

Supplemental Digital Content is available in the text.
Keywords: APG101, CD95, CD95L, glioblastoma, invasion
Abstract
Glioblastoma is a disease characterized by rapid invasive tumour growth. Studies on the proapoptotic CD95/CD95L signalling pathway recently suggested a significant contribution of CD95 signalling towards the high degree of motility in glioma cells. Apogenix has developed APG101, a clinical phase II compound designed to bind and neutralize CD95L, and thus to interfere with CD95/CD95L-based signalling. APG101 has shown clinical efficacy in a controlled randomized phase II trial in patients with recurrent glioma. Because APG101 is not cytotoxic to tumour cells in vitro, we postulated that the anti-invasive function of APG101 is the main mechanism of action for this compound. Using three-dimensional spheroid invasion assays in vitro and in murine brain tissue cultures, we found that knockdown of endogenous CD95L reduced the invasive phenotype in our two glioblastoma model cell lines U87-MG and U251-MG. Invasion was restored in CD95L knockdown cells upon the addition of soluble recombinant CD95L and this effect was inhibited by APG101. We conclude that CD95L from autocrine and paracrine sources contributes towards the invasive phenotype of glioblastoma cells and that APG101 acts as a suppressor of proinvasive signalling by the CD95/CD95L pathway in glioblastoma.
Introduction
Glioblastoma is the most frequent and aggressive form of brain tumours, with a very poor prognosis for affected patients. The highly invasive growth of these tumours often prevents complete surgical resection and, consequently, patients progress shortly after surgery. Over the past decade, the adjuvant administration of irradiation combined with chemotherapy (temozolomide) has evolved as the standard of care, although it leads to minimal improvement in terms of patient survival and is often associated with severe side effects. The highly invasive growth of glioblastoma cells has recently been, at least in part, attributed to nonapoptotic signalling of the CD95/CD95L complex, including activation of the Yes/PI3K/Akt 1,2 and other pathways (reviewed in Steller et al. 3).
CD95 (APO1/FAS) was originally discovered because of its ability to induce apoptosis in cells of the immune system 4, but is now recognized as a multifaceted receptor that also mediates nonapoptotic functions 5. In glioma resistant to CD95-induced apoptosis, autocrine or paracrine stimulation of CD95 on the tumour cell by its cognate CD95 ligand (CD95L/Apo1L/FasL/CD178) leads to increased invasion of tumour cells 6. Neutralization of CD95L should therefore benefit patients with glioblastoma by decreasing tumour spread to the surrounding healthy brain parenchyma and eventually prolong time to progression and overall survival of glioblastoma patients.
Apogenix is developing APG101 for the treatment of glioblastoma. APG101 is a fusion protein comprising the N-terminal ligand-binding domain of human CD95 and the Fc-part of human IgG1. APG101 is designed to interfere with the CD95/CD95L interaction by masking CD95L present on the cell membrane or in soluble form. Apogenix has successfully completed a controlled-randomized phase II trial with APG101 in patients with recurrent glioma, showing positive results for progression-free survival and also overall patient survival, while showing excellent tolerability 7.
Neutralization of the signalling capacity of CD95L has different effects on the tumour characteristic as reported in the current literature. Inhibition of CD95L/CD95-mediated signalling exerts a direct inhibitory effect on the invasive growth of tumour cells as described for glioblastoma 1. In addition, CD95L neutralization exerts a systemic immune-modulatory effect that enables the infiltration of the tumour by cytotoxic T cells in a syngeneic murine model of ovarian cancer 8.
To gain a better understanding of the effects of the mode of action of APG101, here we addressed the question of how neutralization of CD95L by APG101 influences the CD95/CD95L-induced migration of glioblastoma cells in vitro and ex vivo. For this purpose, we carried out three-dimensional in-vitro and ex-vivo studies, comparing the invasive growth of multicellular glioblastoma spheroids in vitro in semisolid matrices 9,10 and an ex-vivo model using murine brain tissue cultures 11. Despite the wide use of two-dimensional transwell assays, we found that these assays were not useful when using CD95L as a proinvasive stimulus because CD95L does not act as an attractant such as FBS or hepatocyte growth factor/scatter factor (C. Merz, unpublished data). Therefore, we chose three-dimensional multicellular spheroid-based assays, also because of the more physiological cellular organization of these microtumours, the ease of implantation of a defined spheroid into the three-dimensional-matrices and the higher level of authenticity in the cell–matrix interplay. Our data suggest that neutralization of CD95L by antagonistic molecules such as APG101 exerts a positive effect on disease progression in glioblastoma not through direct cytostatic/cytotoxic effects, but by inhibition of tumour cell invasion, which is at least in part because of prevention of activation of PI3K by the CD95/CD95L signalling complex.
Materials and methods
Recombinant proteins
Human recombinant trimerized CD95L (APG293) was produced and purified by Apogenix GmbH (Heidelberg, Germany) as described 1. Human recombinant CD95-Fc (APG101) was produced at Celonic GmbH (Basel, Switzerland) using a proprietary GMP process. Human recombinant CD95-R87S-Fc (APG122) is a CD95L-binding defective mutant of APG101 harbouring an R87S amino acid exchange in the extracellular domain of CD95 9. APG122 was produced and purified at Apogenix from transiently transfected CHO-S cells using protein-A affinity chromatography, followed by size exclusion chromatography. APG122 serves as a negative control protein with an impaired ability for CD95L interaction. The purity and identity of all proteins were checked by SDS-PAGE using the precast NuPAGE gel system (4–12% gradient gel) in 1× MES running buffer according to the manufacturer’s protocol (Life Technologies/Thermo Fisher Scientific; Waltham, Massachusetts, USA). Separated proteins were visualized by silver staining.
Enzyme-linked immunosorbent assay-based binding assay for APG101 and APG122
Nunc Maxisorp 96-well microtitre plates (VWR International GmbH, Darmstadt, Germany) were coated with 2.5 μg/ml of APG293 or 1.5 μg/ml mouse anti-CD95 antibody (APO1-mIgG1; Apogenix) as the capture matrix in PBS at 4°C overnight. Wells were then blocked for 30 min at 37°C using Starting Block (Thermo Scientific, Rockford, Illinois, USA) and subsequently APG101 and APG122 were added in serial dilutions. Following incubation for 1 h at 37°C, the enzyme-linked immunosorbent assay (ELISA) was washed with PBS–Tween and probed with purified polyclonal rabbit anti-CD95L and rabbit anti-APG101 as detection antibodies for 1 h at 37°C. Rabbit antibodies against CD95L and CD95 were raised against recombinant APG293 and APG101, respectively, and purified at Apogenix. Goat–anti-human IgG–peroxidase conjugate (Dianova GmbH, Hamburg, Germany) was used for ELISA development using a ready-to-use TMB substrate (TMB One; Kem-En-Tec Diagnostics, Taastrup, Denmark) and subsequent analysis of the optical density at 450 nm.
Bioactivity testing of APG293, APG101 and APG122
A potency assay using Jurkat A3 cells was performed to test the ability of APG101 and APG122 to interfere with a well-described biological function of CD95L, the induction of apoptosis. 1×105 cells were seeded in 96-well microtitre plates and treated with APG293. Apoptotic cell death was assessed by the ability to induce activation of the executioner caspases 3 and 7. Proapoptotic activity of APG293 was titrated by preincubation of 250 ng/ml APG293 with increasing amounts of APG101 or the inactive control protein APG122, respectively, for 30 min at room temperature. As a control for specific APG293/APG101 interaction, recombinant TRAIL was used as a proapoptotic control protein of the tumour necrosis factor superfamily that cannot be neutralized by APG101. After 4 h of treatment, Jurkat cells were lysed in lysis buffer (25 mmol/l HEPES pH 7.5, 5 mmol/l magnesium chloride, 1 mmol/l EGTA, 0.5% Triton X-100, 10 mmol/l DTT, 1 mmol/l AEBSF) for 30 min on ice. 20 μl of cell lysate was mixed with 80 μl of substrate buffer (50 mmol/l HEPES pH 7.5, 1% saccharose, 0.1% CHAPS, 25 mmol/l DTT) containing the fluorogenic caspase substrate Ac-DEVD-AFC at 50 nmol/l concentration. Kinetic measurement of caspase activity was performed for 15 min on a Tecan Infinite F200Pro fluorescence reader (Tecan, Mánnedorf, Switzerland). Recombinant TRAIL was purchased from PeproTech (Hamburg, Germany).
Cell lines and culture media
The cell lines U87-MG and U251-MG were cultured as monolayers at 37°C in RPMI-1640 supplemented with 10% FBS in a humidified incubator with 5% CO2. Cells were passaged by trypsinization when reaching 70–80% confluence, with complete medium change twice weekly. Transfected cell lines were kept under identical growth conditions in medium containing 1 μg/ml puromycin. Jurkat A3 cells for the potency assay were purchased from ATCC (LGC Standards GmbH, Wesel, Germany) (CRL-2570) and cultured in RPMI-1640 medium supplemented with 10% FBS in a humidified incubator with 5% CO2. Jurkat cells were passaged at a split ratio of 1 : 15 twice weekly with complete medium change. All cell lines used within this study were tested routinely for mycoplasma contamination using the VenorGem kit (Minerva Biolabs, Berlin, Germany).
Quantification of endogenous CD95L and CD95 by enzyme-linked immunosorbent assay
Nunc Maxisorp 96-well microtitre plates were coated with 2.5 μg/ml of mouse anti-CD95L (NOK-2; BD Biosciences, Heidelberg, Germany) or mouse anti-CD95 antibody (APO1-mIgG1; Apogenix) as capture antibodies in PBS at 4°C overnight. Wells were then blocked for 30 min at 37°C using Starting Block and, subsequently, cell lysates (50 μg protein per well for the detection of CD95L or 20 μg protein per well for the detection of CD95) were added. Following incubation for 2 h at 37°C, the ELISA was washed with PBS–Tween and probed with rabbit polyclonal anti-CD95L and rabbit anti-APG101 (both antibodies were raised and purified by Apogenix) as detection antibodies for 1 h at 37°C. Rabbit antibodies were detected using a goat–anti-rabbit IgG–peroxidase conjugate with a ready-to-use TMB substrate (TMB One; Kem-En-Tec) and subsequent analysis of the optical density at 450 nm. Amounts of endogenous CD95L and CD95 were calculated using a 4-parameter fit in SigmaPlot on the basis of a calibration curve using APG293 and APG101 as standards.
Cell viability assay (MTS)
Cell viability after treatment with APG293 and APG101 was assessed using the MTS-based assay (Cell Titer 96 Aqueous Non-Radioactive Cell Proliferation Assay; Promega GmbH, Mannheim, Germany). Cell lines were cultured as monolayers, detached using trypsinization and counted. 1×103 cells per well were seeded in triplicate in 96-well microtitre plates in complete growth medium and incubated in humidified incubators at 37°C and 5% CO2. APG293 and/or APG101 were added to final concentrations indicated in the respective figures immediately after plating of cells. Forty-eight hours and 96 h after seeding, MTS was added and optical density was determined at 492 nm. OD492 values of treated cells were used to calculate (%) cell viability against untreated cells and a dose–response curve.
Construction and transfection of vectors encoding shRNAs
The shRNA sequences were cloned into Promega pGeneClip vectors by PCR-based outward full vector amplification, followed by head-to-head ligation with the following PCR primer sequences:
SCRMBL control sequence (5′→3′):
Sense-oligo: tgtca ATAGCTCTAGTAGCGCTAGC gcagtctggagtttcaaaagtagac.
Anti-oligo: ggaag ATAGCTCTAGTAGCGCTAGC gagatcttgggcctctgcc.
CD95L sequence (5′→3′):
Sense-oligo: gagacATTAGGTGAGTTGAGGAGCTAcgcagtctggagtttcaaaagtagac.
Anti-oligo: gagacATTAGGTGAGTTGAGGAGCTAcgagatcttgggcctctgcc.
Sense-oligos were obtained containing a 5′-phosphorylation for ligation. Target sequences (reverse complement) are shown in upper case. All vectors were sequence verified before transfection. Transfection of U87-MG and U251-MG cells with sh[SCRMBL] or sh[CD95L] vectors was performed using Dharmafect Duo (Thermo Fisher Scientific) according to the manufacturer’s protocols. Transfected cells were selected starting 2 days after transfection using increasing amounts of puromycin over a period of 2 weeks and subsequently cultured in RPMI-1640 with 10% FBS and 1 μg/ml puromycin. Endogenous levels of CD95L in the sh[SCRMBL] and sh[CD95L] vector transfected cells were quantified by ELISA analysis of 50 μg whole-cell lysate proteins with an ELISA set-up developed at Apogenix using a calibration curve of APG293.
Spheroid initiation
Single-cell suspensions were membrane labelled using Vybrant DiI (Life Technologies) according to the manufacturer’s protocol. Formation of spheroids was then initiated from 500 cells (U87) or 1000 cells (U251) in 100 μl of complete medium in agarose-coated 96-well microtitre plates (day 0). On day +1, 100 μl of serum-free Neurobasal-A medium (Life Technolgies, Darmstadt, Germany) was added and half of the medium was replaced by fresh Neurobasal-A medium on day +3. After 4 days, the spheroids were picked from the wells and used for invasion assays.
Three-dimensional spheroid invasion assays
An in-vitro spheroid invasion assay method has been described in the studies by Weiler et al. 10 and Pfenning 11 and was used with modifications as follows: growth factor-reduced Matrigel (BD Biosciences) was thawed on ice and diluted to 4 mg/ml (protein concentration) with ice-cold serum-free RPMI-1640. The matrix was then supplemented with 5–10 ng/ml APG293 in the presence or absence of 100 μg/ml APG101 or 100 μg/ml APG122 and briefly vortexed. 50 μl of matrix was added to each well of a 96-well microtitre plate and incubated at 37°C for 1 h to allow gelling. Single spheroids were then implanted into each well containing control or supplemented matrix. Invasion was monitored by taking pictures under a light microscope (Leica EC3; Leica, Wetzlar, Germany) immediately after implantation and after 72 h (magnification 40-fold, resolution 1260×840). Digital images were compiled without further adjustments. Depicted spheroids are representative of three independent experiments. Quantification of invasion was performed as follows: the mean radius of the spheroids was measured at the 0 h time point. The mean radius at 0 h and an even radial grid (20° steps) emanating from the centre of the spheroid was projected on the picture of spheroids after 72 h. The radial distance of the farthest invaded cell along the gridline after 72 h was determined using the Keyence BZ II Analyzer software (Keyence, Neu-Isenburg, Germany). Measured values were imported into Microsoft Excel 2010. The mean radial distance of invaded cells at 72 h/mean radius at 0 h is calculated and shown in figures as ‘fold-increase of mean radius after 72 h’.
Brain slice invasion assay
Five-to-eight week-old C57BL/6 mice were anaesthetized and killed. Brains were removed, embedded in 1% agarose/PBS and cut into 350-μm-thick coronal sections using a Microm HM 650V vibratome (Thermo Fisher Scientific). The brain slices were cultured on Millicell cell culture inserts (Millipore Merck KGaA, Darmstadt, Germany) in MEM medium containing 25% horse serum, 25 mmol/l HEPES [4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid], 1 mmol/l glutamine, 5 mg/ml glucose, 100 U/ml, penicillin and 100 U/ml streptomycin for 2 days at 37°C in a humidified atmosphere (5% CO2) to allow the culture media to form an interface for proper oxygenation and nutrition of the tissue. The slices were pretreated for 12 h with medium only or with medium containing 10 ng/ml APG293 in the presence or absence of 50 μg/ml APG101 or 50 μg/ml APG122. Subsequently, the fluorescence-labelled tumour cell spheroids were implanted into the preincubated brain slices. During the observation period, APG293 was omitted while treatment with APG101 and APG122 was continued. Images were taken immediately after implantation and after 96 h using a Nikon Eclipse Ti microscope (Nikon GmbH, Düsseldorf, Germany). Digital images were adjusted for brightness, contrast and colour balance using Adobe Photoshop CS5.1 (Adobe Systems, München, Germany). Images are shown in false colours.
Results
APG101 was developed to interfere with CD95/CD95L-dependent signalling by binding to and neutralizing CD95L and should therefore block CD95L-induced invasion. Here, we show in-vitro experimental evidence supporting the hypothesized mode of action of APG101 acting on the tumour cell and the tumour microenvironment to inhibit CD95L-induced glioma cell invasion.
APG293 and APG101 are interaction partners in vitro
To assess the biological activity of the compounds used, we purified APG293 and the CD95-Fc fusion proteins APG101 and APG122, the latter being a ligand-binding defective mutant of APG101 serving as an inactive control protein (Fig. 1a). All proteins were purified to homogeneity and analysis by SDS-PAGE and subsequent silver staining showed no contaminating proteins in either of the protein preparations. The identity and binding properties of APG293, APG101 and APG122 were verified by ELISA (Fig. 1b). The identity of APG101 and APG122 was confirmed by detection of the human Fc-part of the molecule after immobilization by an antibody specific for the human CD95 extracellular domain. To assess the respective CD95L-binding properties of APG101 and APG122, APG293 was immobilized to an ELISA plate and used to capture both molecules. As expected, APG101 showed a dose-dependent increase in the ELISA signal, indicating specific binding to APG293. However, even at high protein concentrations, no ELISA signal could be obtained with APG122, thereby confirming the inability of the R87S point mutation to interact with CD95L (Fig. 1b).
Fig. 1.
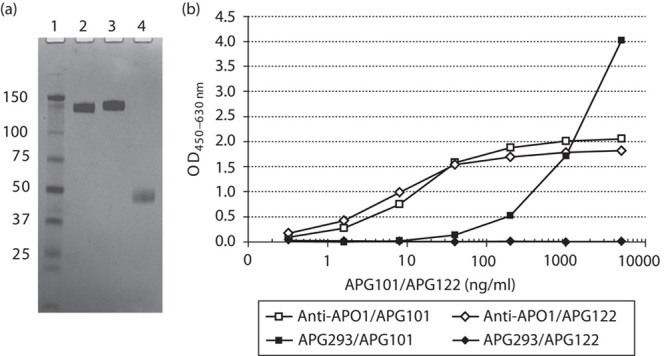
Characterization of the recombinant proteins. (a) Nonreducing SDS-PAGE analysis of recombinant CD95-Fc (APG101, lane 2), CD95-R87S-Fc (APG122, lane 3) and CD95L (APG293, lane 4) showing purity. Lane 1: molecular weight standard. (b) Enzyme-linked immunosorbent assay shows binding of APG293 by APG101, but not the R87S mutant APG122. The control antibody anti-APO1 captures both APG101 and APG122.
APG101 specifically neutralizes the proapoptotic activity of recombinant CD95L
Jurkat A3 cells are used widely for in-vitro studies on the process of apoptosis. Two apoptosis-inducing proteins (death receptor agonists), APG293 and TRAIL, were used in an in-vitro cell death assay. As shown in Fig. 2a, both proteins trigger apoptosis in Jurkat A3 cells, indicated by the activation of caspases in a dose-dependent manner after 4 h of treatment. To examine the specificity and biological activity of APG101, we used a fixed concentration of 250 ng/ml APG293 or TRAIL, and attempted to quench the proapoptotic activity of both molecules by the addition of increasing amounts of either APG101 or APG122 (Fig. 2b). Apoptosis induction by APG293 was dose dependently and completely neutralized by APG101, whereas APG122 exerted no detectable effect. Both compounds did not interfere with TRAIL-induced activation of apoptosis.
Fig. 2.
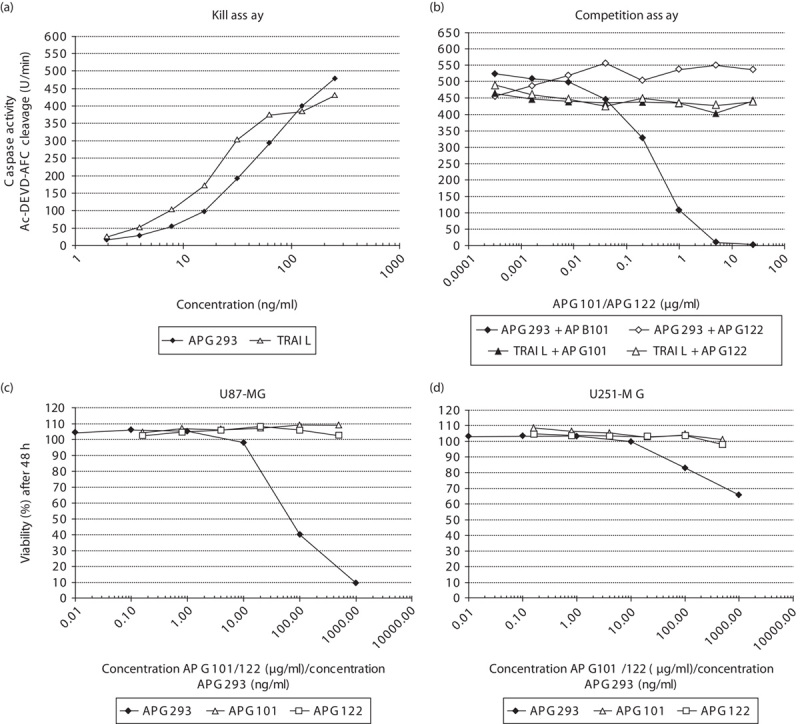
Analysis of apoptosis induction and cell proliferation. (a) Caspase 3/7 assay. Jurkat A3 cells were treated with indicated amounts of APG293 or TRAIL to induce apoptosis. Dose-dependent caspase activation was measured after 4 h of incubation. (b) APG101 or APG122 were used to compete with the proapoptotic activity of APG293 or TRAIL. 250 ng/ml APG293 or TRAIL were coincubated with increasing amounts of APG101 or APG122, respectively, and then added to Jurkat A3 cells. Dose-dependent decrease in caspase activation was measured after 4 h of incubation. (c, d) MTS-based cell viability assay: indicated cell lines were exposed to APG293 (1–1000 ng/ml) and APG101 (16–500 μg/ml) for 48 h before the addition of MTS.
Resistance of glioma cell lines U87-MG and U251-MG to CD95/CD95L induced killing
The aim of this study was to analyse CD95L-induced invasion in vitro using the established glioblastoma cell lines U87-MG and U251-MG. To exclude possible cytotoxic side effects of APG293 or APG101 on U87-MG and U251-MG cells, we assessed cell viability using an MTS assay and in addition analysed the proliferation rates using sulforhodamine-B. In vitro, concentrations of up to 1000 ng/ml of APG293 were used to test for CD95L-induced apoptosis. For APG101, estimated serum levels in patients under APG101 treatment during the phase II study were in the range of 40–300 μg/ml, and we therefore tested concentrations of up to 500 μg/ml of APG101 for in-vitro cell exposure under assay conditions. After 48 h (MTS assay) and 1 week (sulforhodamine-B) of cell culture in the presence of the compounds, no effects of APG101 or APG122 on cell viability or proliferation rates were observed. We observed slightly impaired proliferation and significant reduction in the overall viability of U87-MG cells at APG293 concentrations exceeding 10 ng/ml (Fig. 2c and Supplementary Fig. S1, Supplemental digital content 1, http://links.lww.com/ACD/A105). Proliferation rates of U251-MG cells were not affected by either compound, although we also observed reduced viability at increased APG293 concentrations after 48 h (Fig. 2d and Supplementary Fig. S1, Supplemental digital content 1, http://links.lww.com/ACD/A105). In general, U251-MG cells are more resistant to CD95L-induced killing compared with U87-MG cells. In addition, we analysed the possible effects of APG293 and APG101 on cell cycle progression of U251-MG cells. As shown in Supplementary Fig. S2 (Supplemental digital content 1, http://links.lww.com/ACD/A105) relative quantification of G0/G1-phase, S-phase and G2/M-phase populations of monolayer cell cultures in the log phase by DNA content (7AAD labelling) was performed after 48 h incubation with APG101 or APG293. None of these experiments provided a hint of any impediment during the cell cycle by the treatment applied. On the basis of these results, we chose a concentration of 10 ng/ml APG293 and 50 to 100 μg/ml APG101/APG122 as a ‘safe’ compound dose for the following in-vitro/ex-vivo experiments on CD95/CD95L-induced migration.
Knockdown of CD95L by shRNA moderately affects the proliferation of glioma cells
Endogenous sources of CD95L in the context of cancer can be either the tumour cells or the tumour microenvironment. To first test for the contribution of endogenous CD95L towards the basal invasiveness of glioblastoma cells, we transfected U87-MG and U251-MG cells with vectors encoding either control shRNA sh[SCRMBL] or shRNA targeting CD95L sh[CD95L]. Because of the very low target abundance in non-transfected cells, endogenous levels of CD95L or the corresponding mRNA are not reliably detectable by western blot or RT-PCR, respectively. To verify the knockdown of CD95L, we therefore used a specific and sensitive ELISA set-up using our own CD95L and CD95 antibodies that are capable of detecting as little as 5 pg/ml CD95L or CD95, respectively. ELISA analysis of whole-cell lysates of control-transfected cells confirmed very low levels of CD95L in the control cells (3–4.5 pg/μg total protein), but specific knockdown with shRNA targeting CD95L reduced endogenous CD95L close to the level of detection (Fig. 3a). Endogenous CD95 levels were not altered in CD95L knockdown cells compared with control cells (Fig. 3b and Supplementary Fig. S6 A and B, Supplemental digital content 1, http://links.lww.com/ACD/A105). We then assessed the content of soluble CD95L in the supernatants of respective spheroid cultures. Importantly, knockdown of CD95L in both cell lines did not alter the ability of the cells to form spheroids. Consistent with the ELISA from cell lysates, low levels of CD95L were detected in supernatants of both [SCRMBL] cell lines, but not in [CD95L] knockdown cells (Supplementary Fig. S5, Supplemental digital content 1, http://links.lww.com/ACD/A105). However, we observed slightly decreased proliferation rates in U251[CD95L] cells compared with U251[SCRMBL] cells, but this effect was absent in the corresponding U87[CD95L] knockout cells (data not shown). Third, we used analytical FACS to detect membrane-bound CD95L in monolayer and spheroid cultures of the transfected cell lines (Supplementary Fig. S6 A and B, Supplemental digital content 1, http://links.lww.com/ACD/A105). We found that surface exposure of the transmembrane form of the CD95 ligand was undetectable in all cell samples tested, in contrast to CD95 and TRAIL-R2, both of which were readily detected on cells cultured as monolayers or spheroids. We observed a broadening of the CD95 peak in U251-MG-derived spheroids compared with monolayer cells, which indicates changes in CD95 expression levels of individual cells. Similarly, the surface expression of TRAIL-R2 in U251-MG spheroids was lower compared with monolayer cells. (Supplementary Fig. S6 B, Supplemental digital content 1, http://links.lww.com/ACD/A105). Both effects were, however, absent in U87-MG cells. The knockdown of CD95L did not seem to affect the overall expression of CD95 or TRAIL-R2 in any case.
Fig. 3.
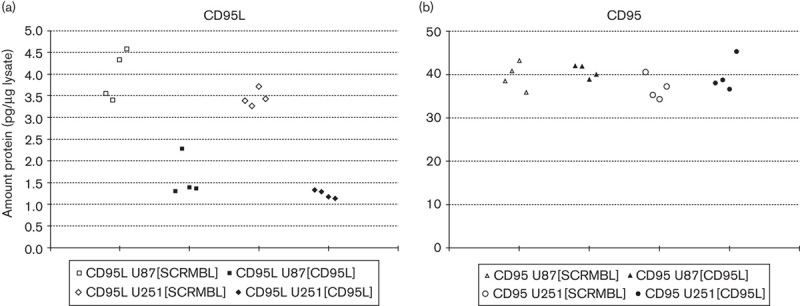
Enzyme-linked immunosorbent assay (ELISA)-based analysis of CD95L-knockdown efficacy. (a) ELISA-based verification of the CD95L knockdown in U87-MG and U251-MG cells. Whole-cell lysates of cells transfected with sh[SCRMBL] or sh[CD95L] vector containing 50 μg of total protein were used to compare the abundance of endogenous CD95L. The calculated amount of CD95L and LOD was derived from a calibration curve using APG293 as the control compound. (b) ELISA-based analysis of CD95 upon knockdown of CD95L. The calculated amount of CD95 was derived from a calibration curve using APG101 as the control compound.
Knockdown of CD95L impairs the invasion of glioma cells in the three-dimensional in vitro assay
Defined single spheroids of shRNA-transfected U87-MG and U251-MG cells were initiated as described in the Materials and methods section. Five days after spheroid initiation, single spheroids were implanted into Matrigel and spheroid outgrowth was monitored over 72 h. Transfected control cells showed high basal invasiveness into the Matrigel and after 72 h, the main body of the implanted spheroid was partially disintegrated because of the massive invasion of the cells from the spheroid body into the matrix (Fig. 4a). In contrast to the control-transfected cells, spheroids that consisted of [CD95L] knockdown cells did not invade the matrix. In addition, spheroid integrity after 72 h was still comparable with the situation observed at the beginning of the experiment (Fig. 4a).
Fig. 4.
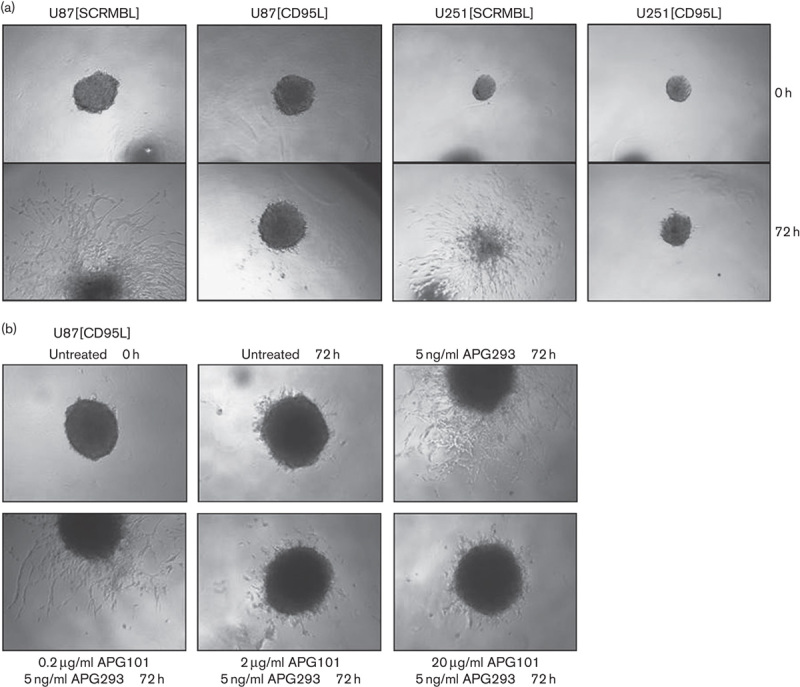
Spheroid invasion assay. (a) Matrigel-based three-dimensional spheroid invasion assay using spheroids of transfected U87-MG cells shown at the start of the assay and after 72 h. Spheroids of the control-transfected cell line U87[SCRMBL] showed invasive behaviour in vitro without stimulation by APG293, whereas their untreated counterparts after CD95L knockdown were refractory. (b) Invasion of U87[CD95L] knockdown cells was induced by the addition of 5 ng/ml APG293 to the semisolid assay matrix. Coincubation of APG293 was performed for 30 min with increasing amounts of APG101. Final concentrations of compounds are indicated in the figure. Spheroids are shown at the start of the assay (0 h) and after 72 h.
To further confirm the influence of CD95L on the induction of cell migration, we next attempted to induce migration of [CD95L] knockdown cells by the addition of APG293 to the Matrigel. As shown in Fig. 4b, the addition of 5 ng/ml APG293 to the matrix rescued invasion of the [CD95L] knockdown cells. These results show that CD95L either from autocrine or from paracrine sources – for example, the tumour microenvironment – can trigger proinvasive CD95 signalling. Hence, we analysed whether the invasion observed in the presence of 5 ng/ml APG293 could be blocked by the concomitant addition of APG101. We added increasing doses of APG101 to the Matrigel invasion assay that contained a constant amount of 5 ng/ml APG293. In this setting, APG101 dose dependently reduced the invasion triggered by APG293. At the highest APG101 concentration tested in this experiment (20 μg/ml), invasion observed after 72 h was reduced to the level of unstimulated [CD95L] knockdown spheroids after 72 h (Fig. 4b and Supplementary Fig. S3, Supplemental digital content 1, http://links.lww.com/ACD/A105). This result clearly shows that APG101 is capable of interfering with CD95-induced migration/invasion of glioma cells.
To confirm the specificity of the inhibitory effect of APG101 on CD95L-induced invasion, we additionally tested the influence of APG101 and APG122 side by side. Spheroids derived from U251[CD95L] and U87[CD95L] knockdown cells were either left untreated for 72 h in Matrigel or invasion was stimulated by the addition of 10 ng/ml APG293. In addition, assays that contained APG293 were supplemented with either 100 μg/ml APG101 or APG122. As shown in Fig. 5, [CD95L] knockdown spheroids from both cell lines show only limited invasion into the Matrigel (see also Supplementary Fig. S8A and B, Supplemental digital content 1, http://links.lww.com/ACD/A105 for quantification). In both cases, invasion into the Matrigel could be induced by the addition of 10 ng/ml APG293. Importantly, abrogation of APG293-induced invasion was only observed after coincubation with APG101, but not when using APG122 (Fig. 5 and Supplementary Fig. S8 A and B, Supplemental digital content 1, http://links.lww.com/ACD/A105). Thus, APG101, but not APG122, could neutralize the proinvasive signal elicited by CD95 activation in vitro.
Fig. 5.
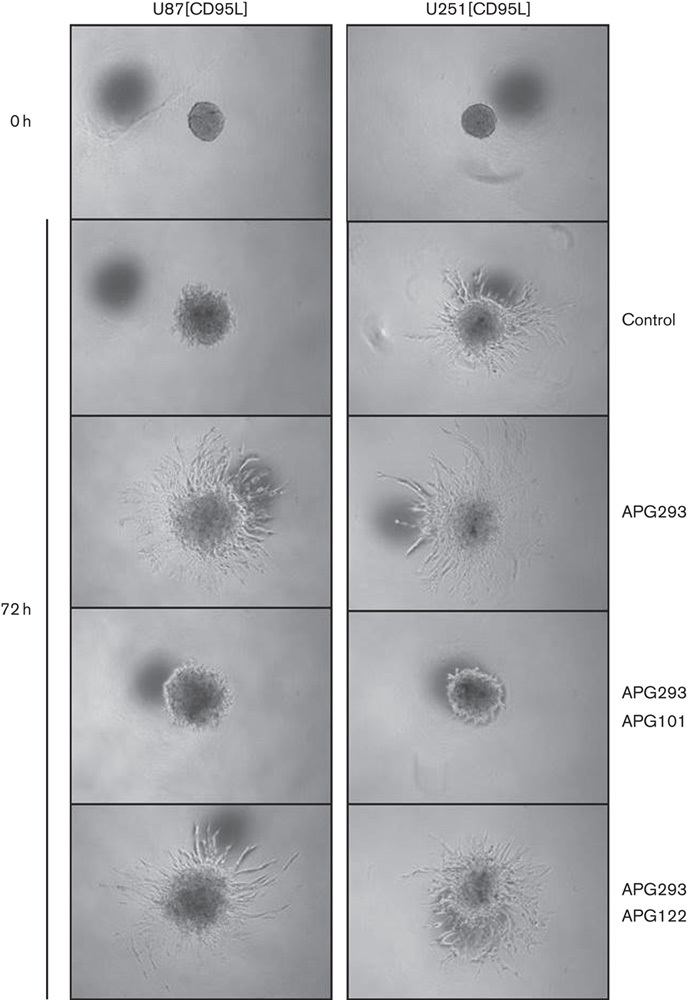
Spheroid invasion assay. Invasion of U87[CD95L] and U251[CD95L] spheroids is shown at the start of the assay and after 72 h. Invasion after CD95L knockdown was induced by the addition of 10 ng/ml APG293 to the assay matrix. Coincubation of APG293 was at room temperature for 30 min with either 100 μg/ml APG101 or 100 μg/ml APG122.
Nonapoptotic CD95 signalling is considered to be executed at least in part by activation of PI3K 1. To test whether the proinvasive effect of CD95L from a paracrine source can be comparably countered by inactivation of PI3K, we performed comparative in-vitro assays using APG293 to stimulate U87[SCRMBL] and U87[CD95L] cells in combination with APG101 or the well-known pan-PI3K-inhibitor LY294002. We observed that both inhibitory compounds efficiently neutralized the ability of APG293 to induce invasion (Supplementary Fig. S9, Supplemental digital content 1, http://links.lww.com/ACD/A105).
We conclude from our in-vitro experiments using CD95L knockdown cells that (i) endogenous CD95L expression is a proinvasive autocrine stimulus for glioma cells, which (ii) can be substituted for by soluble CD95L from paracrine sources in the absence of endogenous CD95 ligand expression, and (iii) the proinvasive function of CD95 depends at least in part on PI3K activity.
Interference with CD95L impairs the migration of glioma cells in murine organotypic brain slices
To analyse tumour cell migration in a context more closely mimicking the environment that glioma cells encounter in vivo, we implanted spheroids derived from U87[SCRMBL], U87[CD95L] and U251[CD95L] cells into organotypic cultures of C57BL/6 mouse brain slices (Fig. 6, left panel, middle panel and right panel, respectively) and followed migration of cells for 4 days as described by Markovic et al. 12, with modifications (for details see the Materials and methods section). To allow quantification of tumour cell migration in the brain slices, additional experiments were conducted which included fixation and optical clearing of the slices 3 days after spheroid inoculation according to the protocol of Ke et al. 13 (for details see Supplementary Methods, Supplemental digital content 1, http://links.lww.com/ACD/A105). Images of one representative experiment are shown in Supplementary Fig. S4 (Supplemental digital content 1, http://links.lww.com/ACD/A105).
Fig. 6.
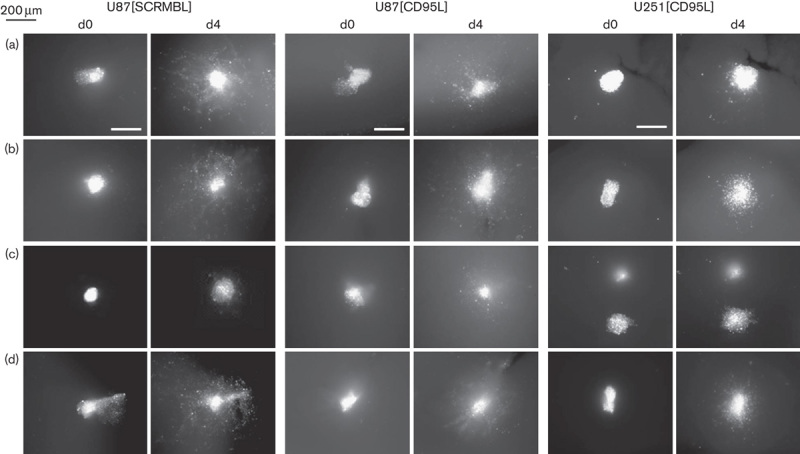
Glioma cell migration in organotypic murine brain slices. Spheroids from DiI-labelled U87[SCRMBL] (left panel), U87[CD95L] (middle panel) and U251[CD95L] cells (right panel) were inoculated into organotypic C57BL/6 mouse brain slices pretreated with (a) control medium or pretreated with medium containing (b) APG293 (10 ng/ml), (c) APG293 (10 ng/ml)+APG101 (50 μg/ml) or (d) APG293 (10 ng/ml)+APG122 (50 μg/ml). Migration of tumour cells was monitored by fluorescence microscopy at the time points indicated. Scale bar=200 μm.
When spheroids derived from U87[SCRMBL] cells were implanted into ex-vivo mouse brain slices, we observed migration of the spheroid cells into the brain tissue (Fig. 6a). When spheroids of U87[SCRMBL] cells were treated with 10 ng/ml APG293 before implantation to mouse brain slices, an additional effect could be observed on migration, whereas there was no significant effect on U251[CD95L] (Fig. 6b, Supplementary Fig. S10, Supplemental digital content 1, http://links.lww.com/ACD/A105). This indicates that endogenous CD95L produced by the U251-MG cells is sufficient for the induction of migration into the brain slice. When APG101 was added in addition to APG293, the invasion of U87[SCRMBL] and U251[SCRMBL] cells into the brain slice was strongly impaired whereas APG122 could not block the effect of APG293 (Fig. 6, left panel, Supplementary Fig. S10, Supplemental digital content 1, http://links.lww.com/ACD/A105). In contrast to the U87[SCRMBL] cells, spheroids derived from knockdown cells U87[CD95L] showed reduced migration into the tissue of the mouse brain slices and this invasion was moderately increased when 10 ng/ml APG293 was added (Fig. 6a and b, middle and right panel, Supplementary Fig. S4, Supplemental digital content 1, http://links.lww.com/ACD/A105 and S10, Supplementary digital content 1, http://links.lww.com/ACD/A105). In contrast to the almost complete lack of invasion observed for [CD95L] knockdown cells in the Matrigel assays, cell migration was not entirely impaired in the mouse brain-slice assay even when left untreated. Consistently, as observed for U87[SCRMBL] cells, the presence of APG101 together with APG293 during pretreatment of the tissue slice before implanting the spheroids from U87[CD95L] and U251[CD95L] cells and throughout the entire observation reduced the migration of the tumour cells into the brain slice (Fig. 6c, Supplementary Fig. S4, Supplemental digital content 1, http://links.lww.com/ACD/A105 and S10, Supplemental digital content 1, http://links.lww.com/ACD/A105), whereas treatment with APG122 was ineffective (Fig. 6d, Supplementary Fig. S4, Supplemental digital content 1, http://links.lww.com/ACD/A105 and S10, Supplemental digital content 1, http://links.lww.com/ACD/A105). In accordance with the effects observed in the in vitro assay, the PI3K-inhibitor LY294002 inhibited migration comparable with APG101 (Supplementary Fig. S4, Supplemental digital content 1, http://links.lww.com/ACD/A105 and S10, Supplemental digital content 1, http://links.lww.com/ACD/A105), corroborating the notion that the ex-vivo assay recapitulates the in-vitro observations in a more complex physiological context. Taken together, the results from the in-vitro and ex-vivo migration analyses confirmed the efficiency of APG101 interference with CD95L to block the migratory behaviour of glioma cells.
Discussion
A hallmark of tumour progression and aggressiveness is fast invasive growth, cell motility and the capability to colonize distant niches, which requires specific survival mechanisms (reviewed in Hanahan and Weinberg 14). There are reports that many tumour cells coexpress CD95 and the CD95 ligand and this is considered to confer increased motility and resistance to CD95-induced apoptosis to these cells 15–17. Glioma cells are well known for their ability to invade the surrounding brain parenchyma, and recently, CD95 signalling has been identified as an important trigger for invasiveness 1,6.
In this preclinical study, we analysed the impact of APG101 on CD95/CD95L-induced invasion. We decided to use two three-dimensional model assays with tumour spheroids derived from the glioblastoma cell lines U87-MG and U251-MG. The Matrigel-based in-vitro spheroid invasion assay and an ex-vivo model of spheroids were implanted on murine organotypic brain tissue cultures. Firstly, to elucidate the contribution of autocrine and paracrine CD95L towards glioma cell invasion, we used semi-stable transfection of shRNA vectors targeting the CD95L gene in U87-MG and U251-MG cells. Knockdown of the CD95L in both cell lines neither had an effect on the overall cell viability nor did it interfere with the ability of the cells to form tumour spheroids. A marginal reduction in the cellular proliferation rate upon CD95L knockdown was observed, an effect that was consistent with the report by Chen et al. 18. Neutralization of endogenous CD95L by APG101 was neither cytotoxic nor did it affect glioma cell proliferation in vitro. The main observed effect of CD95L knockdown was a considerably reduced basal invasiveness of tumour spheroids from U87-MG and U251-MG cells, which became apparent in the in-vitro assays. The addition of a recombinant soluble form of CD95L (APG293) entirely restored invasion of CD95L knockdown cells, indicating that tumour cells that do not express CD95L might also use the ligand from paracrine sources – for example, stromal or immune cells. To further address this question, we decided to use a second ex-vivo assay with murine organotypic brain tissue slices as substrates for spheroid invasion. Murine models have been used extensively during preclinical development of APG101. Compared with the widely established in-vitro transwell assays, the brain-slice model adds several physiological aspects to the system: (i) the presence of cell–cell contacts with noncancer and immune cells in the tissue, (ii) a more complex secretome/microenvironment and cellular context in which the tumour spheroid resides and (iii) morphologic borders directing or inhibiting spheroid outgrowth 11,19. Both experimental models clearly showed the invasive behaviour of the tested glioblastoma-cell-derived tumour spheroids. We observed a stronger residual migration of CD95L knockdown cells in mouse brain slices compared with the migration into Matrigel matrices. This effect is probably because of the presence of endogenous murine CD95L in the brain slices. This also corroborates the notion that CD95L from paracrine sources, in this context cells of the organotypic brain slices, may contribute towards invasion of the tumour cells. Mouse and human CD95L have comparable affinities for human CD95 and, thus, exposure of tumour spheroids to the murine ligand in the brain slices is probably responsible for the residual invasion of [CD95L] knockdown cells. APG101 effectively attenuated the invasive phenotype of the tested cells also in the ex-vivo assay, whereas the CD95L-binding defective control protein APG122 showed no inhibition of cell invasion. We conclude that binding and accompanying neutralization of CD95L is a basis for the pharmacological efficacy of APG101. These findings underline the value of murine organotypic brain slices as a model system to investigate migration of glioma cells in a microenvironment, first more closely related to the situation in vivo and, second, manipulable in terms of treatment with pharmacological inhibitors. Along this line, we also used a small molecule inhibitor of PI3K, which has been described as one intracellular key factor promoting invasion – for example, in glioma cells 1,19. Inhibition of PI3K similarly abrogated invasion induced by exogenous activation of CD95 as did APG101. This raises the question of whether deregulation of PI3K in glioma and other highly invasive tumours can be similarly attenuated by CD95 inhibition through APG101.
In summary, our results have shown that APG101 is an inhibitor of CD95 by sequestering the CD95 ligand in vitro and ex vivo, which markedly reduced the invasiveness of glioma cells. The compound APG101 could neutralize the proinvasive action of CD95L in two different assay systems. Thus, APG101-based inhibition of glioma cell motility very likely represents a mechanism that accounts for the positive effects observed for glioblastoma patients treated with APG101 in the clinical phase II study 7. Evidence exists that APG101 could also provide additional pharmacological effects that might be beneficial for tumour patients. It was shown recently that the CD95/CD95L pathway plays an important immune-modulatory role in the tumour infiltration of cytotoxic T cells 8. Therefore, the overall antitumour effect of APG101 treatment in glioma patients requires further work to evaluate the contribution of these two CD95/CD95L-dependent mechanisms towards the positive effects observed in the clinical phase II study with APG101.
Supplementary Material
Acknowledgements
Conflicts of interest
C.M., A.S., J.S., O.H., H.F. and C.G. are employees of Apogenix GmbH. For the remaining authors there are no conflicts of interest.
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website (www.anti-cancerdrugs.com).
References
- 1.Kleber S, Sancho-Martinez I, Wiestler B, Beisel A, Gieffers C, Hill O, et al. Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell 2008; 13:235–248. [DOI] [PubMed] [Google Scholar]
- 2.Tauzin S, Chaigne-Delalande B, Selva E, Khadra N, Daburon S, Contin-Bordes C, et al. The naturally processed CD95L elicits a c-yes/calcium/PI3K-driven cell migration pathway. PLoS Biol 2011; 9:e1001090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steller EJ, Borel Rinkes IH, Kranenburg O. How CD95 stimulates invasion. Cell Cycle 2011; 10:3857–3862. [DOI] [PubMed] [Google Scholar]
- 4.Krammer PH. CD95’s deadly mission in the immune system. Nature 2000; 407:789–795. [DOI] [PubMed] [Google Scholar]
- 5.Peter ME, Budd RC, Desbarats J, Hedrick SM, Hueber AO, Newell MK, et al. The CD95 receptor: apoptosis revisited. Cell 2007; 129:447–450. [DOI] [PubMed] [Google Scholar]
- 6.Barnhart BC, Legembre P, Pietras E, Bubici C, Franzoso G, Peter ME. CD95 ligand induces motility and invasiveness of apoptosis-resistant tumor cells. EMBO J 2004; 23:3175–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wick W, Fricke H, Junge K, Kobyakov G, Martens T, Heese O, et al. A phase II, randomized, study of weekly APG101+reirradiation versus reirradiation in progressive glioblastoma. Clin Cancer Res 2014; 20:6304–6313. [DOI] [PubMed] [Google Scholar]
- 8.Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med 2014; 20:607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Starling GC, Bajorath J, Emswiler J, Ledbetter JA, Aruffo A, Kiener PA. Identification of amino acid residues important for ligand binding to Fas. J Exp Med 1997; 185:1487–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weiler M, Bähr O, Hohlweg U, Naumann U, Rieger J, Huang H, et al. BCL-xL: time-dependent dissociation between modulation of apoptosis and invasiveness in human malignant glioma cells. Cell Death Differ 2006; 13:1156–1169. [DOI] [PubMed] [Google Scholar]
- 11.Pfenning PN. RGS4, CD95L and B7H3: targeting evasive resistance and the immune privilege of glioblastoma [Dissertation]. 2011; Heidelberg.
- 12.Markovic DS, Glass R, Synowitz M, Rooijen Nv, Kettenmann H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J Neuropathol Exp Neurol 2005; 64:754–762. [DOI] [PubMed] [Google Scholar]
- 13.Ke MT, Fujimoto S, Imai T. SeeDB: a simple and morphology-preserving optical clearing agent for neuronal circuit reconstruction. Nat Neurosci 2013; 16:1154–1161. [DOI] [PubMed] [Google Scholar]
- 14.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646–674. [DOI] [PubMed] [Google Scholar]
- 15.Trauzold A, Röder C, Sipos B, Karsten K, Arlt A, Jiang P, et al. CD95 and TRAF2 promote invasiveness of pancreatic cancer cells. FASEB J 2005; 19:620–622. [DOI] [PubMed] [Google Scholar]
- 16.Malleter M, Tauzin S, Bessede A, Castellano R, Goubard A, Godey F, et al. CD95L cell surface cleavage triggers a prometastatic signaling pathway in triple-negative breast cancer. Cancer Res 2013; 73:6711–6721. [DOI] [PubMed] [Google Scholar]
- 17.Ametller E, García-Recio S, Costamagna D, Mayordomo C, Fernández-Nogueira P, Carbó N, et al. Tumor promoting effects of CD95 signaling in chemoresistant cells. Mol Cancer 2010; 9:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen L, Park SM, Tumanov AV, Hau A, Sawada K, Feig C, et al. CD95 promotes tumour growth. Nature 2010; 465:492–496. Erratum in: Nature 2011; 471 (7337):254. Nature 2011; 475 (7355):254. Nature 2012; 491 (7426):784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miao H, Gale NW, Guo H, Qian J, Petty A, Kaspar J, et al. EphA2 promotes infiltrative invasion of glioma stem cells in vivo through cross-talk with Akt and regulates stem cell properties. Oncogene 2015; 34:558–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.