

Summary
While membrane simulations are widely employed to study the structure and dynamics of various lipid bilayers and membrane proteins in the bilayers, simulations of lipopolysaccharides (LPS) in membrane environments have been limited due to its structural complexity, difficulties in building LPS-membrane systems, and lack of appropriate molecular force field. In this work, as a first step to extend CHARMM-GUI Membrane Builder to incorporate LPS molecules and to explore their structures and dynamics in membrane environments using molecular dynamics simulations, we describe step-by-step procedures to build LPS bilayer systems using CHARMM and the recently developed CHARMM carbohydrate and lipid force fields. Such procedures are illustrated by building various bilayers of Escherichia coli O6 LPS and their preliminary simulation results are given in terms of per-LPS area and density distributions of various components along the membrane normal.
Keywords: lipid A, R1 core, O6 antigen, bilayer, E. coli, glycan, molecular dynamics simulation
1. Introduction
Lipopolysaccharides (LPS), also known as endotoxins, cover a large part of the outer membrane of Gram-negative bacteria (1–4). They are amphiphilic compounds consisting of the three regions, viz., Lipid A, the core region, and the O-antigen polysaccharide. Lipid A has acyl chains and anchors the LPS in the biomembrane. It is the bacterial Lipid A component that is responsible for the toxic effects in mammalian cells by binding to Toll-like receptors with subsequent immunostimulation of the innate immune system and induction of inflammatory cytokines (5). The core region is relatively conserved across species with only a few canonical structures (e.g., five in the case of Escherichia coli) consisting of about 10–12 monosaccharides. Finally, the O-antigen polysaccharide is made up of repeating units that are highly variable in structure.
In E. coli, about 180 different serogroups have been reported and primary sequence information of more than half of their O-antigen polysaccharides have been determined (6). The number of repeats is usually on the order of 10–25, resulting in a molecular mass of up to 25 kDa (7), thereby creating a physical barrier and protective shield at the surface of the bacterium. In some cases, only ‘a single repeat’ is observed leading to what is known as a semi-rough strain (8, 9). These Gram-negative bacteria may also co-express capsular (10) or exo-polysaccharides of even higher molecular mass, but this expression is dependent on, inter alia, environment and growth conditions.
In the LPS of E. coli (Figure 1), the lipid A part consists of two glucosamine residues joined by a β-(1→6)-linkage, six N- and O-linked fatty acids for the canonical form, and phosphoester moieties. The inner core region has a few keto-deoxyoctulosonate (Kdo, eight-carbon sugar) residues, one of which is linked to the Lipid A part, and L-glycero-D-manno-heptose (Hep, seven-carbon sugar) residues; in the outer core, half a dozen hexose (Hex, six-carbon sugar) residues are present. The O-antigen polysaccharide is likewise linked to one of the core sugars via a D-GlcpNAc or a D-GalpNAc residue, and is built by repeating units usually consisting of 3–6 sugars that are linear or branched, sometimes with more than one branch making the three-dimensional (3D) structure highly complex. The structural features of some of these polysaccharides are such that they can act by molecular mimicry (11), meaning that they imitate the glycoconjugate structures of the host that they invade or inhabit in a symbiotic manner. The three regions of the LPS have different functions, and it is mandatory to obtain information on their 3D structure and dynamics in order to understand the interactions of endotoxins in biological and biomedical systems.
Figure 1.

Schematic of the lipopolysaccharide (LPS) of E. coli O6 having an R1 core. The LPS consists of three regions, viz., the lipid A, the core, and the O-antigen polysaccharide built of pentasaccharide repeating units. Note that the N-acetyl-D-glucosamine residue of the first repeating unit is ligated to the core via a β-linkage, whereas in the remaining part of the polysaccharide, it is joined via α-linkages to the subsequent repeating unit. The dashed lines indicate the repeating unit of the polymer and n describes the total number of repeating units in the O-antigen polysaccharide.
While membrane simulations are widely employed to study the structure and dynamics of various lipid bilayers and membrane proteins in the bilayers (reviewed in (12–17)), simulations of LPS molecules in membrane environments (18, 19) have been limited due to its structural complexity, difficulties in building LPS membrane systems, and lack of appropriate molecular force field. We have developed CHARMM-GUI Membrane Builder (20, 21) to standardize and automate the building procedures of various lipid bilayers and membrane protein systems. In this work, as a first step to extend CHARMM-GUI Membrane Builder to incorporate LPS molecules and to explore their structures and dynamics in membrane environments using molecular dynamics (MD) simulations, we describe step-by-step procedures to build LPS bilayers using CHARMM (22) and the modified Membrane Builder procedure. For this work, we have added lipid A and new sugar types (e.g., core region Kdo and Hep residues) to the recently developed CHARMM carbohydrate and lipid force fields (23–26). A LPS molecule, E. coli R1 (core) O6 (antigen) (8), was used as an example in this study and described in the following section. In the Methods, the LPS bilayer building procedures are presented in terms of (i) generation of a LPS molecule, (ii) building of LPS bilayer components, (iii) their assembly, and (iv) equilibration and production.
2. E. coli O6 LPS
In this work, the 3D structure of E. coli O6 LPS was built and simulated. The primary structure, i.e., sugar and lipid components, substituents, anomeric configurations, ring forms, substitution positions, and sequence of sugars, was previously determined using chemical and spectroscopic methods. The structural information comes from two studies. In the first study, the structure of the repeating unit of the O-antigen polysaccharide was determined (27). In the second study, the semi-rough strain Nissle 1917 was investigated for the lipid A, the core region, and one pentasaccharide unit (8). As shown in Figure 1, the lipid A structure of E. coli O6 LPS consists of two D-glucosamine residues joined by a β-(1→6)-linkage, two monophosphoester groups at O1 and O4′, and six amide/ester-linked fatty acids which anchor the LPS in the outer membrane of the bacterium. The R1 core (most common core type reported for E. coli) of E. coli O6 LPS has two Kdo residues and three Hep residues, two of which have a monophosphoester group at their respective O4 positions in the inner core (Figure 1). Nonstoichiometric decoration with ethanolamine or glucosamine may also occur in this region. The outer core consists of five hexopyranoses, D-glucose, and D-galactose, all of which are α-linked, except for the terminal β-linked glucose (Figure 1). The O-antigen polysaccharide of E. coli O6 LPS substitutes the O3 position of the terminal glucosyl residue in the core. The linkage between the reducing end sugar of the pentasaccharide and the core region has the β-configuration. This is in contrast to the corresponding α-(1→3)-linkage between the repeating units. Access to the semi-rough strain also facilitated determination of the biological repeating unit with a 3-substituted N-acetyl-D-glucosamine residue at the reducing end. The additional sugars in the repeating unit are two β-D-mannoses, an N-acetyl-α-D-galactosamine, leading to four sugars in the backbone of the polymer, and a β-D-glucose residue forming a branched structure via its (1→2)-linkage to the second mannose residue.
3. Methods
Figure 2 shows the overall building and simulation scheme of LPS bilayers that is comprised of five main steps. Each step is described in detail in sections 3.1 to 3.4.
Figure 2.
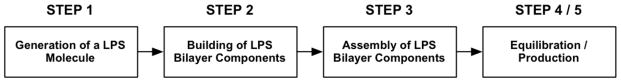
Overall building and simulation procedure of a LPS bilayer.
3.1 Generation of a LPS molecule (STEP 1)
The first step involves the generation of a single LPS molecule with proper sugar types, glycosidic linkages, and phosphorylation, starting from lipid A. For the E. coli O6 LPS molecule (Figure 1), each region (lipid A, R1 core, and O6 antigen) is generated and linked together in CHARMM. This generation step is shown explicitly below to illustrate the complexity of sugar generation procedure with different glycosidic linkage types, unlike the generation of protein, which has identical peptide bonds between residues. Therefore, one needs to be very careful with the glycosidic linkage and sugar types.
lipid A
The molecular topology (LIPA) of Lipid A is initialized in CHARMM and assigned to a segment name of “L1”.
READ SEQUENCE LIPA 1
GENERATE L1 FIRST NONE LAST NONE SETUP WARN
R1 core
The R1 core structure is initialized in this section. First, the sugar residues of the core are initialized and a segment name of “C1” is assigned. Note that the R1 core is consists of 10 residues and the sequence of the residues is not arranged in serial manner due to branches in the sequence. Once initialized, each residue is connected by appropriate glycosidic linkages using PATCH commands.
READ SEQUENCE CARD
* LPS core
*
10
KDOA KDOA HEPA HEPA HEPA
AGLC AGLC BGLC AGAL AGAL
GENERATE C1 FIRST NONE LAST NONE SETUP WARN
PATCH KD24E C1 1 C1 2 SETUP WARN
PATCH KH15A C1 1 C1 3 SETUP WARN
PATCH 13AB C1 3 C1 5 SETUP WARN
PATCH HH17A C1 5 C1 4 SETUP WARN
PATCH 13AB C1 5 C1 6 SETUP WARN
PATCH 13AB C1 6 C1 7 SETUP WARN
PATCH 13BB C1 7 C1 8 SETUP WARN
PATCH 12AB C1 7 C1 9 SETUP WARN
PATCH 12AB C1 9 C1 10 SETUP WARN
PATCH PH4B C1 3 SETUP WARN ! phosphorylation
PATCH PH4B C1 5 SETUP WARN ! phosphorylation
lipid A – R1 link
In this section, the core (segment “C1”) and lipid A (segment “L1”) is joined together using a PATCH command.
PATCH LK26A L1 1 C1 1 SETUP WARN
AUTOGENERATE ANGLE DIHE
O6 antigen
The O-antigen is composed of a varying number of the repeating units, where each one consists of 5 residues in the case of O6. In CHARMM, one can take an advantage of scripting facility in the input level, which allows writing flexible input scripts that can generate a LPS with different number of repeating units without changing the input. In the first half of the input, the first repeating unit of O6 antigen is generated and a segment name of “O1” is assigned. In the second part of the input (starting from the LABEL command), the input goes into a loop and repeats the command in between LABEL and GOTO commands until the condition is met (until the variable “I” is less than the variable “Noantigen”, which is an input number of the O-antigen unit). Inside the loop, an additional repeating unit is generated and different segment names are assigned (“O2”, “O3”, …). Note that “@I” represents the value of the variable “I”.
READ SEQUENCE CARD
* LPS o-antigen unit
*
5
BGLCNA AGALNA BMAN BMAN BGLC
GENERATE O1 FIRST NONE LAST NONE SETUP WARN
PATCH 13BB O1 1 O1 3 SETUP WARN
PATCH 14BB O1 3 O1 4 SETUP WARN
PATCH 13AB O1 4 O1 2 SETUP WARN
PATCH 12BA O1 4 O1 5 SETUP WARN
CALC I = 2
LABEL DOGENER
READ SEQUENCE CARD
* LPS o-antigen unit
*
5
AGLCNA AGALNA BMAN BMAN BGLC
GENERATE O@I FIRST NONE LAST NONE SETUP WARN
PATCH 13BB O@I 1 O@I 3 SETUP WARN
PATCH 14BB O@I 3 O@I 4 SETUP WARN
PATCH 13AB O@I 4 O@I 2 SETUP WARN
PATCH 12BA O@I 4 O@I 5 SETUP WARN
INCREASE I BY 1
IF I .LE. @nOANTIGEN GOTO DOGENER
R1 – O6 link
The core structure and the first unit of O-antigen are connected here.
PATCH 13BB c1 8 o1 1 SETUP WARN
O6 – O6 link
Each repeating unit of O-antigen is connected sequentially here.
CALC I = 2
LABEL DOPATCH
CALC J = @I - 1
PATCH 14AA o@J 2 o@I 1 SETUP WARN
INCREASE I BY 1
IF I .LE. @nOANTIGEN GOTO DOPATCH
Like other typical lipid molecules, LPS molecules in a membrane environment would have various conformations, and unfortunately, no information of average glycosidic bond dihedral angles or their probable distributions are currently available. This implies that the initial LPS 3D structures need to be modeled properly to build a LPS bilayer, and sufficient conformational sampling of the LPS molecules using MD simulations would be necessary to obtain representative (or an ensemble of) LPS structures in the membrane environment. Initial coordinates of the LPS molecule were assigned using internal coordinate information of common glycosidic dihedral angle values (see Table 1) through the “IC BUILD” command in CHARMM. To improve sampling and study the impact of chain length of the O-antigen polysaccharide, we have built four different E. coli O6 LPS molecules and a corresponding bilayer for each LPS. For simplicity, they are denoted as LPS0 (lipid A + R1 core), LPS5 (lipid A + R1 core + 5 units of O6 antigen), LPS10 (lipid A + R1 core + 10 units of O6 antigen), and LPS20 (lipid A + R1 core + 20 units of O6 antigen). Figure 3A shows the 3D structure of a single LPS5 molecule.
Table 1.
Initial glycosidic torsion angles
| Glycosidic linkage1 | Glycosidic torsion angle2
|
||||
|---|---|---|---|---|---|
| ϕ | ψ | ω | ε | ||
| 1′→1″ | β-(1→6) | −65 | −130 | 60 | |
| 2→1′ | α-(2→6) | 68 | 180 | 162 | |
| 3→2 | α-(2→4) | 60 | −86 | ||
| 4→2 | α-(1→5) | 22 | −125 | ||
| 5→4 | α-(1→3) | 65 | −141 | ||
| 6→5 | α-(1→7) | 71 | 120 | −41 | −120 |
| 7→5 | α-(1→3) | 65 | −141 | ||
| 8→7 | α-(1→3) | 65 | −141 | ||
| 9→8 | α-(1→2) | 87 | 115 | ||
| 10→9 | α-(1→2) | 87 | 115 | ||
| 11→9 | β-(1→3) | −130 | −130 | ||
| 12→11 | β-(1→3) | −130 | −130 | ||
| 13→12 | β-(1→3) | −130 | −130 | ||
| 14→13 | β-(1→4) | −130 | 82 | ||
| 15→14 | β-(1→2) | −168 | −130 | ||
| 16→14 | α-(1→3) | 65 | −141 | ||
Residue number for carbohydrates in the LPS molecule (see Figures 1) is used.
The following glycosidic torsion angle definitions are adopted: ϕ = O5′-C1′-Ox-Cx, ψ = C1′-Ox-Cx-C(x−1), ω = Ox-Cx-C(x−1)-C(x−2), and ε = Cx-C(x−1)-C(x−2)-C(x−3), whereas for the (2→6) and (2→4) linkages ϕ = O6′-C2′-Ox-Cx, ψ = C2′-Ox-Cx-C(x−1), ω = Ox-Cx-C(x−1)-C(x−2).
Figure 3.
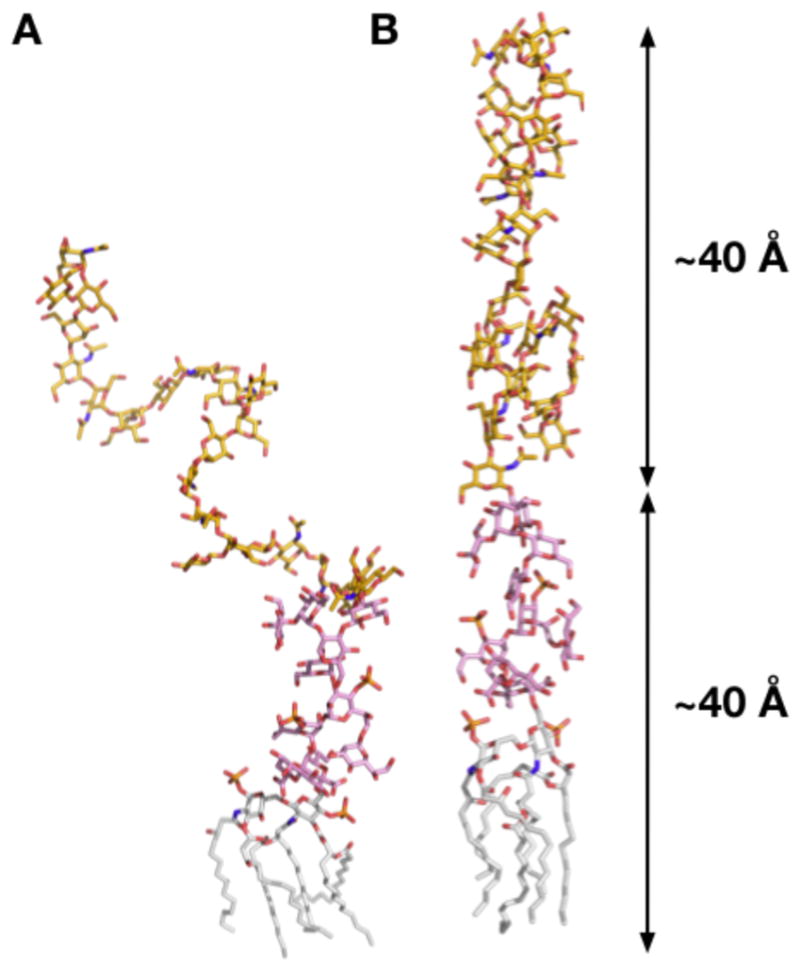
3D structures of a LPS5 (lipid A + R1 core + 5 units of O6 antigen) single molecule, generated by (A) CHARMM IC BUILD and (B) Langevin dynamics with cylindrical restraints: lipid A (gray), R1 core (pink), and O6-antigen (yellow).
Any LPS initial structures generated by “IC BUILD” may not be linear as shown in Figure 3A, and any nonlinear LPS form can cause severe bad contacts when they are assembled to form a membrane. Therefore, to make the building procedure general for any types of LPS molecules, the following procedures are used to make each LPS single molecule to be cylindrical.
Orient each LPS molecule along the Z-axis and place phosphorus atoms (PA and PB) in lipid A at Z = 20 Å; the Z-axis is parallel to the bilayer normal.
Perform Langevin dynamics (LD) with the oriented LPS molecule using a cylindrical restraint potential to retain the LPS molecule to be cylindrical in shape.
We ran a total of 12 sequential LD simulations. Each run was 10 ps, and the target radius for the cylindrical restraint potential starting from a radius of 16 Å was reduced by 1 Å to make the final LPS molecule confined in a cylinder with a radius of 8 Å. A radius of 8 Å roughly corresponds to a per-lipid area of 200 Å2 that was used for an initial guess of the LPS molecule surface area. To avoid any severe bad contacts during the membrane assembly, we used a slightly larger area (200 Å2) than 180 Å2; 180 Å2 is an estimate based on an assumption that each of 6 acyl chains in lipid A has about ~30 Å2 from DMPC (28). The LPS area in the membrane would be converged during the NPT (constant pressure and temperature) dynamics for equilibration and production (STEP 4/5). The resulting LPS5 structure after the LD simulations is shown in Figure 3B.
3.2 Building of LPS bilayer components (STEP 2)
This is the most critical step to build the LPS bilayer components (bilayer itself, ions, and water). First, a LPS bilayer composed of 50 LPS0, LPS5, LPS10, and LPS20 (25 LPSs in each leaflet) was made through the following rigid-body search procedure.
The XY center of PA and PB (two phosphorus atoms of lipid A) was initially placed in a 5 × 5 square grid with random rotations along the Z-axis in one leaflet. The grid spacing was based on an initial surface area of 200 Å2/LPS.
The initial Z position of lipid A PA and PB was set to 20 Å.
To remove bad contacts, we performed a systematic translation and rotational rigid-body search for each LPS molecule: translation with ~1.1 Å spacing for ±2.2 Å search from each LPS’s XY position (5 moves in the X and Y directions, respectively) and rotation with 11° interval for 360° (32 moves). A bad contact is defined when a heavy atom-heavy atom distance is less than 2.5 Å.
The search continued until the average number of bad contacts per lipid reached a minimum. We made 5 independent systems for each LPS molecule using different initial random numbers, and the average number of bad contacts per lipid was 0 (LPS0), 0.03 (LPS5), 0.2 (LPS10), and 0.4 (LPS20). Simulations of multiple independent systems provide better sampling and one can also check the simulation convergence.
To build a bilayer, the lower leaflet was generated based on the minimum-contact upper leaflet by 180° rotation with respect to the X-axis. So, the bilayer center is located at Z = 0. The resulting structure of a LPS5 bilayer is shown in Figure 4A.
Figure 4.
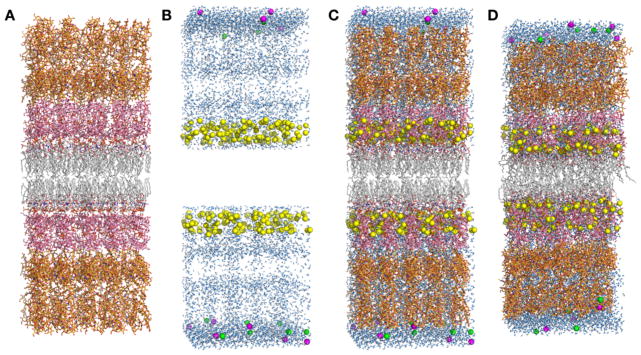
LPS5 bilayer simulation system. (A) An initial bilayer assembly through the rigid-body search to minimize the number of bad contacts. (B) Initial positions of water molecules (light blue) and ions (Ca2+: yellow, K+: magenta, and Cl−: green). (C) An initially assembled structure (B+C). (D) A structure after equilibration.
The next step is to generate ions. The total charge of E. coli O6 lipid A and R1 core is −10e and the O-antigen segment is neutral. Ca2+ was used to neutralize the lipid A and the core, i.e., 125 Ca2+ ions in each leaflet. 150 mM KCl was used for bulk ionic solution, and the number of KCl ions was determined by the system area times the 10 Å bulk region (along the Z-axis), i.e., 10 K+ and Cl− ions. To obtain initial positions of ions, we performed 2000 steps of Monte Carlo (MC) simulations for Ca2+ ions and 2000 steps of MC simulations for K+ and Cl− ions. LPS-ion interaction energies were calculated using a primitive model with the Coulombic interaction scaled by a dielectric constant of 80 and van der Waals interaction. During the MC runs, Ca2+ ions (125 for each leaflet) were restricted to the lipid A and core region, and K+ and Cl− ions to the bulk region. The resulting ion positions in a LPS5 bilayer are shown in Figure 4B. Finally, a water box was made to solvate LPS bilayer systems in the next step. The water box had a dimension of the system area (XY) times 20 Å (Z).
3.3 Assembly of LPS bilayer components (STEP 3)
This step assembles the components and solvates the system with the following procedure.
LPS and ions were assembled.
The water box was sequentially overlaid with LPS and ions from Z = +15 Å to Zmax and from Z = −15 Å to Zmin; Zmax is defined by the maximum Z position of the LPS molecules in the upper leaflet plus the 10 Å bulk water region, and Zmin is defined by the minimum Z position of the LPS molecules in the lower leaflet minus the 10 Å bulk water region. Any water molecule whose oxygen atom had a distance less than 2.8 Å from any LPS heavy atoms or ions was removed during the sequential overlays. The resulting structure of a fully hydrated LPS5 bilayer is shown in Figure 4C.
We built 5 independent systems for each LPS bilayer type by repeating the above steps with different initial random numbers. The number of atoms in each bilayer system is ~48,000 (LPS0), ~84,000 (LPS5), ~136,000 (LPS10), and ~225,000 (LPS20). Due to the system size, we did not herein perform the equilibration and production for the LPS20 system.
3.4 Equilibration and production (STEP 4/5)
After a complex LPS bilayer system is assembled, simulations to equilibrate the system must be performed to relax the uncorrelated initial system. In this work, we have modified/extended the equilibration steps used in the current CHARMM-GUI Membrane Builder for the LPS bilayer system. As shown in Table 2, to assure gradual equilibration of the assembled system, various restraints are applied to the LPS and water molecules, and the restraint forces are gradually reduced during the equilibration. In addition, to warrant the successful equilibration, i.e., to avoid instability of dynamics integrations during equilibration, the NVT dynamics (constant volume and temperature) was used for the first and second steps and the NPT (constant temperature and pressure) dynamics for the rest at 310 K. Each equilibration run was 25,000 steps (1-fs timestep in step 1–3 and 2-fs timestep in step 4–10). The equilibrated structure of a fully hydrated LPS5 bilayer is shown in Figure 4D. Note that there is no unique equilibration procedure, but the aforementioned equilibration steps (also in Table 2) were optimized through many trial-and-error steps.
Table 2.
Various force constants during the equilibration steps.
| step | 4.1 | 4.2 | 4.3 | 4.4 | 4.5 | 4.6 | 4.7 | 4.8 | 4.9 | 4.10 |
|---|---|---|---|---|---|---|---|---|---|---|
| kwater1 | 2.5 | 2.5 | 1 | 1 | 0.5 | 0.1 | 0 | 0 | 0 | 0 |
| khead2 | 2.5 | 2.5 | 1 | 1 | 0.5 | 0.1 | 0 | 0 | 0 | 0 |
| ktail3 | 2.5 | 2.5 | 1 | 1 | 0.5 | 0.1 | 0 | 0 | 0 | 0 |
| kchiral4 | 250 | 100 | 50 | 50 | 25 | 5 | 0 | 0 | 0 | 0 |
| kring5 | 250 | 200 | 150 | 100 | 100 | 50 | 50 | 50 | 25 | 25 |
| kbilayer6 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 |
kwater is the force constant [in kcal/(mol·Å2)] for the repulsive planar restraints to prevent water from entering into the membrane hydrophobic region (|Z| < 12 Å).
khead is the force constant [in kcal/(mol·Å2)] for the planar restraints to hold the position of lipid A PA and PB along the Z-axis (Z = 20 Å for the upper leaflet and Z = −20 Å for the lower leaflet).
ktail is the force constant [in kcal/(mol·Å2)] for the planar restraints to keep the last atoms of lipid A acyl chains in (|Z| < 5 Å).
kchiral is the force constant [in kcal/(mol·rad2)] for the dihedral restraints to maintain the correct chirality in the lipid A.
kring is the force constant [in kcal/(mol·rad2)] for the dihedral restraints to maintain the chair conformation of sugars.
kbilayer is the force constant [in kcal/(mol·Å2)] for the planar restraint to hold the position of lipid A molecules at Z = 0.
The MD production run is basically the continuation of the (equilibration) simulations without any restraints. However, most membrane MD simulation runs require much longer time to fully equilibrate the systems even after the (proposed) equilibration steps. In the LPS bilayer simulation case, we found that it was necessary to keep the restraint potential for the 4C1 sugar conformation (kring) until the system is fully converged. Figure 5 shows the time-series of per-LPS area in the 5 independent LPS5 systems (including the equilibration steps), and Figure 6 shows the heavy atom distributions of the systems along the Z-axis. Clearly, more production time is necessary to guarantee the simulation convergence for each system, but Figures 5–6 were shown to illustrate the type of information one can obtain from the LPS bilayer simulations.
Figure 5.

Time-series of per-LPS area in the 5 independent LPS5 bilayer systems color coded for each run. The dotted line represents the end of the NPT equilibration (step 4.3 to step 4.10).
Figure 6.

Heavy atom distributions of the LPS5 systems along the Z-axis. (A) Water, lipid A, and calcium ion. (B) The inner and outer core. (C) 5 repeating O-antigen units. The distributions were averaged over the 5 independent systems over the production time in Figure 5.
4. Conclusions
We have described the detailed procedure to build the E. coli O6 LPS bilayer systems. While the LPS molecules only exist in the outer leaflet of the outer membrane of E. coli, we used a bilayer system simply to increase the conformational sampling during MD simulations. And, building an asymmetric membrane system should be relatively easy. We expect that long simulations (more than 100 ns) will be required for these complex LPS systems and a critical validation of bilayer properties will be necessary. To this end, NMR studies on the O-antigen using 1H,1H-NOESY experiments and ultimately NMR data on the complete LPS molecule will be measured in order to compare with and validate the MD simulations of these LPS bilayers. We also plan to incorporate the LPS membrane building procedure in CHARMM-GUI Membrane Builder in the future for its wider usage along with the ability to build the biologically-relevant asymmetric bilayer.
Acknowledgments
This work was supported by the University of Kansas General Research Fund allocation #2301388-003, Kansas-COBRE NIH P20 RR-17708, TeraGrid resources provided by Purdue University (NSF OCI-0503992) (to WI) and grants from the Swedish Research Council and the Stockholm Center for Biomembrane Research/Swedish Foundation for Strategic Research (to GW).
References
- 1.Endotoxin in health and disease. Marcel Dekker; New York: 1999. [Google Scholar]
- 2.Bacterial polysaccharides. Structure, chemical synthesis, biogenesis and interaction with host cells. Springer-Verlag; Wien: 2011. [Google Scholar]
- 3.Silipo A, Castro DC, Lanzetta R, Parrilli M, Molinaro A. Lipopolysaccharides. In: König H, Claus H, Varma A, editors. Prokaryotic cell wall compounds. Springer-Verlag; Berlin: 2010. pp. 133–153. [Google Scholar]
- 4.Wang X, Quinn PJ. Endotoxins: lipopolysaccharides of gram-negative bacteria. In: Wang X, Quinn PJ, editors. Endotoxins: Structure, Function and Recognition, Subcellular Biochemistry. 2010/07/02. Springer Science+Business Media B.V; Dordrecht: 2010. pp. 3–25. [DOI] [PubMed] [Google Scholar]
- 5.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 6.Stenutz R, Weintraub A, Widmalm G. The structures of Escherichia coli O-polysaccharide antigens. Fems Microbiol Rev. 2006;30:382–403. doi: 10.1111/j.1574-6976.2006.00016.x. [DOI] [PubMed] [Google Scholar]
- 7.Linnerborg M, Weintraub A, Widmalm G. Structural studies utilizing 13C-enrichment of the O-antigen polysaccharide from the enterotoxigenic Escherichia coli O159 cross-reacting with Shigella dysenteriae type 4. Eur J Biochem. 1999;266:246–251. doi: 10.1046/j.1432-1327.1999.00851.x. [DOI] [PubMed] [Google Scholar]
- 8.Grozdanov L, Zahringer U, Blum-Oehler G, Brade L, Henne A, Knirel YA, Schombel U, Schulze J, Sonnenborn U, Gottschalk G, Hacker J, Rietschel ET, Dobrindt U. A single nucleotide exchange in the wzy gene is responsible for the semirough O6 lipopolysaccharide phenotype and serum sensitivity of Escherichia coli strain Nissle 1917. J Bacteriol. 2002;184:5912–5925. doi: 10.1128/JB.184.21.5912-5925.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knirel YA, Widmalm G, Senchenkova SN, Jansson PE, Weintraub A. Structural studies on the short-chain lipopolysaccharide of Vibrio cholerae O139 Bengal. Eur J Biochem. 1997;247:402–410. doi: 10.1111/j.1432-1033.1997.00402.x. [DOI] [PubMed] [Google Scholar]
- 10.Whitfield C. Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annual review of biochemistry. 2006;75:39–68. doi: 10.1146/annurev.biochem.75.103004.142545. [DOI] [PubMed] [Google Scholar]
- 11.Moran AP, Knirel YA, Senchenkova SN, Widmalm G, Hynes SO, Jansson PE. Phenotypic variation in molecular mimicry between Helicobacter pylori lipopolysaccharides and human gastric epithelial cell surface glycoforms - Acid-induced phase variation in Lewis(X) and Lewis(Y) expression by H. Pylori lipopolysaccharides. J Biol Chem. 2002;277:5785–5795. doi: 10.1074/jbc.M108574200. [DOI] [PubMed] [Google Scholar]
- 12.Ash WL, Zlomislic MR, Oloo EO, Tieleman DP. Computer simulations of membrane proteins. Biochim Biophys Acta. 2004;1666:158–189. doi: 10.1016/j.bbamem.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 13.Gumbart J, Wang Y, Aksimentiev A, Tajkhorshid E, Schulten K. Molecular dynamics simulations of proteins in lipid bilayers. Curr Opin Struct Biol. 2005;15:423–431. doi: 10.1016/j.sbi.2005.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindahl E, Sansom MSP. Membrane proteins: molecular dynamics simulations. Curr Opin Struct Biol. 2008;18:425–431. doi: 10.1016/j.sbi.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 15.Pastor RW, MacKerell AD. Development of the CHARMM Force Field for Lipids. The Journal of Physical Chemistry Letters. 2011;2:1526–1532. doi: 10.1021/jz200167q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feller SE. Molecular dynamics simulations of lipid bilayers. Current Opinion in Colloid & Interface Science. 2000;5:217–223. [Google Scholar]
- 17.Stansfeld Phillip J, Sansom Mark SP. Molecular Simulation Approaches to Membrane Proteins. Structure. 2011;19:1562–1572. doi: 10.1016/j.str.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 18.Lins RD, Straatsma TP. Computer simulation of the rough lipopolysaccharide membrane of Pseudomonas aeruginosa. Biophys J. 2001;81:1037–1046. doi: 10.1016/S0006-3495(01)75761-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Straatsma TP, Soares TA. Characterization of the outer membrane protein OprF of Pseudomonas aeruginosa in a lipopolysaccharide membrane by computer simulation. Proteins. 2009;74:475–488. doi: 10.1002/prot.22165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jo S, Kim T, Im W. Automated Builder and Database of Protein/Membrane Complexes for Molecular Dynamics Simulations. Plos One. 2007;2 doi: 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jo S, Lim JB, Klauda JB, Im W. CHARMM-GUI Membrane Builder for Mixed Bilayers and Its Application to Yeast Membranes. Biophys J. 2009;97:50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brooks BR, Brooks CL, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. CHARMM: The Biomolecular Simulation Program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guvench O, Greene SN, Kamath G, Brady JW, Venable RM, Pastor RW, Mackerell AD., Jr Additive Empirical Force Field for Hexopyranose Monosaccharides. J Comput Chem. 2008;29:2543–2564. doi: 10.1002/jcc.21004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guvench O, Hatcher E, Venable RM, Pastor RW, MacKerell AD., Jr CHARMM Additive All-Atom Force Field for Glycosidic Linkages between Hexopyranoses. J Chem Theory Comput. 2009;5:2353–2370. doi: 10.1021/ct900242e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hatcher E, Guvench O, MacKerell AD., Jr CHARMM Additive All-Atom Force Field for Aldopentofuranoses, Methyl-aldopentofuranosides, and Fructofuranose. J Phys Chem B. 2009;113:12466–12476. doi: 10.1021/jp905496e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr, Pastor RW. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jansson PE, Lindberg B, Lönngren J, Ortega C, Svenson SB. Structural studies of the Escherichia coli O-antigen 6. Carbohydr Res. 1984;131:277–283. doi: 10.1016/0008-6215(84)85250-7. [DOI] [PubMed] [Google Scholar]
- 28.Klauda JB, Kucerka N, Brooks BR, Pastor RW, Nagle JF. Simulation-based methods for interpreting x-ray data from lipid bilayers. Biophys J. 2006;90:2796–2807. doi: 10.1529/biophysj.105.075697. [DOI] [PMC free article] [PubMed] [Google Scholar]