

Abstract
We report the synthesis, thermal stability, and RNase H substrate activity of 2′-deoxy-2′,4′-difluoroarabino-modified nucleic acids. 2′-Deoxy-2′,4′-difluoroarabinouridine (2,′4′-diF-araU) was prepared in a stereoselective way in six steps from 2′-deoxy-2′-fluoroarabinouridine (2′-F-araU). NMR analysis and quantum mechanical calculations at the nucleoside level reveal that introduction of 4′-fluorine introduces a strong bias toward the North conformation, despite the presence of the 2′-βF, which generally steers the sugar pucker toward the South/East conformation. Incorporation of the novel monomer into DNA results on a neutral to slightly stabilizing thermal effect on DNA–RNA hybrids. Insertion of 2′,4′-diF-araU nucleotides in the DNA strand of a DNA–RNA hybrid decreases the rate of both human and HIV reverse transcriptase-associated RNase H-mediated cleavage of the complement RNA strand compared to that for an all-DNA strand or a DNA strand containing the corresponding 2′-F-araU nucleotide units, consistent with the notion that a 4′-fluorine in 2′-F-araU switches the preferred sugar conformation from DNA-like (South/East) to RNA-like (North).
INTRODUCTION
Antisense oligonucleotides (AONs) and small interfering RNA (siRNA) are both recognized as potential therapeutic agents for the silencing of specific genes at the post-transcriptional level.1,2 At present, dozens of AONs and siRNAs are undergoing human clinical trials, with two AONs and one aptamer having been already clinically approved.3 Given the fact that a majority of these are chemically modified, the search for chemical modifications to improve delivery, stability, efficacy, and specificity of AONs- and siRNA-based drugs is a very active area of research.1,4–6
Modification to the 2′ position of nucleosides provides a remarkable level of control over sugar conformational preferences, a physical property that is intimately related to ON binding affinity toward complementary RNA strands. Sugars that adopt the 3′-endo (North) conformation, such as 2′-deoxy-2′-fluoro-RNA (2′-F-RNA) and 2′-O-alkyl RNA, stabilize DNA–RNA and RNA–RNA duplexes by inducing a conformational preorganization for formation of A-form duplexes.7,8 2′-Deoxy-2′-fluoroarabinonucleic acids (2′-F-ANA) have also attracted much attention. The corresponding monomers have a preference for the 2′-endo (South)/4′-endo (East) conformation in solution (Figure 1A).9 In the context of oligonucleotides, 2′-F-ANA adopts East puckers, allowing the formation of internucleosidic C–H···F–C pseudohydrogen bonds at pyrimidine–purine steps and resulting in the stabilizing effect observed when it is incorporated in the DNA strand of DNA–RNA hybrids.10 Additionally, 2′-F-ANA is one of the handful of analogues that support cleavage of modified duplexes by RNase H, an enzyme that cleaves the mRNA in AON–mRNA hybrids.11,12 The features listed above together with the high nuclease resistance13,14 make 2′-F-ANA a very attractive modification for antisense applications.15,16
Figure 1.

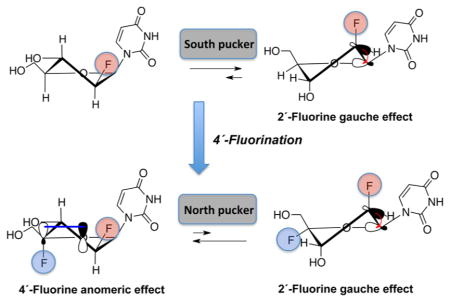
(A) Pseudorotational cycle describing the sugar conformations of nucleosides (E, envelope; T, twist). Superscripts and subscripts indicate the specific atoms in the ribose ring that project away from the plane defined by the remaining ring atoms. Natural nucleosides have characteristic minima in the North (0–36°) and South (144–180°) regions. (B) Anomeric effect (left) favoring the North conformation due to the overlap of a lone-pair orbital O4′(p-type) with the σ*C4′–F4′ antibonding orbital and gauche effect (right) favoring the South conformation due to the interaction between σ*C2′–H2′ bonding orbital and the σ*C1′–O4′ antibonding orbital.
Although the 2′ modification of ONs has been extensively studied, the 4′-substitution has been explored only in the case of 2′,4′-bridged modified nucleic acids.1 Yet, modification to the 4′-position of the furanose sugar can have a profound impact on the conformational preferences and biological properties of 4′-modified nucleos(t)ides and duplexes derived from them. For example, we recently reported the effect of introducing a 4′-fluorine on 2′-deoxy-2′-fluororibouridine.17 The strong gauche and anomeric effects18–21 imparted by both the 2′- and 4′-fluorines, respectively, strongly favor the North conformation, thus locking the sugar in the North region of the pseudorotational cycle. In the case of 2′,4′-difluoroarabinonucleosides, the 2′-fluorine is expected to promote a [O4′–C1′–C2′–F2′] gauche interaction that favors the South sugar conformation, whereas the 4′-fluorine should impart a strong anomeric effect [O4′(p-type)/σ*C4′–F4′] that drives the pseudorotational equilibrium toward the North conformation (Figure 1B). It is of interest to determine how these opposing effects tailor the conformation of 2′,4′-difluoroarabinonucleosides.
Our group has focused on the development of fluorinated nucleoside and nucleic acids encouraged by the conformational changes that fluorine imparts in the ribose and arabinose moiety.9,10,22 As a part of this ongoing program, we chose to place a fluorine at the 4′ position of 2′-F-araU, as this modification was expected to modulate stereolectronic effects on the arabinose sugar moiety. We were especially interested in studying its impact on the sugar pucker, duplex stability, and the susceptibility of DNA–RNA hybrids containing 2′,4′-diF-araU toward RNase H, an enzyme that cleaves the RNA strand of DNA–RNA hybrids. Since RNase H activity is sensitive to subtle structural changes in the sugar–phosphate backbone, any inhibitory effect caused by a nucleotide analogue would suggest incompatible RNaseH–hybrid interaction localized at, or around, the modification. As such, this assay allows for probing DNA-like conformation/flexibility, as previously demonstrated with 2′-F-ANA–RNA hybrids.12,14 Herein, we describe, for the first time, the synthesis, characterization, and conformational analysis of 2′-deoxy-2′,4′-difluoroarauridine (2′,4′-diF-araU). We have also incorporated this novel modification into the DNA strand of DNA–RNA hybrids and studied the stability and susceptibility of such hybrids toward RNase H-mediated RNA cleavage catalyzed by the human enzyme as well as a catalytically active RNase H domain fragment of HIV reverse transcriptase.
RESULTS AND DISCUSSION
We envisaged that the synthetic method developed recently for the preparation of 2′-deoxy-2′,4′-difluororibouridine (2′,4′-diF-rU) could be adapted for the synthesis of its 2′ epimer, namely, 2′,4′-diF-araU (Scheme 1).17 Thus, reaction of commercially available 2′-F-araU (1) with triphenylphosphine and iodine afforded the corresponding 5′-iodo derivative 2 in 93% yield. The 4′,5′-exocyclic double bond in 3 was installed by an elimination reaction of the 5′-iodo with sodium methoxide in 53% yield. In situ generation of iodine fluoride from the reaction of AgF and iodine was found to be an attractive procedure for the introduction of a 4′-fluorine in 4′,5′-unsaturated nucleosides.17,23–26 Indeed, dropwise addition of iodine at 0 °C to a mixture of nucleoside 2 and AgF resulted in the formation of 5′-iodo,4′-fluoro nucleoside 4 with high stereoselectivity in 68% yield. Next, nucleophile substitution of the 5′-iodine was required to obtain the final nucleoside analogue. Because the reactivity of the 5′-iodine is considerably decreased due to the presence of the 4′-fluorine, we placed a 3′-benzoyl group in nucleoside 4 to assist in the substitution reaction.17,25,26 The actual substitution was carried out in the next step by activating the protected 5′-iodo,4′-fluoro analogue 5 to the corresponding hypoiodite with mCPBA in water-saturated dichloromethane with concomitant migration of the benzoyl group from the 3′ to the 5′ position to obtain 5 in 68% yield.17,24 Deprotection of the 5′-O-benzoyl group with methanolic ammonia proceeded in nearly quantitative yields to afford the novel nucleoside monomer 2′,4′-diF-araU (7). The NOESY NMR spectra of 7 show correlations between H6 of the base and the H5′ protons and between H5′ and H3′, thus supporting the desired α-configuration of fluorine at C4′.
Scheme 1.

Synthesis of 2′,4′-diF-araU (7) and the Corresponding Amidite (9)
Next, we embarked on the study of the conformation of 2′,4′-diF-araU (7). In solution, the furanose ring of nucleosides exists in a conformational equilibrium between the North and South puckers. The Karplus equation predicts that a northern conformer of an arabino sugar will have a large 3JH2″H3′ coupling constant, whereas a southern conformer will have a large 3JH1′F2′ value since each H2′/H3′ and H1′/F2′ adopt a nearly antiperiplanar orientation (Figures 1B and 2). Since the 4′-fluorine can affect the magnitudes of 3JH1′H2″ and 3JH2″H3′, thus misleading our analysis,21 their magnitudes were calculated for 2′-F-araU (1) and 2′,4′-diF-ara-U (7) by applying a generalized Karplus equation that takes into account the inductive effect of the 4′-fluorine.27,28 Figure S1 (Supporting Information) shows that the 4′-fluorine has, in fact, no effect on 3JH1′H2″ and 3JH2″H3′ coupling constants. According to the Karplus equation, a JH1′H2″ of ≈7.0 Hz is expected for the North conformers (P = 0–30°) of both 1 and 7 (Figure S1, Supporting Information). Experimental JH1′H2″ for 2′,4′-diF-araU is 6.0 Hz, indicating that the North pucker is highly predominant in solution. However, for 2′-F-araU, JH1′H2″ is 4.0 Hz, suggesting a contribution of Southeastern conformations in which a smaller JH1′H2″ is expected (Figure S1). The experimentally obtained 3JH2″H3′ value of 1 in D2O is 3.0 Hz, whereas that for 7 is 5.5 Hz, indicating a significantly higher North population for the latter (Table 1). A rough estimation of the relative population of North and South conformers can be obtained from theoretical J-coupling values assuming a two-state equilibrium between the two conformers (see Supporting Information for details). In the case of 2′,4′-diF-araU, 1H–1H J-coupling constants are consistent with a ≈ 80% population of the North conformer. In the case of 2′-F-araU, the major conformer is South, with a population of around 60%. Also consistent with this notion is the significantly higher 3JH1′F2′ value (ΔJ = +9.0 Hz) for 7 relative to that for 1. Furthermore, J H3′F4′ is 18.0 Hz (Table 1), consistent with the pseudotrans-diaxial orientation of H3′ and F4′ of a North-puckered nucleoside (Figure 2). As reference, the recently reported North “locked” nucleoside, 2′,4′-diF-rU, has a similar J H3′F4′ value (20.7 Hz) as that of 7 (18.0 Hz). In the South conformer of 1 and 7, H3′ and F4′ are pseudoequatorial and hence a much smaller coupling constant value (JH,F = 4–10 Hz) should be expected (Table 1 and Figure 2).23,29–31
Figure 2.
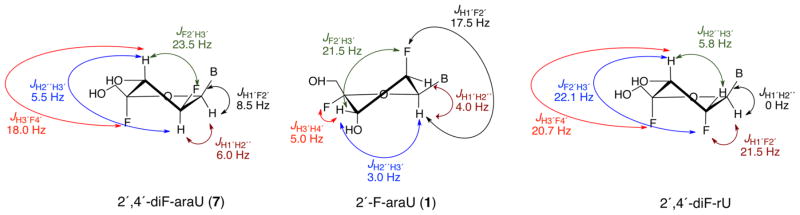
Comparison of sugar ring 1H–1H and 1H–19F coupling constants for 2′,4′-diF-araU (7), 2′-F-araU (1), and 2′,4′-diF-rU represented in the preferred conformation.
Table 1.
1H–1H and 1H–19F Coupling Constant Values for Different Nucleoside Derivatives at 25 °C in D2O (500 MHz)
| J (Hz) (±0.2)a | 2′,4′-diF-araU (7) | 2′-F-araU (1) | 2′,4′-diF-rUc |
|---|---|---|---|
| H1′–H2″b | 6.0 | 4.0 | 0 |
| H1′–F2′ | 8.5 | 17.5 | 21.5 |
| H2″–H3′b | 5.5 | 3.0 | 5.8 |
| H3′–F2′ | 23.5 | 21.5 | 22.1 |
| H3′–X4′ | 18.0 | 5.0 | 20.7 |
X = H for 2′-F-araU; X = F for 2′,4′-diF-araU and 2′,4′-diF-rU.
The proton and the fluorine are labeled as ′′ and ′, respectively, regardless of the relative orientation.
Ref 17.
While this qualitative examination of vicinal 1H–1H and 1H–19F coupling constants is useful, the conclusions can be only approximate because of the rapid interconversion of nucleoside conformers at room temperature. We therefore used quantum mechanical calculations to further explore the impact of 4′-fluorine substitution. We carried out the calculations using Gaussian 09 at the M062x/6-31+G(d,p) level.32 The pseudorotation energetic profiles of 2′,4′-diF-araU, 2′-F-araU, and 2′,4′-diF-rU were calculated by means of a constrained energy optimization (Figure 3A). As previously reported, quantum mechanical calculations indicate a clear preference for the North pucker in 2′,4′-diF-rU with a very high interconversion barrier to the destabilized South conformation (P ≈ 180°).17 For 2′-F-araU, quantum mechanical calculations predict similar energies for the North and South puckers with a low activation barrier.22 The South conformation is shifted toward the East region (P ≈ 140). However, for 2′,4′-diF-araU, the North conformation is clearly the most stable among all possible puckers that can be adopted, and as with 2′,4′-diF-rU, the energy minimum of the South pucker increases significantly relative to the corresponding 2′-F nucleotide. These results match preliminary NMR experiments suggesting that the anomeric effect induced by the 4′-fluorine in 2′,4′-diF-araU is able to overcome the opposing [O4′–C1′–C2′–F2′] gauche effect, hence imparting a bias toward the North pucker. Figure 3B shows the minimized structure for 2′,4′-diF-araU (P = 26°).33
Figure 3.
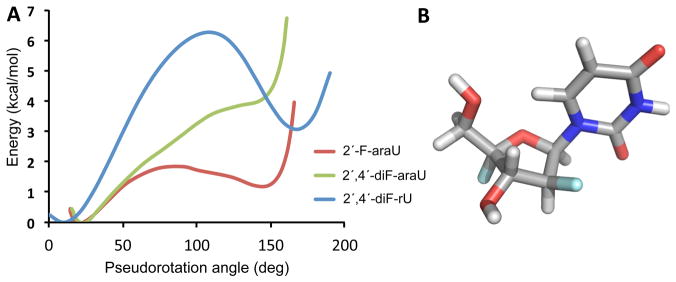
(A) Comparison of the energy profiles of 2′,4′-diF-rU, 2′-F-araU, and 2′,4′-diF-araU. (B) Minimized structure for 2′,4′-diF-araU computed at the M062x/6-31+G(d,p) level using Gaussian 09.
Encouraged by the stabilizing effect observed for 2′-F ONs (2′-F-ANA and 2′-F-RNA) when inserted into oligonucleotide duplexes, we incorporated 2′,4′-diF-araU in the DNA strand of a DNA–RNA duplex. This context is the most relevant for antisense applications in which the AON (a DNA strand) is generally modified to avoid degradation while maintaining a high affinity for the RNA target. With this purpose, we first prepared the 2′,4′-diF-araU phosphoramidite. The 5′-hydroxyl of 7 group was protected using an excess of DMTr chloride to afford 8 in 77% yield. The tritylation reaction proceeded very slowly, probably due to the presence of the electronegative 4′-fluorine, and heating at 40 °C was required. Phosphitylation of tritylated compound 8 using ClP(OCEt)N(iPr)2 in the presence of N,N-diisopropylethylamine gave phosphoramidite 9 in moderate yield and enough purity for standard solid-phase oligonucleotide synthesis (Scheme 1).
To assess the impact 2′,4′-diF-araU on duplex thermal stability, we measured the Tm values of 5′d-GCTTXTTTGCT-3′ (X = 2′,4′-diF-araU, 2′-F-araU, dT) hybridized to an RNA complement (Table 2). This DNA sequence has previously been used to test the impact of other fluorinated modifications on duplex stability.34 For example, it has been reported that CeNA and North-like modifications such as 2′-O-MOE and 2′-F-RNA are destabilizing (ΔTm ≈ −2 °C) when inserted at the same position within this specific heteroduplex.34 Interestingly, in our study, 2′,4′-diF-araU was found to be neutral (A2, ΔTm = −0.1 °C), whereas 2′-F-araU had a small stabilizing effect (A3, ΔTm = +0.6 °C). Given that the 2′,4′-diF-araU nucleoside favors a North pucker, this result was somewhat unexpected. We hypothesize that in the oligonucleotide 2′,4′-diF-araU adopts an alternative backbone conformation (e.g., a different epsilon angle C4′–C3′–O3′–P3′) in a way that minimizes distortions within the hybrid structure as shown here. Preliminary computational studies indicate that, while the North pucker is favored, reorientation of the phosphate linkage (epsilon dihedral angle) may occur as a result of repulsive F4′– O3′ lone-pair interactions (ε = 180–92°). Clearly, high-field NMR analysis on these modified duplexes are needed to gain a better understanding of these effects.
Table 2.
Thermal Stability Measurements of 2′,4′-diF-ANA- and 2′-F-ANA-Modified DNA Strands versus Complementary RNA
| oligomer | modification | sequence (5′–3′)a | Tm (°C)b | ΔTm (°C) | ΔTm/mod (°C) |
|---|---|---|---|---|---|
| A1 | DNA | d(GCGTTTTTTGCT) | 44.1 | ||
| A2 | 2′,4′-diF-araU | d(GCGTTUTTTGCT) | 44.0 | −0.1 | −0.1 |
| A3 | 2′-F-araU | d(GCGTTUTTTGCT) | 44.7 | +0.6 | +0.6 |
| B1 | DNA | d(CUAUAGUAUAC) | 25.2 | ||
| B2 | 2′,4′-diF-araU | d(CUAUAGUAUAC) | 25.8 | +0.6 | +0.15 |
| B3 | 2′-F-araU | d(CUAUAGUAUAC) | 26.8 | +1.6 | +0.4 |
Underlined letter indicates modified residue.
Tm values were measured in 10 mM sodium phosphate buffer (pH 7.2) containing 100 mM NaCl and 0.1 mM EDTA. Sequence of RNA complement: 5′-r(AGCAAAAAACGC)-3′ and 5′-r(GUAUACUAUAG)-3′.
To further study the impact of our modification, we studied another sequence, 5′-d(CUAUAGUAUAC)-3′, and replaced each dU residue with either 2′,4′-diF-araU or 2′-F-araU. In this case, both 2′-F-araU and 2′,4′-diF-araU nucleotides were tolerated, providing small but detectable increases in melting temperatures (Tm = +0.4 °C/mod and +0.15 °C/mod, respectively, Table 2).
The ability of an oligonucleotide to elicit RNase H activity is particularly important since this mechanism potentiates the biological activity of AONs by enabling degradation of the mRNA portion of the AON–mRNA duplex. The subsequent release of the AON allows it to bind to another mRNA molecule to continue the degradation process. Although RNase H cleaves only the RNA strand of the hybrid, this action is very much dependent on the nature of the bound AON strand. RNase H acts upon DNA–RNA hybrids (the native substrate in vivo), but it will not degrade double-stranded RNA. Minor groove dimensions of the hybrid, as well as the conformation and flexibility of the AON, are key determinants in the activation of RNase H.12,35–37 RNA mimics (e.g., North-like 2′-O-alkyl-RNA, 2′-F-RNA) produce duplexes that are too similar to the RNA–RNA duplex and hence are not cleaved by the enzyme.
We have previously shown that RNase H can cleave an oligo-RNA strand that is hybridized to a complementary 2′-F-ANA or a chimeric 2′-F-ANA–DNA strand.38 To determine if chimeric 2,′4-diF-ANA–DNA oligonucleotides can elicit RNase H degradation of target RNA, an assay comprising [32P]-target RNA and test oligonucleotide was carried out with a slight excess of human and HIV-RT RNase H. The corresponding native and 2′-F-ANA-modified oligonucleotides were studied for comparison. The reaction products were resolved by electrophoresis and visualized by autoradiography. The results of such experiments are shown in Figure 4A.
Figure 4.

Ribonuclease H degradation of DNA (B1) and 2′-F-ANA (B2) and 2′,4′-diF-ANA (B3) modified hybrids. An 11 nt 5′-32P labeled target RNA 5′-r(GUAUACUAUAG)-3′ was preincubated with complementary B1–B3 and then added to reaction assays containing either human RNase H (A) or HIV-RT-associated RNase H (B). PAGE image shows representative points for each assay. Aliquots were removed as listed in the figure (in minutes). Reactions were conducted at room temperature and contained 20 nM of each duplex, 200 nM recombinant human RNase H or the catalytically active RNase H domain fragment of HIV RT, and 5 mM MgCl2 (human RNase H) or 5 mM MnCl2 (HIV RT RNase H domain). (C) Schematic representation of the RNase H-mediated cleavage at positions 1 and 2 of the RNA strand.
The data show that all three oligonucleotides are able to form hybrids with target RNA that serve as substrates for human RNase H, as indicated by the smaller sized degradation products of the target RNA in Figure 4A. The overall efficiency of RNA cleavage observed for the 2′,4′-diF-ANA-modified oligomer is reduced relative to that of unmodified DNA and the corresponding 2′-F-ANA-modified strand. The ability of the three hybrids to trigger human RNase H activity followed the order DNA > 2′-F-ANA > 2′,4′-diF-ANA. The locations of cleavage sites of the RNA strand for the 2′,4′-diF-ANA duplex differ from those of the DNA and 2′-F-ANA duplexes (Figure 4A,C). The same trend was observed with the HIV-RT RNase H enzyme; however, in this case, negligible cleavage was observed for the 2′,4′-diF-ANA–RNA hybrid (Figure 4B). The different rates and cleavage patterns suggest structural variations among the various hybrids and are consistent with the introduction of North/South junctions along the antisense ON strand.
CONCLUSIONS
Introduction of a 4′-fluorine in 2′-F-araU switches the preferred sugar conformation from South to North due to a strong anomeric effect, as assessed by NMR and quantum mechanical calculations. The novel 2′,4′-diF-araU modification is well-tolerated when placed in the DNA strand of DNA–RNA hybrids, resulting in a neutral or slightly stabilizing thermal effect. RNase H recruitment studies of a 2′,4′-diF-ANA-RNA modified duplex has been carried out and compared with native DNA and 2′-F-araU. The differences in the cleavage sites and the lower degradation rates of the RNA strand observed for the 2′,4′-diF-ANA duplex support structural changes in the hybrid structure induced by the North pucker adopted by the novel modification. The results presented here warrant further studies on 2′,4′-diF-ANA for gene silencing applications.
EXPERIMENTAL SECTION
Procedures and Experimental Data
1-(2-Deoxy-2-fluoro-5-iodo-β-D-arabinofuranosyl)uracil (2)
Iodine (1.92 g, 7.56 mmol) and triphenylphosphine (2.15 g, 8.19 mmol) were added to a suspension of 1 (1.55 g, 6.30 mmol) in pyridine (6.6 mL) and anhydrous CH3CN (120 mL). After being stirred at room temperature for 48 h, solvents were concentrated under vacuum, and the residue was purified by column chromatography (1–4% MeOH in CH2Cl2) to give 2 as a pale yellow solid (2.1 g, 93%). Rf (10% MeOH/CH2Cl2): 0.41. 1H NMR (MeOH-d4, 500 MHz): δ 3.49 (m, 2H), 4.02 (dt, 1H, JHH = 3.5 Hz, JHH = 6.0 Hz), 4.32 (ddd, 1H, JHH = 2.0 Hz, JHH 3.5 Hz, JHF = 17.5 Hz), 5.05 (ddd, 1H, JHH = 2.0 Hz, JHH = 3.5 Hz, JHF = 17.5 Hz), 5.74 (d, 1H, H-5, JHH = 8.0 Hz), 6.23 (dd, 1H, JHH = 3.5 Hz, JHF = 20.0 Hz), 7.73 (dd, 1H, JHH = 2.0 Hz, JHH = 8.5 Hz). 13C NMR (MeOH-d4, 125 MHz): δ 3.0, 77.0 (d, JCF = 25.0 Hz), 83.8, 84.9 (d, JCF = 16.4 Hz), 95.1 (d, JCF = 189.8 Hz), 100.7, 141.6, 150.3, 164.5. HRMS (ESI+) m/z: calcd for C9H10FIN2NaO4 [M + Na]+, 378.9562; found, 378.9560.
1-(2,5-Dideoxy-2-fluoro-β-D-4-enoarabinofuranosyl)uracil (3)
A commercially available solution of 25% sodium methoxide in MeOH (6.1 mL, 28.2 mmol) was added to a suspension of compound 2 (2.1 g, 5.90 mmol) in anhydrous MeOH (53 mL). The reaction mixture was stirred at reflux for 24 h. MeOH was evaporated, and the residue was filtered over a small bed of silica gel using 10% MeOH/CH2Cl2 as eluent to remove the salts. The obtained solid was then purified by column chromatography (4% MeOH/CH2Cl2) to afford 3 as a white solid (0.72 g, 53%). Rf (10% MeOH/CH2Cl2): 0.38. 1H NMR (MeOH-d4, 500 MHz): δ 4.49 (d, 1H, JHH = 2.5 Hz), 4.60 (m, 2H), 5.06 (dd, 1H, JHH = 2.0 Hz, JHH = 3.0 Hz, JHF = 51.0 Hz), 5.74 (d, 1H, JHH = 8.0 Hz), 6.53 (dd, 1H, JHH = 3.0 Hz, JHF = 19.5 Hz), 7.49 (dd, 1H, JHH = 2.0 Hz, JHH = 8.1 Hz). 13C NMR (MeOH-d4, 125 MHz): δ 72.8 (d, JCF = 27.5 Hz), 85.3 (d, JCF = 15.9 Hz), 86.8, 93.2 (d, JCF = 191.3 Hz), 101.1, 140.9, 150.3, 160.2, 164.4. HRMS (ESI+) m/z: calcd for C9H9FN2NaO4 [M + Na]+, 251.0439; found, 251.0449.
1-(2-Deoxy-2,4-difluoro-5-iodo-β-D-arabinofuranosyl)uracil (4)
A suspension of alkene 3 (253 mg, 1.11 mmol) and silver fluoride (4.43 mmol, 563 mg) in MeCN (15 mL) was vigorously stirred at 0 °C while a solution of iodine (563 mg, 2.22 mmol) in MeCN (9 mL) was added over 15 min. After completion, the reaction mixture was directly filtered over a small bed of silica gel using 50% MeOH/CH2Cl2 as eluent to remove the Ag salts. Fractions containing the product were collected, solvents were evaporated, and the resulting residue was purified by column chromatography (2% MeOH/CH2Cl2) to afford 4 as a white solid (283 mg, 68%). Rf (10% MeOH/CH2Cl2): 0.44. 1H NMR (MeOH-d4, 300 MHz): δ 3.64 (d, 1H, JHH = 7.5 Hz), 3.69 (s, 1H), 4.69 (ddd, 1H, JHH = 5.1 Hz, JHF = 16.8 Hz, JHF = 21.9 Hz), 5.29 (dt, 1H, JHH = 5.7 Hz, JHF = 54.0 Hz), 5.74 (d, 1H, JHH = 8.1 Hz), 5.89 (dd, 1H, JHH = 5.7 Hz, JHF = 9.9 Hz), 7.60 (dd, 1H, JHF = 1.8 Hz, JHH = 8.1 Hz). 13C NMR (MeOH-d4, 125 MHz): δ −0.05 (d, JCF = 32.6 Hz), 76.1 (dd, JCF = 22.7, JCF = 24.9 Hz), 82.5 (d, JCF = 16.9 Hz), 94.3 (d, JCF = 193.8 Hz), 101.6, 113.2 (d, JCF = 229.3 Hz), 141.5, 150.3, 164.2. HRMS (ESI+) m/z: calcd for C9H9F2IN2NaO4 [M + Na]+, 396.9467; found, 396.9465.
1-(3-O-Benzoyl-2-deoxy-2,4-difluoro-5-iodo-β-D-arabinofuranosyl) uracil (5)
Benzoyl chloride (82 μL, 0.705 mmol) was added dropwise to a solution of compound 4 (240 mg, 0.64 mmol), triethylamine (447 μL, 3.20 mmol), and DMAP (0.5 mg, 0.004 mmol) in THF (16 mL). The reaction was stirred for 15 min at room temperature and was then quenched by addition of 3 mL of MeOH. Solvents were evaporated, and the residue obtained was purified by column chromatography (40% AcOEt/hexanes) to give benzoylated compound 5 (242 mg, 79%) as a white solid. Rf (70% AcOEt/Hexanes): 0.57. 1H NMR (acetone-d6, 300 MHz): δ 3.93 (d, 1H, H-5′, JHH = 4.5 Hz), 3.96 (s, 1H), 5.79 (d, 1H, JHH = 8.5 Hz), 5.91 (dt, 1H, JHH = 4.0 Hz, JHH = 5.5 Hz, JHF = 52.5 Hz), 6.19 (ddd, 1H, JHH = 3.4 Hz, JHF = 14.5 Hz, JHF = 22.5 Hz), 6.69 (dd, 1H, JHH = 5.5 Hz, JHF = 12.5 Hz), 7.58 (t, 2H, JHH = 8.0 Hz), 7.72 (t, 1H, JHH = 7.7 Hz), 7.78 (dd, 1H, JHF = 1.8, Hz, JHH = 8.1 Hz), 8.13 (m, 2H). 13C NMR (acetone-d6, 125 MHz): δ 3.2 (d, C-5′, JCF = 32.0 Hz), 78.1 (dd, JCF = 20.8, JCF = 27.6 Hz), 84.9 (m), 94.3 (d, JCF = 195.0 Hz), 103.6, 114.4 (d, JCF = 232.3 Hz), 129.9, 131.2, 135.3, 142.8, 151.4, 163.4, 166.2. HRMS (ESI+) m/z: calcd for C16H13F2IN2NaO5 [M + Na]+, 500.9729; found, 500.9714.
1-(5-O-Benzoyl-2-deoxy-2,4-difluoro-β-D-arabinofuranosyl)uracil (6)
3-Chloroperoxybenzoic acid (mCPBA) (77% purity, 523 mg, 2.33 mmol of mCPBA) was added to a suspension of 5 (282 mg, 0.59 mmol) in CH2Cl2 (16 mL) and H2O (0.6 mL). The mixture was heated at 40 °C for 5 h, after which time solvents were concentrated and the residue was purified by flash column chromatography (40% AcOEt/hexanes) to afford 6 (148 mg, 68% yield) as a white solid. Rf (70% AcOEt/Hexanes): 0.40. 1H NMR (MeCN-d3, 500 MHz): δ 4.64 (dd, 1H, H-5′, JHF = 8.5 Hz, JHH = 12.0 Hz), 4.68 (dd, 1H, JHF = 10.0 Hz, JHH = 12.5 Hz), 4.78 (ddd, 1H, JHH = 5.0 Hz, JHF = 16.5 Hz, JHF = 21.5 Hz), 5.36 (dt, 1H, JHH = 5.5 Hz, JHF = 53.5 Hz), 5.58 (dd, 1H, JHH = 8.0 Hz), 6.53 (dd, 1H, JHH = 6.0 Hz, JHF = 9.5 Hz), 7.39 (dd, 1H, JHF 1.5 Hz, JHH = 8.1 Hz,), 7.55 (t, 2H, JHH = 8.0 Hz), 7.68 (t, 1H, JHH = 7.7 Hz), 8.08 (m, 2H), 9.45 (br s, 1H). 13C NMR (MeCN-d3, 125 MHz): δ 61.7 (d, JCF = 39.4 Hz), 75.1 (dd, JCF = 21.3, JCF = 24.8 Hz), 82.9 (m), 93.6 (d, JCF = 193.1 Hz), 102.1, 113.3 (dd, JCF = 11.0 Hz, JCF = 229.0 Hz), 128.8, 130.3, 129.6, 133.0, 140.8, 150.0, 162.8, 165.4 (Bz). HRMS (ESI+) m/z: calcd for C16H14F2N2NaO6 [M + Na]+, 391.0712; found, 391.0696.
1-(2-Deoxy-2,4-difluoro-β-D-arabinofuranosyl)uracil (7)
Protected nucleoside 6 (148 mg, 0.402 mmol) was treated with 2 M NH3 in MeOH (6 mL), and the mixture was stirred overnight at room temperature and then evaporated to dryness under reduced pressure. Purification by column chromatography (1–10% MeOH/CH2Cl2) gave 7 (106 mg, 99%) as a white solid. Rf (10% MeOH/CH2Cl2): 0.22. 1H NMR (D2O, 500 MHz): δ 3.81 (m, 2H, H-5′), 4.64 (ddd, 1H, H-3′, JHH = 5.5 Hz, JHF = 18.0 Hz, JHF = 23.5 Hz), 5.37 (dt, 1H, H-2′, JHH = 6.0 Hz, JHF = 53.0 Hz), 5.81 (d, 1H, H-5, JHH = 8.0 Hz), 6.49 (d, 1H, H-1′, JHH = 6.0 Hz, JHF = 8.5 Hz), 7.57 (dd, 1H, H-6, JHF = 1.5 Hz, JHH = 8.0 Hz). 13C NMR (MeOH-d4, 125 MHz): δ 59.0 (d, JCF = 40.5 Hz), 73.2 (dd, JCF = 21.5, JCF = 24.2 Hz), 81.9 (d, JCF = 17.3 Hz), 93.8 (d, JCF = 193.4 Hz), 101.4, 115.1 (dd, JCF = 11.6 Hz, JCF = 228.9 Hz), 140.7, 150.5, 164.3. 19F NMR (D2O, 470.35 MHz) δ −121.8 (m, F-4′), −200.9 (ddd, F-2′, JHF = 8.0 Hz, JHF = 23.5 Hz, JHF = 53.1 Hz). HRMS (ESI+) m/z: calcd for C9H10F2N2NaO5 [M + Na]+, 287.0450; found, 287.0448.
1-(2-Deoxy-2,4-difluoro-5′-[4,4′-dimethoxytrityl]-arabinofuranosyl) uracil (8)
Nucleoside 7 (74 mg, 0.28 mmol) and 4,4′-dimethoxytrityl chloride (143 mg, 0.42 mmol) were dried overnight under vacuum. Pyridine (2 mL) and DMAP (catalytic) were added under nitrogen. The reaction was stirred for 2 h at 40 °C. Pyridine was evaporated under vacuum, and the resulting residue purified by column chromatography (1–3% MeOH/CH2Cl2) to afford tritylated nucleoside 8 as a white solid (122 mg, 77%). Rf (10% MeOH/CH2Cl2): 0.38. 1H NMR (MeOH-d4, 500 MHz): δ 3.43 (d, 1H, JHF = 3.0 Hz, JHH = 10.0 Hz), 3.56 (d, 1H, JHF = 3.5 Hz, JHH = 10.0 Hz), 4.78 (ddd, 1H, JHH = 6.5 Hz, JHF = 18.5 Hz, JHF = 24.0 Hz), 5.35 (td, 1H, JHH = 6.0 Hz, JHF = 54.0 Hz), 5.36 (d, 1H, JHH = 8.5 Hz), 6.55 (t, 1H, JHH/F = 6.0 Hz), 6.90 (m, 4H), 7.27–7.41 (m, 9H), 7.77 (d, 1H, JHH = 8.0 Hz). 13C NMR (MeCN-d3, 125 MHz): δ 54.9 (OMe), 60.8 (d, JCF 43.3 Hz), 69.7 (m), 88.3 (d, JCF 221.7 Hz), 92.2 (m), 102.2, 113.2, 117.1 (d, C-4′, JCF 232.5 Hz), 127.1–135.1 (DMTr), 141.8 (C-6), 144.4 (DMTr), 158.9 (DMTr), 149.9 (C-2), 162.8 (C-4). HRMS (ESI+) m/z: calcd for C30H28F2N2NaO7 [M + Na]+, 589.1757; found, 589.1750.
1-(3-[2-Cyanoethoxy(diisopropylamino)-phosphinyl]-2-deoxy-2,4-difluoro-5-[4,4′-dimethoxytrityl)]-β-D-arabinofuranosyl)uracil) (9)
Tritylated nucleoside 8 (111 mg, 0.196 mmol), was dried under vacuum overnight and coevaporated with MeCN four times. Dry THF (2 mL) was added under N2. To the solution was added EtN(iPr)2 (127 mg, 0.98 mmol) and then ClPOCEtN(iPr)2 (51 mg, 0.216 mmol). The reaction mixture was stirred for 2 h at room temperature. The reaction progress was monitored by TLC (Et3N/CH2Cl2/eter, 2:48:50). After the reaction reached completion, the mixture was directly loaded for purification on a silica gel column chromatography (eluent: 40% EtOAc, 60% hexanes) to afford phosphoramidite 9 (76 mg, 50%). The mixture of two diastereomers at phosphorus led to complex 1H and 13C NMR spectra. 31P NMR (81 MHz, acetone-d6): δ 151.8 (d, JPF = 5.6 Hz), 152.1 (d, JPF = 6.2 Hz). HRMS (ESI+) m/z: calcd for C39H45F2N4NaO8P [M + Na]+, 789.2835; found, 789.2851.
Procedure for the RNase H Assays
Recombinant human RNase H1 in pBAD-His plasmid was expressed in BL21 Escherichia coli and purified by affinity chromatography followed by gel permeation. The catalytically active RNase H domain fragment of HIV-1 RT was expressed from plasmid pCSR231 (a generous gift from Dr. Daria Hazuda, Merck, West Point, PA) and purified as previously described.39 RNA template was 5′-radiolabeled with γ-P32-ATP (PerkinElmer) using T4 polynucleotide kinase and annealed with either DNA (B1), 2-F-araU (B2), or 2′,4′-diF-araU (B3) modified substrate. RNase H hydrolysis reactions were conducted at room temperature in 50 mM Tris-HCl, pH 8.0, and 50 mM KCl buffer with 20 nM each duplex and 100 nM enzyme. Reaction was started by adding 5 mM MgCl2 and quenched at different time points by 95% formamide and 10 mM EDTA, pH 8.0, with a trace amount of bromophenol blue dye. Products of reactions were separated using 20% denaturing PAGE and analyzed by phosphorimaging.
Oligonucleotide Synthesis
Standard phosphoramidite solid-phase synthesis conditions were used for the synthesis of all modification and unmodified oligonucleotides on a DNA synthesizer. Each oligonucleotide was synthesized at the 1 μmole scale, using Unylinker CPG as solid support. DNA phosphoramidites were prepared as 0.1 M solutions in acetonitrile, RNA phosphoramidites, as 0.15 M solutions in ACN, and 2′-F-araU phosphoramidite, as 0.15 M in ACN. 2′,4′diF-araU phosphoramidite was prepared as a 0.08 M solution in acetonitrile. Significant vortexing and sonication were required to fully dissolve the compound. 5-Ethylthiotetrazole was used as activator, 3% trichloroacetic acid in dichloromethane was used to detritylate, acetic anhydride in tetrahydrofuran (THF) and 16% N-methylimidazole in THF was used to cap, and 0.1 M I2 in 1:2:10 pyridine/water/THF was used for oxidation. DNA was coupled for 110 s (270 s for G); all other phosphoramidites were coupled for 600 s (900 s for G). 2′,4′-diF-araU phosphoramidite was coupled for 1200 s. Deprotection and cleavage from the solid support was achieved with 3:1 aqueous NH4OH/EtOH for 48 h at room temperature. After decanting to remove the CPG, the deprotection solution was removed under vacuum in a SpeedVac lyophilizer. For RNA-containing oligonucleotides, desilylation was achieved in neat TREAT-HF (150 μL) with shaking at room temperature for 48 h. Purifications were performed by HPLC, using a Protein Pak DEAE 5PW analytical anion-exchange column. A stationary phase of Milli-Q water and a mobile phase of 1 M LiClO4 in water was used for analysis and purification using a gradient of 0–50% over 46 min. Following purification, excess LiClO4 salts were removed using NAP-25 sephadex size-exclusion columns. Oligonucleotides were quantitated by UV (extinction coefficients were determined using the IDT OligoAnalyzer tool (www.idtdna.com/analyzer/Applications/OligoAnalyzer). Extinction coefficients for DNA and RNA were used for oligonucleotides containing 2′-F-araU and 2,′4′-diF-araU inserts, respectively.
Thermal Denaturation Studies
Equimolar amounts of complementary sequences were combined, dried, and rediluted in 10 mM sodium phosphate buffer (pH 7.2) containing 100 mM NaCl and 0.1 mM EDTA (1 mL). They were then transferred into cold cuvettes in a UV spectrophotometer. The samples were heated at 90 °C and then cooled to 5 °C. The change in absorbance at 260 nm was then monitored upon heating from 5 to 90 °C. Melting temperatures were determined using the hyperchromicity method.
Supplementary Material
Acknowledgments
Financial support was provided by the Natural Sciences and Engineering Research Council of Canada (M.J.D), the McGill CIHR Drug Development Training Program (S.M.-M.), a Vanier NSERC Scholarship (G.F.D.), and NIH grants AI100890 and GM103368 (M.A.P.).
Footnotes
Notes
The authors declare no competing financial interest.
1H, 13C, some 19F, and 2D NMR spectra for compounds 2–9. Additional computational data. Thermal denaturation curves. Analytical data for oligonucleotides. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Deleavey GF, Damha MJ. Chem Biol. 2012;19:937–954. doi: 10.1016/j.chembiol.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 2.Watts JK, Corey DR. J Pathol. 2012;226:365–379. doi: 10.1002/path.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGowan MP, Tardif JC, Ceska R, Burgess LJ, Soran H, Gouni-Berthold I, Wagener G, Chasan-Taber S. Plos One. 2012;7:e49006. doi: 10.1371/journal.pone.0049006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bramse JB, Grünweller A, Hartmann RK, Kjems J. Handbook of RNA Biochemistry. 2. Wiley-VCH; Weinheim, Germany: 2014. Using chemical modification to enhance siRNA performance; pp. 1243–1278. [Google Scholar]
- 5.Burnett JC, Rossi JJ. Chem Biol. 2012;19:60–71. doi: 10.1016/j.chembiol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma VK, Sharma RK, Singh SK. Med Chem Commum. 2014;5:1454–1471. [Google Scholar]
- 7.Kawasaki AM, Casper MD, Freier SM, Lesnik EA, Zounes MC, Cummins LL, Gonzalez C, Cook PD. J Med Chem. 1993;36:831–841. doi: 10.1021/jm00059a007. [DOI] [PubMed] [Google Scholar]
- 8.Williams DM, Benseler F, Eckstein F. Biochemistry. 1991;30:4001–4009. doi: 10.1021/bi00230a027. [DOI] [PubMed] [Google Scholar]
- 9.Watts JK, Sadalapure K, Choubdar N, Pinto BM, Damha MJ. J Org Chem. 2006;71:921–925. doi: 10.1021/jo051844+. [DOI] [PubMed] [Google Scholar]
- 10.Anzahaee MY, Watts JK, Alla NR, Nicholson AW, Damha MJ. J Am Chem Soc. 2011;133:728–731. doi: 10.1021/ja109817p. [DOI] [PubMed] [Google Scholar]
- 11.Damha MJ, Wilds CJ, Noronha A, Brukner I, Borkow G, Arion D, Parniak MA. J Am Chem Soc. 1998;120:13545–13545. [Google Scholar]
- 12.Mangos MM, Min KL, Viazovkina E, Galarneau A, Elzagheid MI, Parniak MA, Damha MJ. J Am Chem Soc. 2003;125:654–661. doi: 10.1021/ja025557o. [DOI] [PubMed] [Google Scholar]
- 13.Kois P, Tocik Z, Spassova M, Ren WY, Rosenberg I, Soler JF, Watanabe KA. Nucleosides Nucleotides. 1993;12:1093–1109. [Google Scholar]
- 14.Watts JK, Martin-Pintado N, Gomez-Pinto I, Schwartzentruber J, Portella G, Orozco M, Gonzalez C, Damha MJ. Nucleic Acids Res. 2010;38:2498–2511. doi: 10.1093/nar/gkp1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dudley A, Sater M, Le PU, Trinh G, Sadr MS, Bergeron J, Deleavey GF, Bedell B, Damha MJ, Petrecca K. Oncogene. 2014;33:4952–4960. doi: 10.1038/onc.2013.436. [DOI] [PubMed] [Google Scholar]
- 16.Fortin M, D’Anjou H, Higgins ME, Gougeon J, Aube P, Moktefi K, Mouissi S, Seguin S, Seguin R, Renzi PM, Paquet L, Ferrari N. Resp Res. 2009;10:39. doi: 10.1186/1465-9921-10-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinez-Montero S, Deleavey GF, Kulkarni A, Martin-Pintado N, Lindovska P, Thomson M, Gonzalez C, Gotte M, Damha MJ. J Org Chem. 2014;79:5627–5635. doi: 10.1021/jo500794v. [DOI] [PubMed] [Google Scholar]
- 18.Thibaudeau C, Acharya P, Chattopadhyaya J. Stereoectronic Effects in Nucleosides and Nucleotides and Their Structural Implications. 2. Uppsala University Press; Uppsala, Sweden: 2005. [Google Scholar]
- 19.Altona C, Sundaralingam M. J Am Chem Soc. 1973;95:2333–2344. doi: 10.1021/ja00788a038. [DOI] [PubMed] [Google Scholar]
- 20.de Leeuw FAAM, Altona C. J Chem Soc, Perkin Trans 2. 1982;375–384 [Google Scholar]
- 21.Thibaudeau C, Plavec J, Chattopadhyaya J. J Org Chem. 1998;63:4967–4984. [Google Scholar]
- 22.Martin-Pintado N, Deleavey GF, Portella G, Campos-Olivas R, Orozco M, Damha MJ, Gonzalez C. Angew Chem, Int Ed. 2013;52:12065–12068. doi: 10.1002/anie.201305710. [DOI] [PubMed] [Google Scholar]
- 23.Jenkins ID, Verheyden JPH, Moffatt JG. J Am Chem Soc. 1976;98:3346–3357. doi: 10.1021/ja00427a049. [DOI] [PubMed] [Google Scholar]
- 24.Ivanov MA, Ludva GS, Mukovnya AV, Kochetkov SN, Tunitskaya VL, Alexandrova LA. Russ J Bioorg Chem. 2010;36:488–496. [Google Scholar]
- 25.Guillerm D, Muzard M, Allart B, Guillerm G. Bioorg Med Chem Lett. 1995;5:1455–1460. [Google Scholar]
- 26.Owen GR, Verheyden JPH, Moffatt JG. J Org Chem. 1976;41:3010–3017. doi: 10.1021/jo00880a018. [DOI] [PubMed] [Google Scholar]
- 27.Haasnoot CAG, de Leeuw FAAM, Altona C. Tetrahedron. 1980;36:2783–2792. [Google Scholar]
- 28.de Leeuw FAAM, Van Beuzekom AA, Altona C. J Comput Chem. 1983;4:438–448. [Google Scholar]
- 29.Hall LH, Steiner PR, Pedersen C. Can J Chem. 1970;48:1155–1165. [Google Scholar]
- 30.Hall LH, Steiner PR, Pedersen C. Can J Chem. 1970;48:3937–3945. [Google Scholar]
- 31.Inch TD. Annu Rep NMR Spectrosc. 1969;2:35–82. [Google Scholar]
- 32.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09. Gaussian, Inc; Wallingford, CT: 2009. [Google Scholar]
- 33.The pseudorotation angle for the minimized structure of 2′,4′-diF-araU was calculated using the online tool for pseudorotational analysis Prosit: http://cactus.nci.nih.gov/prosit/.
- 34.Seth PP, Yu JH, Jazayeri A, Pallan PS, Allerson CR, Ostergaard ME, Liu FW, Herdewijn P, Egli M, Swayze EE. J Org Chem. 2012;77:5074–5085. doi: 10.1021/jo300594b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noy A, Luque FJ, Orozco M. J Am Chem Soc. 2008;130:3486–3496. doi: 10.1021/ja076734u. [DOI] [PubMed] [Google Scholar]
- 36.Noy A, Perez A, Marquez M, Luque FJ, Orozco M. J Am Chem Soc. 2005;127:4910–4920. doi: 10.1021/ja043293v. [DOI] [PubMed] [Google Scholar]
- 37.Mangos MM, Damha MJ. Curr Top Med Chem. 2002;2:1145–1169. doi: 10.2174/1568026023393110. [DOI] [PubMed] [Google Scholar]
- 38.Min KL, Viazovkina E, Galarneau A, Parniak MA, Damha MJ. Bioorg Med Chem Lett. 2002;12:2651–2654. doi: 10.1016/s0960-894x(02)00439-0. [DOI] [PubMed] [Google Scholar]
- 39.Gong QG, Menon L, Ilina T, Miller LG, Ahn J, Parniak MA, Ishima R. Chem Biol Drug Des. 2011;77:39–47. doi: 10.1111/j.1747-0285.2010.01052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.