
Pipette 0.5 μL of purified 20S CP or 26S proteasome (1 μM) into 9.5 μL Buffer A, and mix well with 2.5 μL 5x native gel loading buffer. Load the entire sample onto the native gel.
The 3% polyacrylamide stacking gel is very soft, so sample wells are easily distorted if the comb is pulled out carelessly. We recommend adding the native gel running buffer to the assembled gel cassette until the buffer covers the top of the gel before taking out the comb. -
Perform gel electrophoresis at 100 V at 4 °C for about 2.5 hours or until the xylene cyanol dye front reaches the bottom of the gel.
To get a better separation between doubly-capped 26S (RP2CP) and singly-capped 26S (RPCP) proteasomes, perform gel electrophoresis for 3 hours or more. Carefully disassemble the glass plates, leave the gel on one of the glass plates. Cut off the stacking gel and discard it. Add 10 mL of Buffer A to a clean tray. Carefully dislodge the resolving gel from the glass plate into the tray. Rinse the gel with Buffer A.
Decant the buffer from the tray carefully as the gel may slip out of the tray. Add 10 mL of developing buffer to the tray. Incubate the gel at 30 °C for 30 minutes in a shaker with slow agitation (~30 rpm).
-
Transfer the gel from the tray to a UV trans-illuminator and expose the gel at 365 nm wavelength in the gel documentation set-up (e.g., G-box) (Figure 2).
The 4% resolving gel is very soft and easy to tear. Handle with care. See Critical Parameters for additional advice. -
Put the gel back in the tray. Add 10 μL of 20% SDS to the Developing buffer from step 6 to monitor the 20S CP activity. The final concentration of SDS in the developing buffer is 0.02%. Incubate the native gel with developing buffer at 30 °C for 30 minutes with slow agitation (~30 rpm).
The entry channel in isolated 20S proteasome (CP) is primarily in a closed state, so 0.02% SDS is added to locally denature the substrate channel gate, allowing unfettered entry of peptide substrates. 26S proteasomes do not require SDS to activate their activity because association of the RP with the CP opens the gate of the CP entry channel. Visualize the gel again under a UV illuminator at 365 nm wavelength in the gel documentation set-up (Figure 2).
Table 2.
Recipe to make one 4% 1.5 mm native-PAGE gel.
| Resolving gel, 4% acrylamide | Stacking gel, 3% acrylamide | ||||
|---|---|---|---|---|---|
| [Final] | [Stock] | 10 mL | [Final] | [Stock] | 5 mL |
| H2O | 6.337 mL | H2O | 2.365 mL | ||
| 90 mM Tris-borate, pH 8.3 | 0.9 M | 1000 μL | 50mM Tris-HCl, pH 6.8 | 1 M | 250 μL |
| 2.5% Sucrose | 25% | 1000 μL | 2.5% sucrose | 25% | 500 μL |
| 4% acrylamide | 40% | 974 μL | 3% acrylamide | 40% | 300 μL |
| bis-acrylamide | 2% | 519 μL | bis acrylamide | 2% | 1500 μL |
| 5 mM MgCl2 | 1 M | 50 μL | 5mM MgCl2 | 1 M | 25 μL |
| 1mM ATP | 1 M | 10 μL | 1mM ATP | 1 M | 5 μL |
| APS | 10% | 100 μL | APS | 10% | 50 μL |
| TEMED | 10 μL | TEMED | 5 μL | ||
Figure 2.
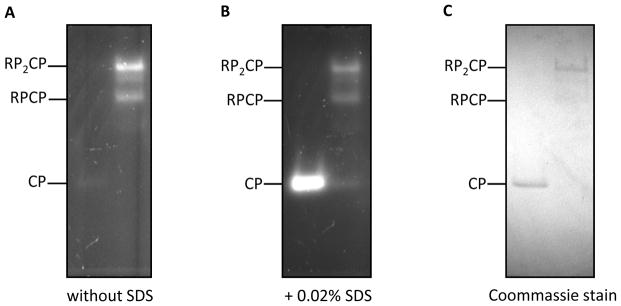
In-gel peptidase acitivity assay to measure 20S and 26S proteasome activity. In-gel peptidase activity assay was performed as described in Basic Protocol 4. (A) A 4% native polyacrylamide gel incubated with the developing buffer without SDS. (B) A 4% native polyacrylamide gel incubated with the developing buffer containing 0.02% SDS. (A) and (B) are visualizations of same samples before and after adding 0.02% SDS. Left lane: 0.10 μg of 20S proteasome (CP). Right lane: 0.37 μg of 26S proteasome (RP2CP and RPCP). (C) A 4% native gel stained with Gelcode Blue (Coomassie Blue). Left lane: 0.30 μg 20S proteasome. Right lane: 1.1 μg 26S proteasome. Proteasome species RP2CP, RPCP and CP are labeled in the figure. In Figure 2A and Figure 2B, there are one or two very weakly stained bands with chymotryptic activity detected in between RP2CP and RPCP, and one or two additional species in between RPCP and CP. The exact compositions of these complexes were not examined. They presumably contain known substoichiometric proteasome-binding proteins such as Hul5, Ubp6, Ecm29 or Blm10, in addition to the RP and CP.
BASIC PROTOCOL 5: In-solution peptidase activity assay for 20S and 26S proteasomes
As noted above, proteasome activities can be measured in solution by monitoring the hydrolysis of AMC from a suc-LLVY-AMC peptide substrate. Compared to the in-gel peptidase activity assay (Basic Protocol 4), the advantage of the in-solution peptidase activity assay is that it is simple, rapid, and can be used to measure quantitatively the effects of proteasome inhibitors and activators. However, it cannot distinguish the peptidase activities of doubly-capped 26S proteasome vs. that of the singly-capped 26S proteasome or 20S proteasome.
Materials
1M Tris-HCl pH 7.5 (see recipe)
1 M MgCl2 (see recipe)
1 M ATP (see recipe)
10 mM suc-LLVY-AMC, Sigma #S6510 (see recipe)
Purified 20S CP and/or 26S proteasomes, 1 μM
1% SDS (w/v) in diH2O
Developing buffer (see recipe)
Fluorimeter
Protocol steps
-
Dilute purified 20S or 26S proteasomes to 100 μL with developing buffer. The final concentration of 20S CP/26S proteasome is ~35 pM.
To measure 20S proteasome (CP) activity, add SDS to 0.02% to open the gate of the substrate channel. We add SDS at the last step before assaying activity. Incubate the reaction at 30 °C for 30 minutes.
Stop the reaction by adding 1 mL 1% SDS.
-
Measure the fluorescence of AMC using a fluorimeter set with an excitation wavelength of 380 nm and an emission wavelength of 460 nm.
Enzyme kinetics can be determined at different substrate concentrations by taking readings at several timepoints after addition of the substrate and plotting the fluorescence vs. time.
BASIC PROTOCOL 6: Measuring degradation of polyubiquitinated Sic1PY
Whereas unstructured proteins or small peptides (Basic Protocol 4 and Basic Protocol 5) can be degraded by the isolated CP, degradation of folded, polyubiquitylated proteins requires the deubiquitinating and ATP-dependent unfoldase activities of the RP. A conveniently synthesized (albeit somewhat heterogenous) substrate that is dependent on these activities has been described, polyUb-Sic1PY. This substrate and its synthesis have been described in great detail elsewhere, so for the production of this substrate we refer the reader to that protocol (Saeki et al., 2005). Here, we describe a basic assay to measure the time-dependent degradation of T7-tagged, polyubiquitinated Sic1PY. Purified proteasomes are mixed with the substrate, and loss of the T7 signal from the sample, indicative of substrate degradation, is measured by Western immunoblotting.
Materials
Purified 26S proteasomes, 1 μM
Polyubiquitinated T7-Sic1PY
Buffer A (see recipe)
10 mM dithiothreitol in diH2O
10x ATP regenerating system (see recipe)
ATP, Grade I (≥99%), Sigma catalog #A2383
Anti-T7 antibody, EMD Millipore catalog #69522
PVDF membrane, EMD Millipore catalog #IPVH00010
Temperature-controlled heat block or water bath, set to 30 °C
Temperature-controlled heat block or water bath, set to 100 °C
Mini gel electrophoresis system (Bio-Rad) or similar
Electroblotting apparatus (Bio-Rad) or a similar electroblotting system
Chemiluminescence imaging equipment (G-Box, Syngene) or a similar imaging system
Protocol steps
Prepare the following mixture on ice: 8 μL Buffer A, 2 μL 10x ATP-regenerating system, 2 μL 10 mM dithiothreitol, 2 μL 20 mM ATP, 4 μL polyubiquitinated T7-Sic1PY.
Add 2 μL of 1 μM 26S proteasomes, mix by vortexing, and immediately place in the 30 °C water bath.
-
Take 4 μL aliquots at 0, 2, 5, and 10 minute timepoints. Add 4 μL Buffer A and 2 μL of 5x SDS loading buffer, and immediately boil the mixture for five minutes to denature the proteins and stop the reaction.
It can be technically difficult or impossible to take the zero minute sample without allowing significant degradation to occur. It may instead be desirable to take an aliquot of the reaction mixture prior to addition of the 26S proteasomes to serve as the zero minute sample. Separate 4 μL of each sample by electrophoresis through a 10% SDS-polyacrylamide gel.
Electrotransfer the proteins to a PVDF membrane using standard blotting conditions.
Perform Western immunoblotting using the anti-T7 antibody at a 1:2000 dilution. Quantify the chemiluminescence signal using the imaging station.
-
Quantify the loss of the T7 signal over time and calculate the percent remaining at each timepoint.
It is important that the chemiluminescence signal is within the linear detection range of the imaging equipment for accurate quantitation.
REAGENTS AND SOLUTIONS
YPD agar plates
6 g yeast extract
12 g peptone
12 g dextrose
12 g agar
1.2 mL of 2% adenine in 0.1 M NaOH
add diH2O to 600 mL
Sterilize by autoclaving. Allow to cool to approximately 50 °C before pouring plates.
Makes approximately 25 10-cm dishes. Store at 4 °C.
YPD liquid medium
10 g yeast extract
20 g peptone
20 g dextrose
2 mL of 2% adenine in 0.1 M NaOH
Dissolve into 900 mL deionized H2O and add deionized H2O to 1 L. Sterilize by autoclaving.
Can be stored for at least 6 months at room temperature.
1 M Tris, pH 7.5
Dissolve 121.14 g Tris into 900 mL deionized H2O, adjust pH with concentrated hydrochloric acid to pH 7.5 and fill up with deionized H2O to 1 L. Can be stored at room temperature indefinitely.
5 M NaCl
Dissolve 292.2 g NaCl into 600 mL deionized H2O and add deionized H2O to 1 L. Can be stored at room temperature indefinitely.
1 M MgCl2
Dissolve 95.21 g MgCl2 into 800 mL deionized H2O and add deionized H2O to 1 L. Can be stored at room temperature indefinitely.
Buffer A
50 mM Tris-HCl, pH 7.5
150 mM NaCl
5 mM MgCl2
10% glycerol
Can be stored at room temperature or pre-cooled at 4 °C indefinitely.
Buffer A500
50 mM Tris-HCl, pH 7.5
500 mM NaCl
5 mM MgCl2
10% glycerol
-
Can be stored at room temperature or pre-cooled at 4 °C indefinitely.
500 mM ATP in Tris, pH 7.0 25 mL ATP, disodium salt hydrate 6.889g 2 M Tris base 15mL
Dissolve ATP as much as possible in 15mL of Tris base. Continue to add Tris base dropwise with stirring until ATP is completely dissolved. Determine the pH, and adjust to 7.0 with additional Tris base or HCl until pH = 7.0. Bring final volume to 25 mL using diH2O. Make aliquots and store at −80°C; can be stored for at least six months.
10x ATP-Regenerating System
500 μg/mL creatine kinase
25 mM creatine phosphate
Dissolve in Buffer A, aliquot, and store at −80°C. Can be used for at least six months.
20S lysis buffer
50 mM Tris-HCl, pH 7.5
500 mM NaCl
5 mM MgCl2
Can be stored at 4 °C indefinitely.
20S washing buffer
50 mM Tris-HCl, pH 7.5
500 mM NaCl
5 mM MgCl2
0.2% Triton-X100
Can be stored at 4 °C indefinitely.
0.9 M Tris-boric acid, pH 8.3
Dissolve 108.99 g Tris and 55.65 g boric acid into 700 mL deionized H2O, and add deionized H2O to 1 L. Can be stored at room temperature for up to 6 months.
1x native gel running buffer
90 mM Tris-boric acid, pH 8.3
5 mM MgCl2
1 mM ATP
Buffer can be made without ATP and store at 4 °C for up to 6 months. Add ATP immediately before use.
5x native gel loading buffer
250 mM Tris-HCl, pH 7.5
25 mM MgCl2
50% glycerol
5 mM ATP
1.5 μg Xylene cryanol
Buffer can be made without ATP and stored at room temperature indefinitely. Add ATP immediately before use.
10 mM Suc-LLVY-AMC
Add 1.3 mL DMSO to 10 mg suc-LLVY-AMC to dissolve the compound. Make 100 μL aliquots. Store at −20°C.
Developing buffer
50 mM Tris, pH 7.5
150 mM NaCl
5 mM MgCl2
1 mM ATP
100 μM Suc-LLVY-AMC
Buffer can be made without ATP and suc-LLVY-AMC and stored at room temperature indefinitely. Add ATP and suc-LLVY-AMC immediately before use.
COMMENTARY
Background Information
The 26S proteasome is a 2.5 MDa protease complex containing at least 33 different subunits (Tomko and Hochstrasser, 2013). It is composed of two subcomplexes: the 20S core particle (CP) and the 19S regulatory particle (RP). The barrel-shaped CP consists of four stacked heteroheptameric rings: two α-rings on the ends that sandwich a pair of β-rings in between. The six proteolytic active sites are housed within the β-rings (three per ring). The β1 subunits bear the caspase-like active sites, the β2 subunits the trypsin-like sites, and the β5 subunits the chymotrypsin-like sites. Entry of substrates into the CP is controlled by the RP, which can be further divided into two subcomplexes, the lid and base. The lid contains at least nine different subunits and aids in substrate recognition and protein deubiquitination. The base contains a hexameric ATPase ring that stacks on the end of the CP and is responsible for opening the gate of the CP substrate channel. The proteasome is highly conserved from yeast to humans. In humans, however, additional forms of the CP can be formed. These alternative forms, called immunoproteasomes and thymoproteasomes contain, respectively, the interferon-inducible subunits β1i, β2i and β5i (for immuno-proteasomes) or β5t (for thymoproteasomes), which occupy the positions normally filled by β1, β2, and β5.
Proteasomes degrade proteins via two broad mechanisms (Ben-Nissan and Sharon, 2014). One is ubiquitin-dependent, whereas the other is ubiquitin-independent. Most proteins are degraded via the ubiquitin-dependent 26S proteasome degradation pathway. The target protein is first modified with the small protein ubiquitin, usually at one or more lysine residues, by a multi-enzyme pathway. Additional ubiquitin molecules can be ligated onto the initial one to form a polyubiquitin chain. A polyubiquitin chain with four or more ubiquitin molecules is typically sufficient to deliver the protein to the 26S proteasome. The RP recognizes the polyubiquitin chain, removes it from the substrate, unfolds the substrate and delivers it into the CP, where the substrate is cleaved into short peptides. ATP hydrolysis provides energy both for ubiquitin conjugation to the substrate and for substrate unfolding at the proteasome. Nucleotide binding by the RP is also required for its stable association with the CP.
In addition to the ubiquitin-dependent pathway, some proteins are degraded via ubiquitin-independent pathways in which either the 26S proteasome or CP can serve as the proteolytic enzyme. A common feature of proteins degraded through this pathway is that they contain an unstructured region, either naturally or induced by stress. The detailed mechanisms used for ubiquitin-independent proteasomal degradation are known to only a limited degree and are expected to vary among different substrates (Erales and Coffino, 2014).
Initially, proteasomes were purified from yeast (and other organisms) using conventional chromatography methods, including ion exchange chromatography and gel filtration (Glickman and Coux, 2001). The affinity purification described here is substantially faster and simpler. With the option of many commercially available epitope tags and the flexibility afforded by the ease of genetic manipulation in yeast, distinct tags can be linked onto different proteasome subunits, thus allowing purification or detection of different proteasome subcomplexes from a single yeast lysate.
Critical Parameters and Troubleshooting
Thorough cell lysis is critical to each of the purification protocols. Generation of a very fine cell powder, similar in consistency to milled flour, will maximize subsequent protein extraction. Cell lysis can be checked by taking a small amount of the cell powder, thawing it completely in Buffer A or water, and examining it with a phase-contrast microscope. Properly lysed cells will appear largely as hollow “ghosts” due to cell membrane breach. We have also utilized French Press lysis, spheroplast lysis and glass bead beating to generate extracts for proteasome purifications, but we prefer cryogenic lysis in most cases because it is easily scalable and helps to preserve native protein complexes during lysis.
After centrifugation of the lysate, a layer of yellowish lipids will sometimes appear at the top of the supernatant. We have found that removing these lipids, either via pipetting or by pouring the supernatant through 2–3 thicknesses of cheesecloth, improves the overall quality of the purification and may preserve the quality of the FLAG resin during repeated use. Finally, we have found that the Bradford assay for estimating protein concentration on yeast lysates is not always accurate when following our procedure for cryogenic lysis, so we instead recommend using the BCA assay, which has been very reliable in our hands.
The key to perform a successful in-gel peptidase assay is to generate an undistorted native acrylamide gel separation and keep the gel intact during the assay. Since the 4% native gel is very soft, it is best to minimize the handling of the gel after dislodging it from the glass plate. While transferring the gel from the tray to the trans-illuminator for imaging, we typical wet the gel releaser with the developing buffer, and then use it to fold the gel over upon itself and push the gel onto the imaging plate. It is important to dip the gel releaser into the developing buffer before touching the gel or the gel will likely to stick to the gel releaser. We also spray some distilled H2O on the imaging plate in the G-box to help maneuver the gel and prevent it from adhering to the surface and tearing while manipulating it.
Anticipated Results
Typically, the purification described in Basic Protocol 1 yields between 400 and 600 μg of purified proteasomes from a 2 L yeast culture. The purification in Basic Protocol 2 yields approximately 200 to 300 μg of RP from a 2 L yeast culture. The purification in Basic Protocol 3 yields approximately 200 to 300 μg of CP from a 2 L yeast culture. An example of purified 26S proteasomes (RP2CP and RPCP) and 20S proteasomes (CP) visualized on a 12% SDS-PAGE gel is shown in Figure 1. An example of the in-gel peptidase activity assay (Basic Protocol 4) is shown in Figure 2. Proteasomes and subcomplexes typically are at >95% purity following the above protocols, as estimated from SDS-PAGE. Common contaminants include the heavy and light chains of the FLAG antibody if the purified species are not completely separated from the FLAG affinity resin after elution. These would be anticipated to migrate at positions corresponding to ~23 kDa and ~50 kDa on an SDS-PAGE gel. As noted above, there are several proteasome-interacting proteins that are found substoichiometrically in proteasomes purified as described here; these may include Ubp6 (57 kDa), Hul5 (106 kDa), Ecm29 (210 kDa), and Blm10 (246 kDa). None of these are encoded by essential genes, and each gene can be readily deleted using standard yeast genetics should it be necessary. Additionally, insufficient washing may allow some nonspecific impurities to remain. To remove these contaminants, additional purification steps can be employed, such as gel filtration chromatography. The exceptionally large sizes of the proteasome, the 19S RP, and the 20S CP allow for them to be readily separated from many other proteins on the basis of size.
In Basic Protocol 5, the fluorescence of the AMC released by 20S or 26S proteasome from a fluorogenic substrate can be read from a fluorimeter. If desired, one can calculate the absolute amount of AMC released from a proteolytic reaction by generating an AMC standard curve with various known concentrations of free AMC.
In the polyubiquitinated T7-Sic1PY degradation assay described in Basic Protocol 6, the substrate typically appears as a high molecular weight smear in the T7 blot, and is near-completely destroyed within 10 minutes (Saeki et al., 2005). Typically, no accumulation of deubiquitinated substrate or substrate intermediates is observed, as deubiquitination occurs en bloc, and is enzymatically coupled to the degradation of the substrate.
Time Considerations
Basic Protocols 1–3 (26S proteasome, 19S RP, and 20S CP purifications): 4–5 days are required to obtain yeast pellets. 20–30 minutes are required to grind one yeast sample. 5–6 hours are required to complete the affinity purification step.
Basic Protocol 4 (in-gel activity assay): 5–6 hours.
Basic Protocol 5 (in-solution activity assay): 40 minutes.
Basic Protocol 6 (degradation assay): 1–2 days.
Acknowledgments
Work on the proteasome in MH’s laboratory is currently supported by NIH grant R01 GM083050.
LITERATURE CITED
- Ben-Nissan G, Sharon M. Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules. 2014;4:862–884. doi: 10.3390/biom4030862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erales J, Coffino P. Ubiquitin-independent proteasomal degradation. BBA-Mol Cell Res. 2014;1843:216–221. doi: 10.1016/j.bbamcr.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman M, Coux O. Purification and characterization of proteasomes from Saccharomyces cerevisiae. In: Coligan John E, et al., editors. Current Protocols in Protein Science/Editorial Borard. Unit 21.5. Chapter 21. John Wiley & Sons, Inc; Hoboken, NJ, USA: 2001. [DOI] [PubMed] [Google Scholar]
- Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- Leggett DS, Glickman MH, Finley D. Purification of proteasomes, proteasome subcomplexes, and proteasome-associated proteins from budding yeast. Methods Mol Biol. 2005;301:57–70. doi: 10.1385/1-59259-895-1:057. [DOI] [PubMed] [Google Scholar]
- Saeki Y, Isono E, Toh EA. Preparation of ubiquitinated substrates by the PY motif-insertion method for monitoring 26S proteasome activity. Methods Enzymol. 2005;399:215–227. doi: 10.1016/S0076-6879(05)99014-9. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Finley D. Regulation of proteasome activity in health and disease. BBA-Mol Cell Res. 2014;1843:13–25. doi: 10.1016/j.bbamcr.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomko RJ, Hochstrasser M. Molecular architecture and assembly of the eukaryotic proteasome. Annu Rev Biochem. 2013;82:415–445. doi: 10.1146/annurev-biochem-060410-150257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wu P, Hu Y. Clinical and marketed proteasome inhibitors for cancer treatment. Curr Med Chem. 2013;20:2537–2551. doi: 10.2174/09298673113209990122. [DOI] [PubMed] [Google Scholar]